

Abstract
Amyloid proteins aggregate into polymorphic fibrils that damage tissues of the brain, nerves, and heart. Experimental and computational studies have examined the structural basis and the nucleation of short fibrils, but the ability to predict and precisely quantify the stability of larger aggregates has remained elusive. We established a complete classification of fibril shapes and developed a tool called CreateFibril to build such complex, polymorphic, modular structures automatically. We applied stability landscapes, a technique we developed to reveal reliable fibril structural parameters, to assess fibril stability. CreateFibril constructed HET-s, Aβ, and amylin fibrils up to 17 nm in length, and utilized a novel dipolar solvent model that captured the effect of dipole-dipole interactions between water and very large molecular systems to assess their aqueous stability. Our results validate experimental data for HET-s and Aβ, and suggest novel (to our knowledge) findings for amylin. In particular, we predicted the correct structural parameters (rotation angles, packing distances, hydrogen bond lengths, and helical pitches) for the one and three predominant HET-s protofilaments. We reveal and structurally characterize all known Aβ polymorphic fibrils, including structures recently classified as wrapped fibrils. Finally, we elucidate the predominant amylin fibrils and assert that native amylin is more stable than its amyloid form. CreateFibril and a database of all stable polymorphic fibril models we tested, along with their structural energy landscapes, are available at http://amyloid.cs.mcgill.ca.
Introduction
Amyloid proteins are believed to be associated in either partial causality or complete aggravation with the severity of neurodegenerative diseases such as Alzheimer’s, Parkinson’s, Huntington’s, and Type II diabetes (1,2). These misfolded proteins form stable aggregates, known as fibrils, that damage tissues of the brain, nerves, and heart, leading to symptoms of severe memory loss, deterioration of cortical neurons, fatigue, muscular rigidity, and depression (3–8). Apart from their common central cross-β spinal core (9), fibrils assemble in polymorphic structures and pack in several orientations, giving rise to different biological functions (10,11) and toxicity levels in cells (12,13). Studies have shown that fibrils are capable of propagating their specific polymorphisms to daughter fibrils (14,15) to preserve their destructive potential. Moreover, it has been hypothesized that proteins of all kinds can self-assemble into amyloid fibrils under optimal conditions (16). Some of the best-known fibrils were observed in cryo-transmission electron microscopy (cryo-TEM) reconstruction of insulin fibrils (17), TEM analysis of amylin (18), cryo-EM analysis of HET-s (12), and TEM analysis of Aβ (19).
Recent experimental studies were able to describe with high atomic resolution the molecular structures of HET-s and Aβ fibrils (20,21), but to date, computational modeling and simulation studies have been limited to analysis of the nucleation phase, which involves only a few polypeptide chains (22–24). It follows that our ability to predict and understand the aggregation process remains limited. More importantly, although the importance of water for forming and stabilizing fibrils is widely acknowledged, current simulation frameworks are restricted to implicit solvent models with limited performance. Computational simulation of amyloid fibrils is challenged by issues involving both scalability and complexity. The ability to model and precisely quantify the stability of the molecular structure of polymorphic amyloid fibrils is of key importance for understanding the toxicity potential and the self-propagation mechanisms of these proteins.
In this work, we used CreateFibril, a computational framework we developed, to build polymorphic fibrils of amyloid proteins and explore their stability by means of stability landscapes. We developed these landscapes to reveal reliable fibril structural parameters and assist CreateFibril in building realistic structures. After the fibrils were created, their structural stability in water was assessed by a novel dipolar solvent model that captures the effect of dipole-dipole interactions and computes the hydration shell that forms around proteins—an insight that cannot be gained with implicit solvent models. More specifically, we expanded the AQUASOL framework (25,26) to compute the solvation, Coulomb, and van der Waals energies of molecular systems with up to 36,180 atoms. CreateFibril explored the architectural landscape of HET-s, Aβ, and amylin proteins, and captured with great accuracy the properties of helical pitch, packing distance (PD) for multimeric polymorphs, and hydrogen (H)-bond distance on β-sheets between amyloid monomers as they aggregated. We predicted correct structural parameters for HET-s fibrils, revealed and characterized all Aβ fibril polymorphs, and developed a fibril model for the most common amylin polymorph. We discovered that native amylin is more stable than its amyloid form.
Materials and Methods
We aimed to analyze the stability and dynamics of large polymorphic amyloid fibrils using force-field calculations. Three-dimensional (3D) models of these fibril structures were required for analysis, but unfortunately, the current protein databases provide only very short fibril segments of a few monomers in length. Thus, we designed an automated tool to bridge this gap and create longer fibril models from amyloid fragments and simulate the structure of various polymorphic fibrils. The tool resorted to the computational technique of rigid affine transformations (27) to construct the fibril models. Fig. S4 in the Supporting Material presents a flowchart of CreateFibril’s pipeline, as described below.
Construction and classification of polymorphic fibrils
Our automated tool, called CreateFibril, produced an ensemble of stable polymorphic fibril structures from a monomer amyloid in the Protein Data Base (PDB) (28). Single fibrils (Fig. 1) were constructed by assembling copies of a monomer amyloid side by side to mimic the oligomerization result and elongation of fibrils. This assemblage was stabilized by alignment of the monomers’ H-bonds and β-sheets that twist around a helical fibril axis, emulating the natural assembly of amyloids (29). Higher-order structures (Rings, Stacks and Polygons) included harmonic combinations of Single fibrils packed in different orientations and distances. The key to create good structures was to pick parameter values that build architecturally stable fibrils. Our tool provides the following parameters to create fibrils:
-
1.
Protein PDB file of an amyloid fragment
-
2.
Fibril class: Ring, Polygon, or Stack
-
3.
Number of filaments and their PD perpendicular to fibril axis
-
4.
Fibril axis location and direction
-
5.
Rotation angle of amyloid monomers along the fibril axis
-
6.
H-bond distance of β-sheets along the fibril axis
-
7.
Length of fibril
Figure 1.
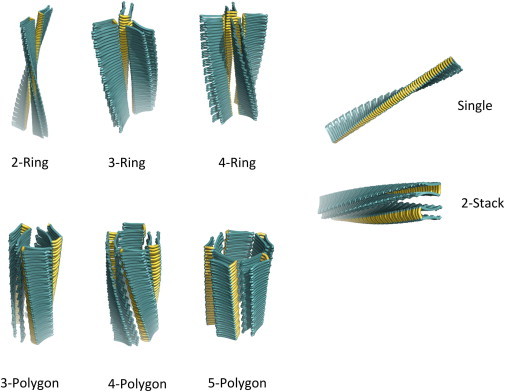
Classification of the polymorphic fibril structures produced by CreateFibril. Ring fibrils pack and join at turns, Polygon fibrils join to create a polygon-shaped core, and Stack fibrils pack laterally.
The algorithm that builds polymorphic amyloid fibrils performs numerous translation and rotation affine transformations, and is explained in detail in the Supporting Material. Although the algorithm may seem complex, CreateFibril’s interface is built to be user-friendly and intuitive. Moreover, CreateFibril is currently the only automated tool available for building fibrils.
Energy minimization to filter fibril structures
It is expected that only a specific set of parameter values can create realistic fibril models. A certain choice of parameter values might assemble monomers too close to one another or too far from one another, whereas another choice might assemble monomers that rotate abruptly with respect to the main fibril axis. We were interested in finding parameter values that CreateFibril could use to create stable conformations. We exhaustively built structures with reasonable distances and angles to search for proper parameter values for HET-s, Aβ, and amylin. An energy function, , based on light energy minimization runs and stability landscape results for each parameter value guided the search. The parameter values that returned the most stable structures were returned for each fibril type (n-Polygon, n-Ring, and n-Stack) and were kept for further analysis by the dipolar water model.
We performed light energy minimization after a structure was created, for two reasons. First, we performed minimization to relieve the system from any clashes that might have occurred during the fibril elongation process due to our rigid placement of monomers. Second, we used the minimization data to gather stability information about the structure. Through minimization, we calculated the Lennard-Jones (LJ) potential and Coulomb forces of the structure before, during, and after minimization, and used them to construct stability landscapes and assess the quality of the parameter values with which we started. Structures that drifted significantly from their initial configuration and contained high LJ and Coulomb energies were not believed to live in local minima, and hence the initial choice of parameter values did not produce stable conformations.
To perform energy minimization and potential calculations, we used the GROMACS (30) molecular dynamics (MD) and energy minimization package with the following parameters: CHARMM force field and the SPC model (31), and a TIP3P box (46,47) with a minimum distance of 15 Å from any edge of the box to any amyloid atom. The box was kept empty of water, and simulations were performed in a vacuum for two reasons. First, water molecules tend to restrict significant conformational changes of unstable fibrils; they added noise to our calculations by stabilizing all systems. Second, minimization to pinpoint the unstable structures was achieved orders of magnitude faster without explicit water molecules in the simulation box. Because we were not interested in a full MD run, water was not of utmost importance here. The systems were energy minimized for 1000 steepest gradient descent steps with an energy step of 0.01. Long-range electrostatic interactions were calculated using particle mesh Ewald with a cutoff of 1.0 Å for all simulations (32,33).
Dipolar water model
At this stage, we had parameter values that structurally encoded an ensemble of the most stable fibril structures of each type of Polygon, Ring, and Stack class. To determine the most stable polymorphs likely to form in nature, we needed to assess the stability of these structures and compare them in water. The main challenge in estimating the energy of large polymers was calculating of the solvation energy, i.e., the energy associated with the formation of a structure in water. For this, we extended the development of a fast and detailed dipolar water model introduced by AQUASOL (25,26), the first tool to succeed in computing the solvation free energy of molecules by solving the dipolar nonlinear Poisson-Boltzmann-Langevin equation. Our model took into account dipole-dipole interactions and treated water explicitly to accurately calculate the solvation energy and the Coulomb and van der Waals forces of our fibrils, as shown in Eq. 1. CreateFibril used the result of adding these terms as a measure to assess the total stability of the fibrils it created. The lower the sum of these three values, the more stable is the fibril. Our method was orders of magnitude faster than full MD simulations.
Once a solution was found by AQUASOL, the free energy of the system was computed a posteriori. Nevertheless, a problem inherent to all grid approaches is the emergence of an artifactual term in the energy, the so-called grid energy. To correct for this term and get the true free energy , one needs to compute the free energy of the system with the solvent concentration defined at a given value and subtract the free energy of the system with the solvent concentration set to zero . This leads to minimization of the artifactual term but also takes out the Coulomb energy of the solute, which needs to be brought back afterward. Finally, one must subtract the solvent energy term , which is linear with the number of water molecules within the grid and can also be computed analytically. If one is interested in the solvation free energy , then the Coulomb energy must not be brought back.
The results we obtained allowed us to compare the stabilities of the best structures across all polymorphic types we designed. In particular, we compared the solvation energies and the total energies of all polymorphs at different fibril lengths to analyze the stability behavior of the polymorphs.
| (1) |
where
The atomic charges and radii were assigned with PDB2PQR using the CHARMM force field at neutral pH. Optimization of H-bonding was performed. The following parameters were used in AQUASOL: grid of 257 points per edge spaced by 1 Å; temperature 300 K, surface definition, solvent-accessible surface (Rprobe = 1.4 Å); trilinear interpolation protocol for projection of fixed charges on the grid; lattice grid size for the solvent, a = 2.8 Å; solvent made of dipoles of moment = 3.00 D at a concentration of = 55 M; no salt added to the solution; electrostatic potential set at zero at the boundaries; and stopping criteria for residual, (when possible).
It is important to note that the solvent and small ions are correctly treated in all of our calculations, as far as enthalpy and entropy are concerned. Because we use frozen models for proteins, two scenarios arise: First, if the protein is very well structured, we neglect the small contribution of vibrational entropy. Second, if the protein has unstructured parts, such as loops, we neglect the conformational entropy of these unstructured parts. However, as far as we know, there is no rigorous way to take conformational entropy into account other than by performing long MD simulations and thermodynamic integrations, which is a hopeless approach for the problem we are interested in here. Therefore, we check to ensure that the monomers under study are well structured, with minimal unstructured parts.
Implicit water model
The second method we used to assess the stability of the polymorphs is based on an implicit model of water molecules in solution. We used the fast adaptive multigrid boundary element (FAMBE)-pH tool (34) to calculate the solvation energy and enthalpy of our structures. FAMBE-pH solves the Poisson equation with an optimized FAMBE method that implicitly models water.
The energy values returned by the implicit model and our explicit model naturally differed in magnitude. The explicit model returned solvation energy values much higher than the implicit model because it considered dipole-dipole and atomic interactions with explicit water molecules in its calculations. Because the water volume was kept relatively constant in all of the explicit simulations, we were able to compare the energies among different fibrils modeled explicitly, and compare their trends with the implicit simulations.
Results and Discussion
Amyloid fibrils are known to grow by monomer addition (29). Monomers aggregating at fibril ends usually create a helical structure of a single fibril filament, but are capable of creating higher-order fibrils composed of multiple single filaments packed closely by H-bond interactions on β-sheets. These higher-order fibrils can be composed of two, three, or even more filaments (n-filaments). While designing the different possible geometric forms into which an n-filament fibril can assemble, we observed three main configurations that characterize the possible interactions of fibril filaments. We classify these various shapes in the categories of: Rings, Polygons, and Stacks: Ring fibrils consist of filaments that pack together at their β-sheet turns, creating a hollow ring shape in the main axis of a fibril. Polygon fibrils consist of filaments that pack together at their β-strands, creating a hollow n-Polygon shape through the main axis of a fibril. Finally, Stack fibrils are comprised of filaments that pack together at their β-strands and pack laterally, creating planar fibril sheets. All of these kinds of fibrils have been recorded experimentally throughout the years, but a formal classification for fibrils has never been formalized. We will use this nomenclature throughout this work to discuss the various polymorphic fibrils we observed. A visual representation of this proposed novel classification is provided in Fig. 1. In the following section, we describe the polymorphic preferences predicted by CreateFibril.
Stability landscape
We developed a tool called CreateFibril (see Materials and Methods) to build energetically stable fibrils out of single experimentally validated amyloid monomers. We were interested in obtaining structural values of distances and rotations that characterize the architecture of stable fibrils. These structural values constitute the numerous fibril degrees of freedom and are given as input parameter values to our tool to assemble monomers together and construct stable conformations (see Fig. 2 for an illustration of these parameters). CreateFibril seeks to find suitable parameters that characterize stable structures (i.e., structures that will not diverge during MD simulations).
Figure 2.
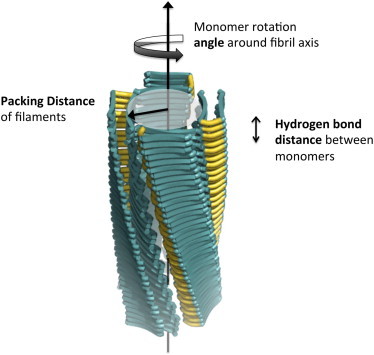
Different parameters used by CreateFibril to build structures. The parameters drawn are the fibril axis location and direction, rotation angle of amyloid monomers along the fibril axis, H-bond distance of β-sheets along the fibril axis, and PD of filaments perpendicular to the fibril axis. Parameters not drawn include the protein PDB structure file, fibril class type, and length of fibril.
One brute-force approach to find suitable parameter values is to run MD simulations on a starting configuration, attempt to remove any atomic clashes, minimize the energy of the structure, and reshape the fibril into a more stable conformation. However, such simulations are computationally expensive in resources and time, are prone to numerical imprecision, and do not guarantee a final stable conformation. For this reason, we introduce a new, to our knowledge, strategy that searches for suitable parameter values by generating fibril stability landscapes. Starting with an accurate crystal or NMR amyloid monomer, we first define a range of naturally possible values for the various fibril degrees of freedom characterized by rotation angles, PDs, and β-strand proximities, as shown in Fig. 2. Second, CreateFibril utilizes this range of values to construct all possible fibril structures using rigid affine transformations (see Supporting Material). Third, we perform light runs of energy minimization (a few hundred steps) on each generated structure to assess its initial stability sensitivity by calculating any enthalpy drift between the final and initial conformations. This step creates the fibril stability landscape by exhausting all suitable parameter values. We then search the landscape for values that construct the most stable initial conformation. These parameter values result in structures with the lowest enthalpy drift and lowest initial LJ and Coulomb terms. Structures with low enthalpy drifts suggest stable conformations (local minima on the structural energy landscape of fibrils), and structures with high-energy drifts suggest parameters that produce unstable conformations. We applied this approach to explore the structures of HET-s, Aβ, and amylin fibrils by exhaustively searching the fibril degrees of freedom for values that produce maximum stability. Parameter mining of the three proteins are summarized in Table 1. Fig. 3 provides an enthalpy drift plot describing the rotation angle versus β-strand distance landscape for the Single HET-s fibril and PD for the 3-Polygon HET-s.
Table 1.
Predicted structural parameters for HET-s, Aβ, and amylin fibrils produced by CreateFibril
| Type | Rotation angle (degrees) | H-bond (Å) | Packing distance (Å) | Max-length model (Å) | Helical pitch 360° (Å) |
Experimentally observed | ||
|---|---|---|---|---|---|---|---|---|
| model | literature | |||||||
| HET-s | Single | 22 | 4.7 | NA | 800 | 402 | 410 | yes (12) |
| 2-Stack | 16 | 4.7 | 8 | 406 | 656 | - | - | |
| 2-Ring | 27 | 4.7 | 3 | 412 | 451 | - | - | |
| 3-Ring | 17 | 4.7 | 7 | 238 | 668 | 1005–1083 | yes (12) | |
| 4-Ring | 14 | 4.7 | 8 | 199 | 738 | - | - | |
| 3-Polygon | 16 | 4.7 | 9 | 290 | 637 | - | - | |
| 4-Polygon | 13 | 4.7 | 16 | 203 | 773 | - | - | |
| Aβ | Single | 6 | 4.7 | NA | 694 | 925 | - | - |
| 2-Stack | 4 | 4.7 | 9 | 373 | 1268 | 1140–1760 | yes (10) | |
| 2-Ring | 4 | 4.7 | 7 | 379 | 1390 | 1620–2980 | yes (10) | |
| 3-Ring | 4 | 4.7 | 7 | 248 | 1334 | - | - | |
| 4-Ring | 4 | 4.7 | 9 | 183 | 1236 | - | - | |
| 3-Polygon | 2 | 4.7 | 16 | 240 | 2748 | 2000–2800 | yes (21) | |
| 4-Polygon | 3 | 4.7 | 21 | 179 | 1684 | - | - | |
| Amylin | Single | 8 | 5 | NA | 789 | 664 | 242–833 | yesa (45) |
| 2-Stack | 9 | 5 | 3 | 396 | 591 | - | yesb (43,44) | |
| 2-StackE | - | - | - | 244 | - | 486 | yesb (43) | |
| 2-Ring | −8 | 5 | 5 | 419 | 624 | - | yesb (42) | |
| 3-Ring | −7 | 5 | 8 | 269 | 870 | - | yesb (18) | |
| 4-Ring | −3 | 5 | 12 | 195 | 1090 | - | yesb (18) | |
| 3-Polygon | 6 | 5 | 16 | 262 | 998 | - | yesb (18) | |
| 4-Polygon | 4 | 5 | 28 | 190 | 1324 | - | yesb (18) | |
Note that the parameters produce structures with helical pitches very similar to those seen in fibrils in nature; hence, the H-bond lengths, angles, and packing distances are accurate predictions. Highlighted rows are the polymorphs that CreateFibril predicted to form in the greatest abundance and possessed the lowest total energies.
A Single fibril model was built out of partial EPR distance measurements.
It is ambiguous as to whether the Ring or Stack structures actually formed.
Figure 3.
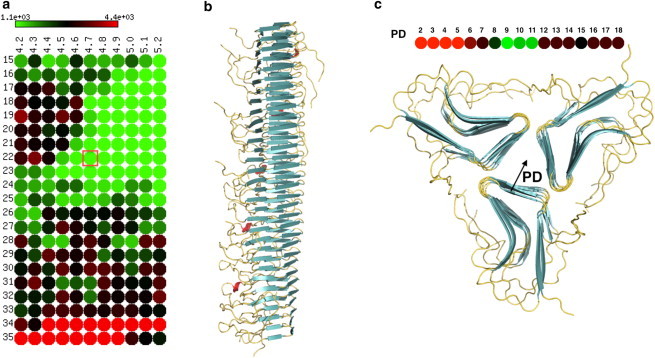
HET-s Single fibril parameter findings. (a) Heatmap representation of the stability landscape of Single HET-s structures, exploring the rotational angle θ (y axis) and β-sheet bonding distance d (x axis). Green circles indicate stable structures with low enthalpy drift, red circles indicate unstable structures with high enthalpy drift, and black circles are intermediately stable structures. (b) Single HET-s fibril built by CreateFibril with the values of θ = 22° and d = 4.7 Å taken from the best result in panel a enclosed by the red square. (c) Stability landscape of the PD of HET-s 3-Polygon. Energy values are in kJ/mol.
HET-s
HET-s (PDB ID: 2RNM) is a fungal prion that is involved in the programmed cell death of the filamentous fungus Podospora anserina. HET-s fibrils have been studied extensively due to the availability of their high-resolution atomic structures (20). CreateFibril explored the stability landscape of HET-s and simulated an ensemble of polymorphic fibril models that confirm the structural properties observed in experimental data.
Capturing 3-ring and Single HET-s fibril structures
We modeled seven different HET-s fibrils up to 80 nm in length. To our knowledge, this large-scale modeling of fibrils is the first of its kind to provide models on the scale of structures observed experimentally (5–10 nm). The enthalpy drift calculations shown in Fig. 3 suggest that a left-handed swirling orientation of the HET-s amyloids around their fibril axis is more favorable than a right-handed twist. The Single and 3-Ring structures are well known in the literature as the predominant forms of HET-s (12). The Single fibrils come together and pack to form 3-Ring structures at pH values < 3. From CreateFibril’s structural findings for HET-s in Table 1, we verify that the HET-s Single fibril helical pitch of 410 Å, β-sheet aggregation of 4.8 Å, and left-handed twist of the fibril reported by Mizuno et al. (12) all fall in the range of the most energetically favorable structural parameters of HET-s. Furthermore, the 3-Ring structures form with a packing radius of 7 Å and an axial repeat of 66.8 nm. Due to the large size of the HET-s protein and the many steric clashes that form during aggregation, it was expensive to run full EM to relax the fibrils in preparation for calculating the total energies of the different polymorphs by our dipolar solvent model—a task not intended for the purposes of this fast method. Fig. 4, a and b, show the predominant HET-s fibrils in nature. The Single fibril hides the hydrophobic regions in its core, whereas the 3-Ring fibril uses the branching residues of each fibril filament to further cover hydrophobic areas.
Figure 4.
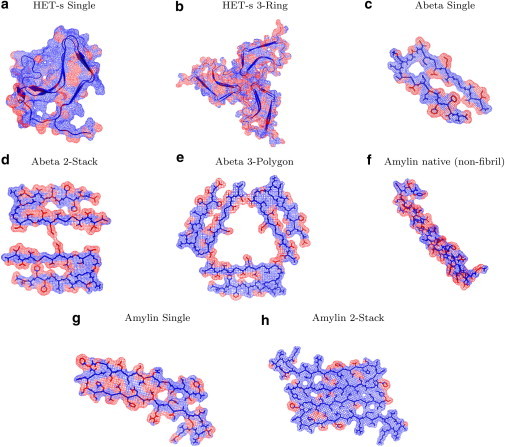
Cross-sectional view of the hydration-shell effect on the hydrophobicity of the predominant HET-s, Aβ, and amylin fibrils produced by AQUASOL. Blue regions represent hydrophilic residues and red regions represent hydrophobic residues. (a and b) HET-s Single fibrils possess a hydrophobic core around which they aggregate (a), and the branching residues of the Single fibrils help hide hydrophobic residues of their neighboring fibril when packed in the 3-Ring structure (b). (d and e) Aβ 2-Stack and 3-Polygon fibrils aggregate, creating a hydrophobic core. (f and g) The native amylin contains many hydrophobic residues (f) but possesses a lower energy than its amyloid counterpart (g). (h) To hide their hydrophobic residues, amylin amyloids aggregate in the 2-Stack polymorph.
Aβ
β-amyloid peptide (Aβ), which is found in excess in patients with Alzheimer’s disease, is believed to lead to neurodegeneration in humans (35). This protein aggregates into various fibril shapes that form neuritic plaques and neurofibrillary tangles (36–38). Aβ molecules are known to form into the 3-Polygon (21), 2-Stack, and 2-Ring polymorphic shapes (10,39). CreateFibril structurally characterized Aβ’s polymorphic fibrils and computationally assessed their stability in solution.
CreateFibril matched the helical pitches of Aβ polymorphs in nature
Using rigid affine transformations and enthalpy drift measurements, CreateFibril built several potential polymorphs for Aβ. In Table 1, we report the best structures along with their structural parameters. The helical pitches of the fibrils we modeled are in line with those reported in the literature (10). Although the structure of Aβ (PDB ID: 2BEG) was missing residues 1–16, CreateFibril was still able to choose the right rotation angles, θ; fibril PDs; and β-sheet aggregation distances, d, for all polymorphs to reproduce the structures that have been observed in nature (see Table 1).
2-Stack, 3-Polygon, and Single Aβ polymorphs predicted to form
Fig. 5 a describes the effect of water solvent on the aggregation of the Aβ polymorphs modeled with our dipolar water formalism. Up to a length of 27 monomers, water favors the formation of the Single Aβ fibril. At a length of 27 monomers, the 2-Stack and 3-Polygon polymorphs start to outperform the Single in aggregation. This could be explained by the packing of Single fibrils to produce 2-Stack and 3-Polygon structures. In Fig. 5 b, the energies of all polymorphs grow negatively, implying favorable aggregation with regard to solvation and enthalpy. At a length of ∼32 monomers, the figure suggests that the 2-Stack structure is the most stable polymorph in the set, followed by the 3-Polygon and the Single fibrils, all of which have been observed experimentally. Fig. 4, c–e, shows the effect of the hydration shell on the monomers that make up these fibrils, and how aggregation attempts to hide hydrophobic regions. Fig. S1, a and b, present the results of the implicit water model in calculating solvation energy and total free energy of Aβ polymorphs. This model proposes the emergence of the 4-Polygon and 4-Ring fibrils, which were never experimentally observed.
Figure 5.
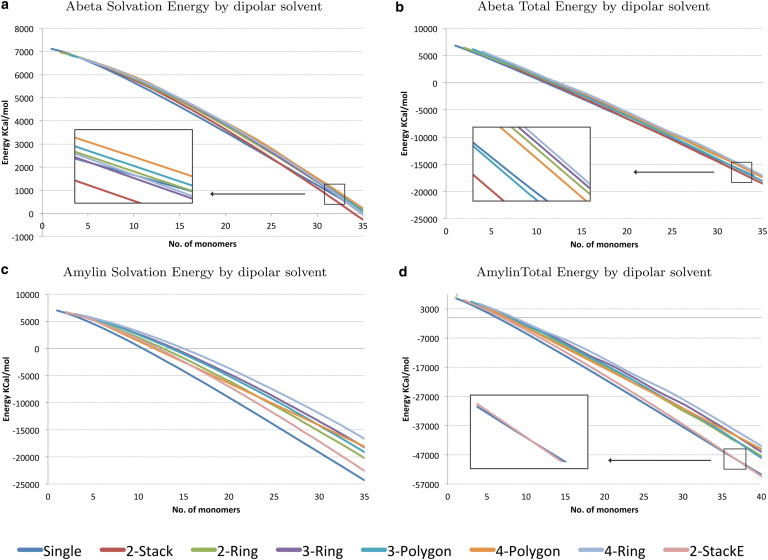
Energies of Aβ and amylin fibrils as they aggregate. (a and c) Solvation energy by dipolar solvent. (b and d) Free energy by dipolar solvent.
CreateFibril characterizes the stability landscape of wrapped Aβ fibrils, and wrapped structures can further stabilize 2-Stack fibrils
Stroud et al. (39) used x-ray powder diffraction to observe that some Aβ fibrils are likely composed of laterally associated fibril filaments that twist around internal helical axes. These internal axes wrap around a common superhelical axis in a geometry that the authors termed wrapping. When a filament is wrapped around a helical axis in this manner, it obtains a twist that is in phase with the fibril helix. Stroud et al. showed that higher crossing angles are related to greater curvature and increasingly large holes in fibrils, suggesting that Aβ fibril toxicity may be related to their potential for forming pores. In Fig. 6 c, we show a stability landscape plot for Aβ wrapped fibrils with crossing angles between 0° and 88° and rotation angles (with respect to the main fibril axis) between −13° and 13°. The wrapped structures we modeled did not contain runaway domain swapping. Two stable wrapped structures suggested by the stability landscape had crossing angles of 8° and 59° and rotation angles of −3° and −11°, respectively. As shown in Fig. 6 d, the first wrapped structure obtained a more stable conformation than the 2-Stack model, validating that some 2-Stack Aβ fibrils are indeed wrapped. Fig. S2 expands the stability landscape results by showing the enthalpy energies obtained before and after minimization runs.
Figure 6.
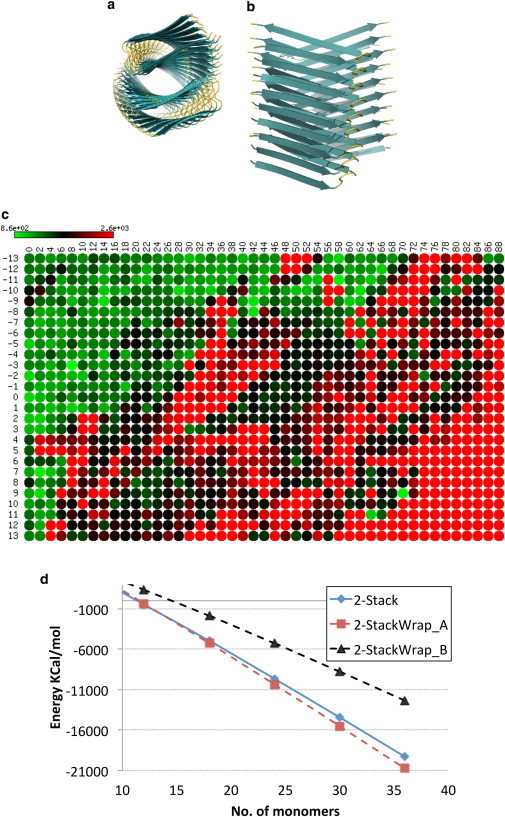
Aβ wrapped fibrils. (a and b) Cross-sectional top view (a) and side view (b) of the wrapped Aβ fibril. (c) Heatmap representation of the stability landscape of Aβ wrapped fibrils characterized by rotational angle θ (y axis) and crossing angle ω (x axis). Green circles indicate stable structures with low energy, red circles indicate unstable structures with high energy, and black circles are intermediately stable structures. Energy values are in kJ/mol. (d) The 2-StackWrap_A structure with a crossing angle of 8° and rotation angle of −3° is more stable than a nonwrapped 2-Stack structure. 2-StackWrap_B has a crossing angle of 59° and rotation angle of −11°.
Amylin
Deposits of islet amyloid polypeptide (amylin) in the pancreas are toxic and believed to be a contributing factor to Type II diabetes (40,41). Amylin fibrils were successfully polymerized in vitro and showed a diverse ensemble of polymorphic shapes (18,42). To reconstruct the polymorphs computationally, CreateFibril required the atomic structure of the amyloid form of amylin. Unfortunately, to date, no one has been able to crystallize full-length human amylin. Instead, many models for the monomeric form of amyloid amylin have been proposed, and among the most prominent structures are ones proposed by Wiltzius et al. (43), Luca et al. (44), and Bedrood et al. (45). In 2007, Luca et al. (44) proposed a full atomic model of a single striated ribbon amylin polymorph based on constraints from solid-state NMR, which opened the door for further amylin experimental model development. Interestingly, we have observed that the amylin structures proposed by Bedrood et al. (45) resemble the wrapped Aβ fibrils containing swapping runaway domains recently discovered by Stroud et al. (39). Although the model of Bedrood et al. gives new insight into a possibly novel amylin conformation depicted by EPR distance measurements, they built the model by only considering a single stack of peptides and ignored additional restrictions that would rise from the packing of multiple fibril filaments around each other. Hence, the model presented by Bedrood et al. may represent a special form an amylin monomer can take, and not the predominant conformation. The final prominent atomic model of amylin proposed by Wiltzius et al. (43) was built using biochemical and structural data along with the fibril NNFGAIL and SSTNVG crystallized regions of amylin to formulate a structure with atomistic details for the protein in the form of a 2-Stack structure. It is crucial to start CreateFibril with a very realistic amyloid monomer, because this is the basis for determining polymorphic shapes and predicting accurate likelihoods of formation. Perturbations at the monomer level can alter the fibril pitch and PDs, which might introduce artifacts when calculating likelihoods of formation. It is important to start with an amyloid crystal structure, when possible, or a precise model that is not very different from the actual amyloid (root mean-square deviation (RMSD) ≈ 2 Å; see Fig. S3 for convergence analysis). An RMSD difference of 2 Å is quite large for small monomers such as Aβ and amylin. MD simulations and minimization techniques should be used to improve the model quality when necessary. It is our understanding that the Wiltzius model is the best model in the literature regarding amylin in its monomeric amyloid form, and we refer to it as 2-StackE in Fig. 5 and Table 1. We extracted one amylin protein from this fibril model and used it as a starting template to build the other polymorphic fibrils. Having verified that our approach generated valid structural results for Aβ and HET-s, and valid solvation results for Aβ, we endeavored to gain insight into amylin polymorphs.
2-Stack and Single amylin polymorphs have the lowest total energies
Fig. 5 d graphs the total energy of the amylin polymorphs and suggests that amylin Single and 2-Stack polymorphs compete in solution. The Single structure dominates in lowest total energy until a 35 amyloid fibril is reached, after which the 2-Stack polymorph becomes more abundant. This figure also shows that the other polymorphs cluster together and are higher in energy. Assuming that the model we started with is the actual model of amylin in amyloid form, we can suggest that the 2-Ring, 3-Ring, 3-Polygon, 4-Ring, and 4-Polygon structures are unlikely to form. Although Fig. 5 c suggests that solvation generally prefers the emergence of the Single structure in solution over the 2-Stack structure, the total stability and dominance of structure is determined by a combination of enthalpy and solvation. The dipolar water model in Fig. 5 d suggests that a 2-Stack structure (43,44) should exist in greater abundance and possess the greatest stability among polymorphs. Because Single fibrils initially are more energetically favorable than the 2-Stack structure and may contribute to its emergence, we propose that finding a way to limit the aggregation of the Single polymorph could greatly diminish all the fibrils that form in solution. This would be of critical importance in the search for therapeutics to combat the growth of amylin fibrils in the β-cells of the pancreas (41). Fig. 4, f–h, show the effects of the hydration shell on the hydrophobicity of amylin when aggregated in the Single and 2-Stack fibril form and when left in the native form. We observe that the 2-Stack model structure hides most hydrophobic regions, which may be one explanation for its abundance in solution. The implicit water model shown in Fig. S1 c suggests that the 2-Stack amylin structures are high in energy and are not likely to exist for a long period of time. This result contradicts the experimental findings of Goldsbury et al. (18,42), which suggested that two-polymorph amylin structures are dominant in vitro. However, the dipolar water model confirms these findings and furthermore suggests that the dominant two-polymorph fibrils would take on a 2-Stack conformation, and any emerging three-polymorph fibril would take on a 3-Polygon conformation similar to that of Aβ.
Native amylin is predicted to be lowest in energy
It has been unclear whether amyloid monomers form because they are lower in energy than their native counterparts or because their aggregation produces lower-energy structures compared with an accumulation of unbound native proteins. Using our dipolar solvent model, we found that the total energy of one amylin monomer in native form (PDB ID: 2KB8) was 6504.7 kcal/mol and the total energy of one amylin monomer in amyloid form was 6660.7 kcal/mol. Assuming we started with an accurate amylin model, this result reveals that the native form of amylin is initially favored in nature over the amyloid form. As amyloids find each other and aggregate, the energy of an aggregate structure of k monomers becomes lower than the energy of k native molecules floating in solution for all . This could explain the rapid aggregation phenomenon observed in amylin fibrils.
Online tool and fibril database
In this work, we have shown how to build amyloid fibrils with various geometries by using rigid affine transformations, starting from the amyloid structure of a single prion form of the protein. We computed the free energy of these structures in water by adding together the LJ, Coulomb, and solvation energies. The latter is a crucial component in the stability of fibrils and was computed with the use of the AQUASOL program, which yields free energies in good agreement with those computed from long runs of MD. These computed free energies in turn allowed us to assess the stability of the various proposed structures and to classify their abundance in amyloid solutions. Our results are in very good agreement with current experimental findings, and in some cases predict the existence of stable forms of aggregates that have not yet been observed. Table 1 summarizes the stability landscape exploration parameters used by CreateFibril to build the most stable fibril structures for HET-s, Aβ, and amylin proteins. All of the polymorphic fibril structures generated here are available in an open database at http://amyloid.cs.mcgill.ca, and CreateFibril is provided as a free online application on the same site.
Acknowledgments
M.R.S. was supported by a fellowship from the Canadian Institutes of Health Research System Biology Training program at McGill University. J.W. was supported by a Discovery grant from the Natural Science and Engineering Research Council of Canada. The French Embassy in Canada provided travel funds.
Supporting Material
References
- 1.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Uversky V.N., Fink A.L. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim. Biophys. Acta. 2004;1698:131–153. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Landrigan P.J., Sonawane B., Droller D. Early environmental origins of neurodegenerative disease in later life. Environ. Health Perspect. 2005;113:1230–1233. doi: 10.1289/ehp.7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirkwood S.C., Su J.L., Foroud T. Progression of symptoms in the early and middle stages of Huntington disease. Arch. Neurol. 2001;58:273–278. doi: 10.1001/archneur.58.2.273. [DOI] [PubMed] [Google Scholar]
- 6.Golden S.H., Williams J.E., Brancati F.L., Atherosclerosis Risk in Communities study Depressive symptoms and the risk of type 2 diabetes: the Atherosclerosis Risk in Communities study. Diabetes Care. 2004;27:429–435. doi: 10.2337/diacare.27.2.429. [DOI] [PubMed] [Google Scholar]
- 7.Goldfarb L.G., Brown P., Mitrovà E., Cervenáková L., Goldin L., Korczyn A.D., Chapman J., Gálvez S., Cartier L., Rubenstein R. Creutzfeldt-Jacob disease associated with the PRNP codon 200Lys mutation: an analysis of 45 families. Eur. J. Epidemiol. 1991;7:477–486. doi: 10.1007/BF00143125. [DOI] [PubMed] [Google Scholar]
- 8.Mendez M.F., Selwood A., Frey W.H., 2nd Pick’s disease versus Alzheimer’s disease: a comparison of clinical characteristics. Neurology. 1993;43:289–292. doi: 10.1212/wnl.43.2.289. [DOI] [PubMed] [Google Scholar]
- 9.Fändrich M. On the structural definition of amyloid fibrils and other polypeptide aggregates. Cell. Mol. Life Sci. 2007;64:2066–2078. doi: 10.1007/s00018-007-7110-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meinhardt J., Sachse C., Fändrich M. Aβ(1-40) fibril polymorphism implies diverse interaction patterns in amyloid fibrils. J. Mol. Biol. 2009;386:869–877. doi: 10.1016/j.jmb.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chamberlain A.K., MacPhee C.E., Davis J.J. Ultrastructural organization of amyloid fibrils by atomic force microscopy. Biophys. J. 2000;79:3282–3293. doi: 10.1016/S0006-3495(00)76560-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizuno N., Baxa U., Steven A.C. Structural dependence of HET-s amyloid fibril infectivity assessed by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA. 2011;108:3252–3257. doi: 10.1073/pnas.1011342108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seilheimer B., Bohrmann B., Döbeli H. The toxicity of the Alzheimer’s β-amyloid peptide correlates with a distinct fiber morphology. J. Struct. Biol. 1997;119:59–71. doi: 10.1006/jsbi.1997.3859. [DOI] [PubMed] [Google Scholar]
- 14.Diaz-Avalos R., King C.Y., Caspar D.L. Strain-specific morphologies of yeast prion amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:10165–10170. doi: 10.1073/pnas.0504599102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamaguchi K.I., Takahashi S., Goto Y. Seeding-dependent propagation and maturation of amyloid fibril conformation. J. Mol. Biol. 2005;352:952–960. doi: 10.1016/j.jmb.2005.07.061. [DOI] [PubMed] [Google Scholar]
- 16.Dobson C.M. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 17.Jiménez J.L., Nettleton E.J., Saibil H.R. The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2002;99:9196–9201. doi: 10.1073/pnas.142459399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldsbury C., Goldie K., Aebi U. Amyloid fibril formation from full-length and fragments of amylin. J. Struct. Biol. 2000;130:352–362. doi: 10.1006/jsbi.2000.4268. [DOI] [PubMed] [Google Scholar]
- 19.Goldsbury C.S., Wirtz S., Frey P. Studies on the in vitro assembly of a β 1-40: implications for the search for a β fibril formation inhibitors. J. Struct. Biol. 2000;130:217–231. doi: 10.1006/jsbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- 20.Wasmer C., Lange A., Meier B.H. Amyloid fibrils of the HET-s(218-289) prion form a β solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 21.Paravastu A.K., Leapman R.D., Tycko R. Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hills R.D., Jr., Brooks C.L., 3rd Hydrophobic cooperativity as a mechanism for amyloid nucleation. J. Mol. Biol. 2007;368:894–901. doi: 10.1016/j.jmb.2007.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fawzi N.L., Yap E.H., Head-Gordon T. Contrasting disease and nondisease protein aggregation by molecular simulation. Acc. Chem. Res. 2008;41:1037–1047. doi: 10.1021/ar800062k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halfmann R., Alberti S., Lindquist S. Opposing effects of glutamine and asparagine govern prion formation by intrinsically disordered proteins. Mol. Cell. 2011;43:72–84. doi: 10.1016/j.molcel.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koehl P., Delarue M. AQUASOL: an efficient solver for the dipolar Poisson-Boltzmann-Langevin equation. J. Chem. Phys. 2010;132:064101. doi: 10.1063/1.3298862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Azuara C., Orland H., Delarue M. Incorporating dipolar solvents with variable density in Poisson-Boltzmann electrostatics. Biophys. J. 2008;95:5587–5605. doi: 10.1529/biophysj.108.131649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shirley P., Ashikhmin M., Marschner S. A. K. Peters, Ltd; Natick, MA: 2009. Fundamentals of Computer Graphics. [Google Scholar]
- 28.Berman H.M., Westbrook J., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collins S.R., Douglass A., Weissman J.S. Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol. 2004;2:e321. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hess B., Kutzner C., Lindahl E. Gromacs 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 31.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., Hermans J. Interaction models for water in relation to protein hydration. In: Pullman B., editor. Intermolecular Forces. D. Reidel Publishing Company; Dordrecht: 1981. pp. 331–342. [Google Scholar]
- 32.Darden T., York D., Pedersen L. Particle mesh Ewald—an n.log(n) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 33.Essmann U., Perera L., Pedersen L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 34.Vorobjev Y.N., Vila J.A., Scheraga H.A. FAMBE-pH: a fast and accurate method to compute the total solvation free energies of proteins. J. Phys. Chem. B. 2008;112:11122–11136. doi: 10.1021/jp709969n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hardy J.A., Higgins G.A. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 36.Khachaturian Z.S. Diagnosis of Alzheimer’s disease. Arch. Neurol. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 37.Mirra S.S., Heyman A., Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 38.Esiri M.M., Pearson R.C., Powell T.P. A quantitative study of the neurofibrillary tangles and the choline acetyltransferase activity in the cerebral cortex and the amygdala in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry. 1990;53:161–165. doi: 10.1136/jnnp.53.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stroud J.C., Liu C., Eisenberg D. Toxic fibrillar oligomers of amyloid-β have cross-β structure. Proc. Natl. Acad. Sci. USA. 2012;109:7717–7722. doi: 10.1073/pnas.1203193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marzban L., Park K., Verchere C.B. Islet amyloid polypeptide and type 2 diabetes. Exp. Gerontol. 2003;38:347–351. doi: 10.1016/s0531-5565(03)00004-4. [DOI] [PubMed] [Google Scholar]
- 41.Westermark P., Andersson A., Westermark G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 2011;91:795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- 42.Goldsbury C.S., Cooper G.J., Kistler J. Polymorphic fibrillar assembly of human amylin. J. Struct. Biol. 1997;119:17–27. doi: 10.1006/jsbi.1997.3858. [DOI] [PubMed] [Google Scholar]
- 43.Wiltzius J.J.W., Sievers S.A., Eisenberg D. Atomic structure of the cross-β spine of islet amyloid polypeptide (amylin) Protein Sci. 2008;17:1467–1474. doi: 10.1110/ps.036509.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luca S., Yau W.M., Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bedrood S., Li Y., Langen R. Fibril structure of human islet amyloid polypeptide. J. Biol. Chem. 2012;287:5235–5241. doi: 10.1074/jbc.M111.327817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jorgensen W., Chandrasekhar J., Klein M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 47.Mahoney M., Jorgensen W. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 2000;112:8910–8922. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.