

Abstract
Microscope cytometry provides a powerful means to study signaling in live cells. Here we present a quantitative method to measure protein relocalization over time, which reports the absolute fraction of a tagged protein in each compartment. Using this method, we studied an essential step in the early propagation of the pheromone signal in Saccharomyces cerevisiae: recruitment to the membrane of the scaffold Ste5 by activated Gβγ dimers. We found that the dose response of Ste5 recruitment is graded (EC50 = 0.44 ± 0.08 nM, Hill coefficient = 0.8 ± 0.1). Then, we determined the effective dissociation constant (Kde) between Ste5 and membrane sites during the first few minutes when the negative feedback from the MAPK Fus3 is first activated. Kde changed during the first minutes from a high affinity of <0.65 nM to a steady-state value of 17 ± 9 nM. During the same period, the total number of binding sites decreased slightly, from 1940 ± 150 to 1400 ± 200. This work shows how careful quantification of a protein relocalization dynamic can give insight into the regulation mechanisms of a biological system.
Introduction
Cells need to respond appropriately to external stimuli presented by a complex and changing environment. These responses are mediated by signal transduction processes that carry the information through the plasma membrane (PM) to the inside of the cell, resulting in changes in physiological state that often involve changes in gene expression. For most signals, the first step of these pathways involves the activation of a surface receptor that in turn activates components on the inner side of the PM. From there, information about the signal crosses the cytoplasm and reaches the nucleus via many different mechanisms. A recurrent motif involves the activation of one or more cytoplasmic components by their relocalization to the membrane, bringing them into close proximity to their activating partners. In addition, relocalization to the membrane greatly increases the local concentration of signaling molecules and thereby facilitates signal transduction (1). Examples of activation by membrane recruitment include STAT proteins in response to cytokines (2), Akt1 in response to endothelial growth factor (3), protein kinase C in response to different stimuli (4–6), and Sos in response to growth factors (7).
Another well-documented example of membrane recruitment occurs in the yeast Saccharomyces cerevisiae pheromone response pathway, a prototypical G protein coupled receptor (GPCR) and mitogen-activated protein kinase (MAPK) cascade signal transduction system (Fig. 1 A). When α-factor mating pheromone binds its receptor (Ste2) at the PM of haploid MATa yeast, Ste2 activates and causes the exchange of GDP for GTP in the associated Gα (Gpa1) subunit of the G protein. In turn, this exchange causes the dissociation of the heterotrimeric G protein. Membrane-anchored Gβγ (Ste4:Ste18) can then bind and thus recruit to the membrane the scaffold protein Ste5. This scaffold binds the MAPKKK Ste11, the MAPKK Ste7, and the MAPK Fus3, and localizes them at the PM, where the PAK family kinase Ste20 initiates the phosphorylation cascade. Activated Fus3 and Kss1 move to the nucleus, where they activate transcription factors and other targets that prepare the cell for mating (8,9).
Figure 1.
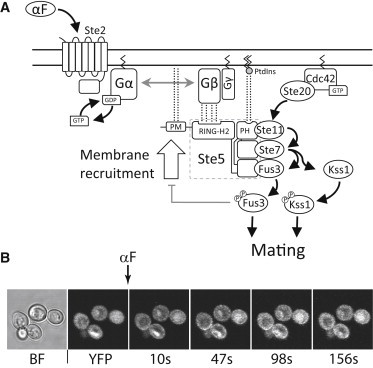
(A) Schematic representation of the pheromone pathway. When α-factor binds its receptor Ste2, the GEF activity of this GPCR is activated, causing the dissociation of the heterotrimeric G protein. Free Gβγ can recruit the scaffold protein Ste5 to the membrane, initiating the MAPK cascade. Phosphorylated Fus3 and Kss1 can phosphorylate other targets that initiate the mating response. (B) Image montage of YFP-Ste5 membrane recruitment. Saturating amounts of α-factor was added at time zero (indicated by the arrow) to strain YAB3770, and changes in fluorescence distribution were followed by time-lapse confocal microscopy. BF, bright field.
The recruitment of Ste5 to the membrane is both required and sufficient for signaling (10), and it is mainly mediated by the interaction of Ste5’s RING-H2 domain with Ste4 (10,11). Nevertheless, other domains of Ste5 are also involved in its membrane recruitment. An amphipathic N-terminal domain of Ste5 called the PM domain interacts directly with membrane phospholipids and is required for efficient recruitment (12). A pleckstrin homology (PH) domain that partially overlaps with the Ste11-binding region and has affinity for phosphoinositides is also important for membrane recruitment (13). Other indirect interactions may contribute to Ste5’s membrane localization as well. For example, Ste5 interacts with Bem1 (14,15), a scaffold protein that binds Ste20, the small rho-like GTPase Cdc42, and its guanine-nucleotide exchange factor (GEF) Cdc24 (16), which are involved in the establishment of cell polarity (17).
Control of Ste5 activity is a key regulatory point of the pathway. Ste5 is the component of the pathway with the lowest abundance, with only 500 molecules on average per cell, and its abundance sets a trade-off between the maximal pheromone response and the dynamic range of the system (18). Ste5 recruitment to the membrane may be the target of a negative feedback loop mediated by Fus3 that modulates the sensitivity of the system to the incoming pheromone signal (19). Although the point of regulation of Fus3 is not known, it is has been reported that both Ste5 and Ste4 (Gβ) are phosphorylated by Fus3 in response to pheromone (11,20–22). Phosphorylation of Ste5 by Fus3 modulates the affinity between Ste5 and Fus3, which in turn modifies the system’s output (21). Binding between Ste5 and Fus3 induces autophosphorylation of Fus3, which also modulates the system’s output (23). Ste5 membrane localization is cell-cycle-regulated and is inhibited by late G1 CDK activity due to the phosphorylation of eight sites flanking the PM domain of Ste5, which reduces its affinity for phospholipids by electrostatic repulsion (24). Degradation of Ste5 via proteasome has been reported to occur in a cell-cycle-regulated manner (25); therefore, Ste5 is an important point of cell-cycle regulation of the mating response. The membrane localization of Ste5 and its bound MAPK cascade also seem to modulate signal transduction, promoting a graded (as opposed to a more switch-like) response of the pathway (26).
Given the regulatory importance of Ste5, we sought to study its initial activation by membrane recruitment in more detail. In terms of measurements, membrane recruitment of Ste5 has several advantages over other signaling steps. It can be measured by fluorescence microscopy in vivo and within seconds of pheromone addition. Furthermore, it is the actual biological process of interest, and not an external, potentially interfering reporter, and thus results in a direct and real-time measurement of pathway activation.
Previous studies measured membrane recruitment of different proteins by employing time-lapse fluorescence microscopy in different cell cultures (4–6,27). In these studies, a fusion between the protein of interest and a fluorescent protein (FP) was usually overexpressed and its localization was followed through time. Usually, conclusions were based on the qualitative change in fluorescence distribution that was apparent in the images. In other studies, investigators quantified recruitment manually based on acquired images, such as by calculating the ratio between membrane and cytoplasmic density of fluorescence (6), or using line scans of the cell (27). In another approach, the concentration of FPs was estimated by comparing the intensity levels with a calibration curve obtained with purified recombinant FP, under the same imaging conditions (28,29). These approaches require a manual definition of the regions of interest, which limits the throughput of the analysis. Furthermore, they require a large signal/noise ratio of the image and consequently are restricted to abundant or overexpressed proteins.
In yeast, we previously measured the recruitment of Ste5 at mildly overexpressed levels (18,19). This was done in an automatic and high-throughput manner, but the measure was not calibrated (i.e., it was given in arbitrary units of a measured quantity), and the relationship between the measured quantity and the actual fraction of membrane-bound Ste5 molecules was not proven to be linear. Nonlinearities in this relationship may modify the apparent response times and dose-response (D-R) curves. A properly quantitative measurement would allow the estimation of relevant system parameters, such as the EC50 and Hill coefficient of the D-R curve, and the affinity constants and relative abundances between binding partners. There are no current estimates for these parameters; for example, values ranging from 48 nM to 1 μM have been used as dissociation constants for the Ste5-Gβγ interaction in different models of the pathway (30–32).
The recruitment of Ste5 to the membrane has a characteristic peak and decline dynamics, due to the Fus3-dependent negative feedback loop (19). This decline may be caused by a reduction in the effective binding affinity, a decrease of the available binding sites, or a combination of both effects. Identifying which of these parameters are modulated by the negative feedback may help us understand the regulation of the system and find the target of Fus3 that is responsible for this modulation.
Here we present a quantitative and calibrated measure of membrane recruitment that reports the absolute fraction of membrane-bound Ste5 molecules. The method is of general utility and may be applied not only to other systems in which membrane recruitment is relevant but also to relocalization events in other subcellular compartments, such as the nucleus and the mitochondria. The method is sensitive enough to measure activation of cells with fluorescently tagged Ste5 expressed at endogenous levels.
We used this method to measure the D-R relationship of Ste5 recruitment, and found that it is graded and exhibits no ultrasensitivity. Our data also indicate that, contrary to our hypothesis, the Fus3 negative feedback does not alter the sensitivity to pheromone at this step, but regulates the amplitude of the response. Finally, using this approach, we calculated the effective binding affinity and effective amount of binding sites for Ste5 at the PM at various times after pheromone exposure.
Materials and Methods
The S. cerevisiae strains were of the W303a genetic background. All strains were derived from ACL379 (33) (MATa, Δbar1) by standard nucleic acid and yeast manipulation procedures (34). ACL379 was used as the reference for autofluorescence calculation. Strains used for the effective dissociation constant determination experiment were constructed by replacing the endogenous STE5 by one or several integrations of a PSTE5-YFP-STE5 construct, under control of STE5 native promoter. TCY3126 (W303a Δbar1 ste5::YFP-STE5, the 1X strain) was cotransformed with two linearized integrative plasmids (one with the URA3 marker and one with the TRP1 auxotrophic marker) harboring the PSTE5-YFP-STE5 expression cassette (35) and selected in synthetic defined medium minus uracil or tryptophan, or both. Large numbers of transformant colonies were screened, and colonies with different numbers of integrations, as assessed by yellow FP (YFP) fluorescence levels, were selected.
Confocal microscopy cytometry was based on protocols described elsewhere (35,36). Briefly, we placed exponential growing cells in 96-well or 384-well glass-bottom plates. To affix cells, we pretreated the wells with 1 mg/ml of concanavalin A (Sigma-Aldrich) and washed them twice with water. We acquired images using an Olympus FV1000 confocal module mounted on a Olympus IX-81 microscope, with an Olympus UplanSapo 63× objective (NA = 1.35). For YFP measurements, we excited at 515 nm and collected between 530 and 630 nm. Laser power was adjusted before each experiment to optimize the signal while maintaining low levels of photobleaching. We stimulated the cells with chemically synthesized α-factor (Yale Small Scale Peptide Synthesis, New Haven, CT) at the indicated concentrations. We prepared the different doses by serial dilutions in synthetic complete (SC) medium (BSM formulations, BIO-101, Qbiogene, Irvine, CA) with 40 μg/ml of casein (Roche Applied Science) to block unspecific binding to tips and tubes, as previously described (33). When cells were stimulated with saturating concentrations of pheromone (1 μM), the casein was not required. Strains with analog-sensitive FUS3 allele FUS3-Q93A (RY2013b) were preincubated for 10 min with 10 μM of 1-NM-PP1 (Caymen Chemicals, Ann Arbor, MI) or the carrier (0.1% DMSO), and these concentrations were maintained after stimulation with pheromone (see Fig. S5, D and E, in the Supporting Material). For the calibration procedure we used FM4-64 (Molecular Probes) at a final concentration of 0.25 μg/ml in SC.
Image segmentation and quantification were done with Cell-ID (35) and the resulting dataset was analyzed with R (36). During this work, the R package Rcell (available at http://cran.r-project.org/) was developed for analysis and visualization of the data (A. Bush, R. Espada, A. Chernomoretz, and A. Colman-Lerner, unpublished).
Model fitting was performed and confidence intervals (CIs) for the parameters were calculated using a χ2 statistic as described in Cedersund and Roll (37).
Results
We acquired time-lapse confocal images of yeast strains with the endogenous STE5 gene replaced by one or several chromosomally integrated PSTE5-YFP-STE5 constructs. We stimulated cells with saturating doses of pheromone at time zero (Fig. 1). To analyze the images in a high-throughput manner, we used our Cell-ID software (35) for segmentation, and defined cell boundaries based on a bright-field image. This approach is advantageous because no fluorescence channel is required for segmentation, and it avoids potential bias in the segmentation process due to differences in fluorescence levels.
With the cell boundaries defined, we searched for a statistic (a quantity calculated from the fluorescence image and the cell boundary) that robustly measures the biological magnitude of interest, i.e., the amount of PM-associated fluorescence. The ideal statistic would be linear with the fraction of membrane-recruited fluorescence, not affected by photobleaching, and independent of the total fluorescence, illumination intensity, and cell shape.
Constructing the recruitment statistic
To gain insight into the behavior of the different candidate statistics, we made numeric simulations of the image acquisition process and studied the dependency of each statistic on the changes in fluorescence distribution within the cell (Supporting Material). To make the statistic independent of total fluorescence and illumination intensity, we decided to normalize the fluorescence in the boundary of the cell by total fluorescence. When we used the fluorescence integrated in the entire area corresponding to a cell for this normalization, the resulting statistic was not linear with the simulated fraction of recruitment (Fig. S2 B). This is because this estimation of total fluorescence is affected by the subcellular localization of the fluorescence: when the distribution changes from cytoplasmic to membrane associated, part of the fluorophores move to out-of-focus regions (above and below the focal plane). Due to confocality, light that originates in these regions does not reach the detector, and thus the total fluorescence estimated in this way decreases with recruitment (Fig. 2 B and Fig. S2 C).
Figure 2.
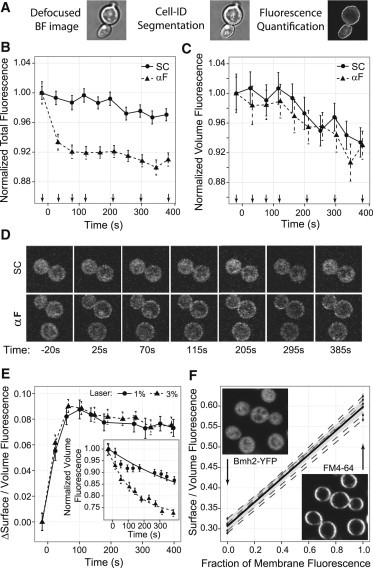
Membrane recruitment statistic. (A) Purposely defocused BF images (left) were processed by Cell-ID for image segmentation. Boundaries and IDs are overlaid in white on the BF image (center) and the fluorescence image (right). (B) Total YFP-Ste5 fluorescence in the focal plane versus time for cells treated with 100 nM α-factor (αF, triangles and dashed line) or SC medium (circles and solid line). Arrows on the x axis indicate times of the images in D. (C) Volume fluorescence versus time for the same cells as in B. (D) Image montage shows a representative cell for each treatment at the indicated times. (E) Increase in membrane recruitment statistic calculated as the ratio of surface to volume fluorescence versus time, for α-factor stimulated cells and two laser intensities that result in different photobleaching rates and total fluorescence. The inset shows the evolution of the volume fluorescence for these same cells, along with the best fit to exponential decay functions. The photobleaching rates are 1.7 ± 0.1 10−3s−1 for 1%, and 6.3 ± 0.3 10−3s−1 for 3% laser power. (F) Calibration of the recruitment statistic using cells with cytoplasmic fluorescence distribution of YFP (Bmh2-YFP) and membrane distribution of FM4-64. Each dashed line is an independent repetition of the calibration. The solid line (shaded region) represents the mean (standard error (SE)) of the calibration y = 0.317 ± 0.013 + x∗(0.291 ± 0.016).
To construct a measure of total fluorescence that does not depend on its distribution, we calculated the volume fluorescence from an image focused at the equatorial plane of the cell. We calculated the fluorescence intensity at each point of the 3D volume of the cell by assuming an ellipsoidal morphology, and then integrated the fluorescence in all of the volume of the cell. This is equivalent to rotating the image of the equatorial plane of the cell around the major axis of the cell and integrating the 3D object obtained (see details in Supporting Material). The volume fluorescence measured this way is independent of changes in the fluorescence distribution, as shown by numeric simulations (Fig. S3 E) and by the fact that cells that were stimulated with α-factor and had clear membrane recruitment showed no difference in the value of this statistic as compared with unstimulated cells in which the fluorescence remained cytoplasmic (Fig. 2, C and D).
In an analogous way, we calculated the surface fluorescence based on the fluorescence of the boundary pixels of the equatorial plane image and the estimated surface of the cell (Supporting Material). Thus, to construct a recruitment statistic (Srec) that satisfies the desired conditions, we divided the surface fluorescence by the volume fluorescence (calculated using Eqs. S5 and S4, respectively). Because this statistic is a ratio, it does not depend on the total fluorescence level, illumination intensity, or photobleaching. Accordingly, an increase in the laser power, which increases the photobleaching rate, did not affect the dynamics of the recruitment statistic (Fig. 2 E). Importantly, because the denominator of the recruitment statistic is independent of the fluorescence distribution, the recruitment statistic has a linear relationship with the fraction of membrane-associated fluorophores (Fig. S2 D).
Calibrating the recruitment statistic
Because of the diffraction-limited optical resolution of the fluorescence microscope, fluorescence that originates in the cytoplasm can reach pixels associated with the membrane and, vice versa, fluorescence from the membrane can reach internal pixels of the cell. Consequently, the measured ratio between the surface and volume fluorescence needs to be corrected for this effect.
To do this calibration, we acquired images of cells with known distributions of fluorescence, with an all-cytoplasmic or all-membrane distribution (Fig. 2 F). As a 0% recruitment calibration point, we used paraformaldehyde-fixed cells expressing Bmh2-YFP, an abundant protein with nuclear and cytoplasmic distribution. To obtain a 100% recruitment calibration point, we stained yeast with FM4-64, a lipophilic dye that in fixed cells labels only the PM. This dye is excited with the same laser used for YFP, but emits in a longer wavelength and thus can be easily separated. In this manner, we acquired images with extreme recruitment levels (0% and 100%) in the same cells in different channels. Because the recruitment statistic is linear with the fluorescence at the membrane, we can use these two extreme points to calibrate it (Fig. 2 F). We obtained the same result when we measured overexpressed YFP-Ste5 instead of Bmh2-YFP.
We calculated the fraction of membrane fluorescence (fmem) from the value of the recruitment statistic (srec) according to Eq. 1:
| (1) |
Here fauto is the fraction of the total fluorescence due to autofluorescence, and ρ is the slope of the calibration curve. The autofluorescence is caused by endogenous fluorophores of the cell that have a cytoplasmic localization. This localization is not affected by pheromone treatment (Fig. S5 A), and thus the recruitable fraction of fluorescence is given by 1 − fauto. We calculated the mean value of fauto by comparing the fluorescence intensities of the YFP-Ste5 expressing strain and its parental strain with no FP (Fig. S4 C). Note that the autofluorescence distribution is very broad and therefore it is possible to obtain recruitment levels greater than 100% for cells with low autofluorescence. In some cases, due to culture-to-culture variability in autofluorescence, the mean recruitment can also be slightly greater than 100%.
Note that we measure the increase in the recruitment statistic after stimulation. This approach has the advantage of eliminating the variability observed in the intercept of Fig. 2 F, and thus the uncertainty of the calibration curve is given only by the variability in the slope ρ, estimated as 0.291 ± 0.016. It also has the advantage of eliminating artifactual cell-to-cell variability in the basal level of the recruitment statistic (Fig. S4). The basal level of the recruitment statistic shows no significant difference between wild-type (WT) and Δste4 strains (Fig. S5 B), and therefore there is no detectable Ste4-dependent basal recruitment of Ste5.
The measurement of membrane recruitment is sensitive to the exact focal plane that is acquired. If this plane does not coincide with the equatorial plane of the cell, a bias will be introduced (Fig. S3). To make the measurement robust to changes in the focal plane, we acquired short Z stacks and then selected the image that corresponded to the equatorial plane (see Supporting Material for details).
Measurement of Ste5 recruitment D-R
It was previously shown that the D-R of the pheromone response pathway measured at the transcriptional level is graded (26,33,38), with no ultrasensitivity (steep D-R). In fact, the transcriptional D-R matched the fraction of occupied receptor at the membrane fairly well (19,39). However, at the Fus3 activation level, the D-R was slightly shifted to lower pheromone doses compared with the receptor occupancy (19). Therefore, using the described technique, we now measured the D-R relationship between pheromone levels and Ste5 recruitment. To do this, we stimulated yeast harboring two integrated copies of the PSTE5-YFP-STE5 construct with a range of pheromone concentrations and measured the amount of Ste5 that was recruited over time for the first 6 min (Fig. 3 A, left). The recruitment level and dynamics were dose dependent, with higher doses resulting in more and faster recruitment than lower doses. Measured at the steady-state (SS) level (∼6 min), the D-R had an EC50 (dose required for 50% recruitment) of 0.44 ± 0.08 nM and a Hill coefficient of 0.8 ± 0.1 (Fig. 3 B, solid line). This result indicates that recruitment is graded, with no ultrasensitivity, and that the D-R is more sensitive (shifted to lower pheromone doses) than the receptor occupancy.
Figure 3.
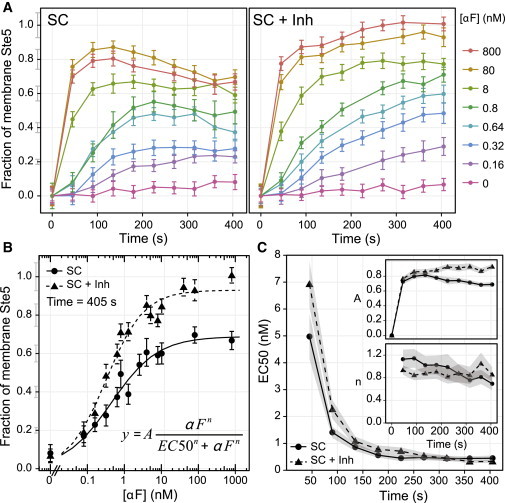
Time-dependent D-R curve for Ste5 membrane recruitment. (A) Fraction of membrane Ste5 versus time for the indicated doses of α-factor, in strain RY2013b (2x YFP-STE5, FUS3-Q93A) with (right) and without (left) 10 μM of the inhibitor 1-NM-PP1. Error bars of the data points indicate the 95% CI of the mean, and the error bars of the ticks of the y axis represent the uncertainty of the calibration (see details of error estimation in Supporting Material). (B) D-R curves of the fraction of membrane Ste5, 405 s after pheromone addition, with (dashed line, triangles) or without (solid line, circles) inhibitor. The curves represent the best fit to a Hill function by nonlinear least squares. Error bars as in A. (C) Time evolution of the parameters of the fits EC50 (inset: amplitude A and Hill coefficient n) for the curves with (dashed line, triangles) and without (solid line, circles) inhibitor. The shaded regions represent the SE of the estimated parameters.
We previously showed that a Fus3-mediated negative feedback was important for maintaining the D-R relationship at the Fus3 activation step. This was evidenced by a 20-fold shift to the left (more sensitive) of the D-R of phosphorylated Fus3 when Fus3 kinase activity was inhibited (19). To determine whether the relevant Fus3 feedback is the one acting on Ste5 recruitment, we repeated our measurement of the D-R in strains expressing a mutant Fus3 (Fus3-Q93A), which is sensitive to the ATP analog 1NM-PP1 (40). These cells behave as WT in SC medium (Fig. S5 D), but inhibition of Fus3 abolished the decline phase in recruitment that was evident in uninhibited cells after the initial peak (Fig. 3 A), confirming that the inhibitor was effective. However, treatment with inhibitor did not alter the D-R, and instead only resulted in higher recruitment when the highest doses of pheromone were used (Fig. 3, B and C). These data suggest that the Fus3 feedback on Ste5 is not the feedback that controls sensitivity to pheromone.
Measurement of effective binding affinity
We then sought to measure the effective binding affinity of Ste5 with its membrane-associated binding sites, and how it changed over time during the first few minutes of the pheromone response, when the Fus3-mediated negative feedback is first established. We reasoned that measuring the recruitment level at different concentrations of Ste5 would provide information about the total amount of binding sites and the binding affinity. For example, if the affinity were very high, virtually all of the available Ste5 would be recruited until all binding sites became occupied. Then, at saturation, the number of Ste5 molecules at the membrane would be virtually equivalent to the total amount of binding sites. On the other hand, if affinity were low, the amount of membrane-bound Ste5 would increase slowly with the total amount of Ste5, reaching saturation at higher total concentrations of Ste5.
To modify the amount of total Ste5, we used a series of strains with different numbers of integrations of the PSTE5-YFP-STE5 construct, which resulted in different abundances of YFP-Ste5 (18) (Fig. S1). We measured Ste5 membrane recruitment in these strains using a saturating amount of pheromone (Fig. 4 A).
Figure 4.
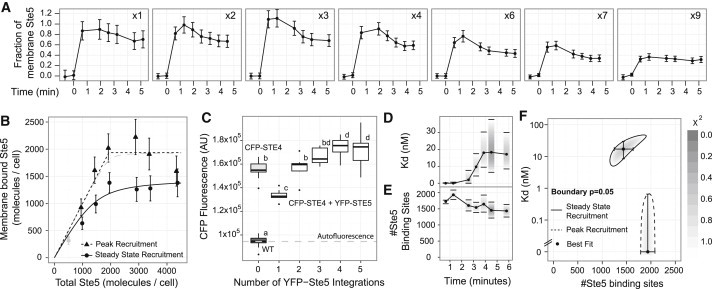
Measurement of effective binding affinity. (A) Membrane recruitment dynamics for strains with the indicated integration number of the PSTE5-YFP-STE5 construct. Error bars represent the 95% CIs for the mean plus the calibration uncertainty (see Supporting Material for details). (B) Bound amount of YFP-Ste5 versus total amount of YFP-Ste5, assuming a concentration of 484 molecules of YFP-Ste5 per integration, considering the recruitment at 4 min (circles) or the maximal level (triangles). The solid line is the best fit of a bimolecular association model for the recruitment level at 5 min, resulting in Kd = 17 ± 9 nM. For the peak recruitment level, only an upper bound for Kd = 0.65 nM (gray dashed line) can be determined, because the best fit results in a Kd of zero (black pointed line). (C) Mean CFP-STE4 membrane fluorescence for different strains, ordered by YFP-STE5 integration number. For each strain, nine images were acquired. The box-whisker represents the distribution of the averages of all cells within an image, the thick black line represents the median, and box boundaries represent the first and third quantiles. Whiskers extend to the most extreme value within 1.5 times the interquantile range, measured from the box boundary. Shared letters (a–d) between two strains indicate nonsignificant differences as computed by Tukey’s honest significance difference test, with α = 0.05. The dashed gray line represents the autofluorescence level. (D) Time evolution of Kd. The best fit of the bimolecular association model at each time is marked with a dot. The horizontal lines correspond to the 95% CI of the Kd. The bars are shaded according to the χ2 cost of the fit for the corresponding value of Kd (see Supporting Material for details). (E) Evolution of the total amount of Ste5 binding sites. Dots, lines, and bars as in panel D. (F) Regions of acceptable model parameters (Kd and number of Ste5 binding sites), for the SS (solid line) and peak (dashed line) recruitment. The area within these regions is shaded according to the χ2 cost of the fit. The dots represent the best fits, and the error bars indicate the 95% CIs as presented in D and E.
Because of the peak and decline dynamics (19), there are two interesting points at which Ste5 recruitment can be measured: the peak (or maximal) recruitment and the SS recruitment. Our previous measurements of Ste5 abundance by Western blot resulted in an estimated 484 ± 61 molecules per cell (18,35). Assuming a linear relationship between the YFP signal observed in each of the strains we constructed and the number of molecules of YFP-Ste5 per cell (Fig. S1), we can calculate the absolute amount of membrane-bound YFP-Ste5 for each level of total YFP-Ste5 (Fig. 3 B).
The number of Ste5 molecules that were recruited at the time of the peak increased linearly with the total amount of Ste5 up to 2000 molecules (four integrations), where it reached a plateau (dashed line in Fig. 4 B). This is consistent with a high-affinity binding and ∼2000 available binding sites for Ste5. Interestingly enough, our previous measure of the abundance of Gβγ and Ste5 by quantitative Western blot indicated that there are on average four Gβγ for every Ste5 (18). Note as well that the decline phase in the membrane-recruitment dynamics is less pronounced at high doses of Ste5 (Fig. 3 A).
To help us interpret our data and extract values of the relevant parameters, we used a simple bimolecular association model:
| (2) |
Here G represents the binding sites and Kd is the dissociation constant, which can be interpreted as the cytoplasmic concentration of Ste5 at which half of the binding sites are occupied.
The model assumes that the total amount of binding sites does not vary with the amount of Ste5. However, recent studies showed that the basal pathway activity (in the absence of pheromone) is affected by Ste5 abundance (18,41), and it is known that some components of the system are induced by the pathway (9). Therefore, we decided to test experimentally whether the amount of Gβ (the main known binding site for Ste5) varies with Ste5 abundance. To that end, we constructed strains expressing different levels of YFP-Ste5 in which the endogenous STE4 (Gβ) was tagged at the N-terminus with cyan FP (CFP). Except for the 1X YFP-Ste5 strain, the levels of CFP were fairly constant (Fig. 4 C). Because of this result, the 1X strain was not included in the subsequent analysis (gray points in Fig. 4 B), although doing so does not change the results.
The model further assumes that the binding reaction is much faster than the modulation of the available binding sites (G) or the binding affinity (Kd), i.e., that the binding is in quasi-SS. Note that the modulation to which we refer is not the initial one just after pheromone addition that causes the fast membrane recruitment (within seconds), but rather the slow modulation that causes the decline dynamics, which occurs in a timescale of minutes (Fig. 3 A). Binding reactions normally reach equilibrium within seconds, and it only takes a fraction of a second for a molecule like Ste5 to diffuse through the cytoplasm to the membrane (Supporting Material). Furthermore, because Ste5 is recruited to the membrane a few seconds after pheromone addition, and recruitment involves several steps including Ste5 diffusion and binding, the diffusion and binding of Ste5 must take at most a few seconds.
Fitting this model to the SS data results in an estimated amount of binding sites of GtotSS = 1400 ± 200 (best fit ± 95% CI) molecules and an effective binding affinity of KdeSS = 17 ± 9 nM (solid line in Fig. 4 F). To our knowledge, this is the first measurement of binding affinity between Ste5 and PM binding sites (likely mostly Gβγ).
The same analysis done on the peak recruitment levels instead of the SS ones (dashed lines and triangles in Fig. 4 B) resulted in a total amount of sites of Gtotpeak = 1940 ± 150 molecules, which is consistent with the abundance of Ste4 and Ste18 (Gβ and Gγ) as measured by quantitative Western blots (18). In this case, we could only determine an upper bound for the effective binding affinity, of Kdepeak < 0.65 nM, because any value lower than that would have resulted in an acceptable fit to the data (Fig. 4, B and F).
Finally, we determined the dynamics of the change in Kde and in total binding sites from peak to SS by fitting data of the intervening time points to the binding model. In Fig. 4 D, we show that the dynamics of the Kde follows a roughly sigmoidal curve, beginning to increase after 2 min and reaching a maximum value at ∼4 min. On the other hand, the total number of binding sites peaks after the first minute and then slowly decreases to a plateau at ∼5 min (Fig. 4 E).
Discussion
In this work, we developed a quantitative method to measure relocalization of a protein to the PM. We have shown that our measurement is linear with the biological quantity of interest and is calibrated. The resulting measurement gives the absolute fraction of membrane-associated fluorescence.
Using the described method, we were able to measure the population average change of localization of as few as 300 mature fluorescent molecules per cell (35). This sensitivity allowed us to measure Ste5 recruitment dynamics at endogenous levels of expression. Changes in the abundance of Ste5 alter key parameters in the performance of the system (18,41), which makes it very important to measure Ste5 activation at physiological levels of expression.
Here we show that the D-R relationship at the Ste5 recruitment step is graded, in agreement with previous work that showed that the pheromone response pathway measured at various points down the signaling cascade is not ultrasensitive (switch-like) (19,26,33,38,39). The D-R was more sensitive (left-shifted) than receptor occupancy and gene expression, suggesting that downstream of Ste5 recruitment, the pathway is able to realign (right-shift) the D-R. Surprisingly, removal of negative feedback from Fus3 did not sensitize (left-shift) the D-R of Ste5 recruitment, in contrast to the strong effect this action has on the D-R at the Fus3 activation step. This result shows that other negative feedbacks, downstream of Ste5 recruitment, must exist to maintain the proper external signal sensitivity. Candidate feedback targets include the Fus3 phosphatase Msg5 (42), the Fus3 kinase Ste7 (43), and Ste5 itself, which has multiple Fus3 sites that modulate signaling downstream of recruitment (21,23).
With our method and a simple bimolecular association model, we were able to determine the effective affinity between Ste5 and its membrane-associated binding sites (presumably Gβγ). To our knowledge, this constitutes the first experimental estimate of this parameter. The total amount of binding sites estimated by this method is in very good agreement with the abundances of Gβγ as measured by quantitative Western blots (18), suggesting that G protein dissociation is nearly 100% at this saturating concentration of pheromone. In this case, it further suggests that binding to Gβγ is constantly necessary to remain in the membrane (i.e., Ste5 is probably unable to remain attached to the membrane via other interactions for any significant time in the absence of Gβγ).
The membrane recruitment of Ste5 shows a characteristic peak and decline dynamics (19). This behavior allowed us to measure the change in binding affinity of Ste5 during the initial response to pheromone, at the time when the MAPK negative feedback is first established. The total amount of binding sites remained fairly constant, and slowly decreased by only 28% from the peak to the SS level. In contrast, the binding affinity decreased at least 10-fold (Kde increased from <0.65 nM to ∼10 nM).
The turnover rate of Ste5 polarized at the tip of the mating projection was previously measured by fluorescence recovery after photobleaching and resulted in a half-time of τ1/2 = 8.2 ± 1.3 s (44). Assuming that this value, measured >1 h after pheromone treatment, is valid at the SS reached in the first 5 min of the response, using it in combination with our Kde, we calculate a Koff = 0.085 ± 0.013 s−1 and Kon = 0.005 ± 0.003 nM−1 s−1 for the binding and unbinding rates of Ste5 with its binding sites.
The binding reaction equilibrates much faster than the characteristic time of the decline phase of Ste5 membrane recruitment; therefore, we assume that at each time the amount of membrane-bound Ste5 is in equilibrium with the amount of binding sites (quasi-SS approximation). This means that all of the complexity of the system (e.g., negative feedback loops) is captured by the evolution of the parameters of the binding model (i.e., the amount of binding sites and the affinity), which eliminates the need to model these complex regulations explicitly. The decline phase in Ste5 membrane recruitment dynamics depends on the activity of the MAPK Fus3, indicating that a negative feedback loop exists in this system (19). This negative feedback acts downstream of G protein dissociation, and at or upstream of Ste5 recruitment. Our results suggest that a Fus3-mediated reduction of Ste5 binding affinity is the main reason for the decline in the recruitment levels. This suggests that the negative feedback may be due to Fus3 phosphorylation of either Ste5 or Gβγ, which would result in loss of affinity. In agreement with this hypothesis, Fus3 phosphorylation of Ste5 was recently reported (21,23). An alternative explanation for the observed decrease in effective binding affinity would be a competitive inhibition, i.e., the binding of second molecule at or nearby the Ste5 binding sites. This is analogous to the effect of a competitive antagonist in a ligand-receptor interaction, which decreases the apparent affinity but not the maximal response (45). Note, however, that this interpretation requires the interaction between the competitive inhibitor and Ste5’s binding sites to be dependent on the activity of Fus3. The decline phase of the membrane recruitment curves tends to disappear at high doses of Ste5. This is consistent with the notion that the decline is due to a decrease in affinity or a competitive inhibition, and can therefore be compensated for by an increase in the dose of Ste5. If the decline were due to a decrease in the amount of binding sites, it should be seen at all levels of scaffold protein.
To correctly quantify the subcellular fluorescence distribution, one must take the 3D geometry of the cell into account. In general, this can be achieved with 4D imaging (time course of Z stacks) and sophisticated image-processing techniques such as surface reconstruction (46–48). This method uses fluorescence intensity levels to define boundaries between subcellular compartments. Once the surfaces are defined, the total fluorescence in each compartment can be calculated by integrating the gray level of the corresponding voxels (3D analogs of pixels). Note that a strong fluorescence mark is required to define the boundary; therefore, to measure membrane recruitment using this method, one must use a membrane marker in another channel to define the boundary when the recruitment level of the protein of interest is low. Furthermore, enough z-slices to span the entire cell are needed, which increases the total number of images acquired.
To reduce quantification errors due to the limited resolution of the fluorescence microscope (especially in the z axis), the images may be deconvolved (49). This image processing technique uses information about the microscope optics, described by the point spread function (PSF), to correct the original images by reducing the bleed-through between contiguous voxels. One can measure the PSF by imaging subresolution particles such as fluorescence beads; however, this approach gives limited precision because the sample itself is part of the optical system and thus distorts the PSF (50). To overcome this problem, investigators have developed blind deconvolution (50,51) methods that estimate both the deconvolved image and the PSF from the original images. These methods are computationally intense.
The segmentation of yeast cells can be done with transmission images, thereby sparing a fluorescence channel to be used to measure other processes of interest. Because yeast cells have a simple geometry, there is no need to do complex surface reconstructions; instead, symmetry considerations can be used to calculate the volume fluorescence from a single image. This means that fewer images have to be acquired, which reduces photobleaching and phototoxicity, and permits a higher sampling rate. The calibration procedure presented here accounts for the light that bleeds through subcellular compartments and thus eliminates the need for image deconvolution. Furthermore, because the calibration was done in the context of our sample, any distortion of the PSF is automatically taken into account.
The described method produces single-cell data, allowing one in principle to study the population heterogeneity that is otherwise hidden in the population average. For the single-cell measurements to be reliable, a stronger fluorescence signal relative to the autofluorescence than the one obtained in this study is required. This could be obtained by using a brighter FP or tandem repeats of FP tags.
Modifications of the method presented in this work can be used to measure other proteins that change localization between different subcellular compartments. To that end, one must follow four key steps: 1), segment the cells and the subcellular compartments of interest (for some cell types and subcellular compartments, fluorescence markers may be required); 2), develop a measure of total fluorescence that is independent of the fluorescence localization (for cell types with regular geometry, this can probably be achieved from one or a few images; for cells with irregular shape, surface reconstruction techniques may be required); 3), use this measure of total fluorescence to normalize the signal of the compartment of interest; and 4), use cells marked with extreme distributions of fluorescence to calibrate the statistic.
Acknowledgments
We thank G. Pesce, A. Chernomoretz, A. Ventura, R. Brent, G. Vasen, V. Repetto, R. Baltanás, L. Durrieu, M. Blaustein, A.V. Grande, L. Acerenza, E. Klipp, and V. Levi for discussion and/or comments on the manuscript.
This work was supported by grants PICT2005-33624, PICT2007-847, and PICT2010-2248 from the Argentine Agency of Research and Technology, and grant 1R01GM097479-01, subaward 0000713502 from the National Institute of General Medical Sciences, National Institutes of Health.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Kholodenko B.N., Hoek J.B., Westerhoff H.V. Why cytoplasmic signalling proteins should be recruited to cell membranes. Trends Cell Biol. 2000;10:173–178. doi: 10.1016/s0962-8924(00)01741-4. [DOI] [PubMed] [Google Scholar]
- 2.Leonard W.J., O’Shea J.J. Jaks and STATs: biological implications. Annu. Rev. Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- 3.Fayard E., Tintignac L.A., Hemmings B.A. Protein kinase B/Akt at a glance. J. Cell Sci. 2005;118:5675–5678. doi: 10.1242/jcs.02724. [DOI] [PubMed] [Google Scholar]
- 4.Sakai N., Sasaki K., Saito N. Direct visualization of the translocation of the gamma-subspecies of protein kinase C in living cells using fusion proteins with green fluorescent protein. J. Cell Biol. 1997;139:1465–1476. doi: 10.1083/jcb.139.6.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oancea E., Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- 6.Pinton P., Tsuboi T., Rutter G.A. Dynamics of glucose-induced membrane recruitment of protein kinase C beta II in living pancreatic islet beta-cells. J. Biol. Chem. 2002;277:37702–37710. doi: 10.1074/jbc.M204478200. [DOI] [PubMed] [Google Scholar]
- 7.Aronheim A., Engelberg D., Karin M. Membrane targeting of the nucleotide exchange factor Sos is sufficient for activating the Ras signaling pathway. Cell. 1994;78:949–961. doi: 10.1016/0092-8674(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 8.Herskowitz I. MAP kinase pathways in yeast: for mating and more. Cell. 1995;80:187–197. doi: 10.1016/0092-8674(95)90402-6. [DOI] [PubMed] [Google Scholar]
- 9.Dohlman H.G., Thorner J.W. Regulation of G protein-initiated signal transduction in yeast: paradigms and principles. Annu. Rev. Biochem. 2001;70:703–754. doi: 10.1146/annurev.biochem.70.1.703. [DOI] [PubMed] [Google Scholar]
- 10.Pryciak P.M., Huntress F.A. Membrane recruitment of the kinase cascade scaffold protein Ste5 by the Gbetagamma complex underlies activation of the yeast pheromone response pathway. Genes Dev. 1998;12:2684–2697. doi: 10.1101/gad.12.17.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng Y., Song L.Y., Elion E.A. Functional binding between Gbeta and the LIM domain of Ste5 is required to activate the MEKK Ste11. Curr. Biol. 1998;8:267–278. doi: 10.1016/s0960-9822(98)70108-3. [DOI] [PubMed] [Google Scholar]
- 12.Winters M.J., Lamson R.E., Pryciak P.M. A membrane binding domain in the ste5 scaffold synergizes with gbetagamma binding to control localization and signaling in pheromone response. Mol. Cell. 2005;20:21–32. doi: 10.1016/j.molcel.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 13.Garrenton L.S., Young S.L., Thorner J. Function of the MAPK scaffold protein, Ste5, requires a cryptic PH domain. Genes Dev. 2006;20:1946–1958. doi: 10.1101/gad.1413706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leeuw T., Fourest-Lieuvin A., Leberer E. Pheromone response in yeast: association of Bem1p with proteins of the MAP kinase cascade and actin. Science. 1995;270:1210–1213. doi: 10.1126/science.270.5239.1210. [DOI] [PubMed] [Google Scholar]
- 15.Lyons D.M., Mahanty S.K., Elion E.A. The SH3-domain protein Bem1 coordinates mitogen-activated protein kinase cascade activation with cell cycle control in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996;16:4095–4106. doi: 10.1128/mcb.16.8.4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slaughter B.D., Smith S.E., Li R. Symmetry breaking in the life cycle of the budding yeast. Cold Spring Harb. Perspect. Biol. 2009;1:a003384. doi: 10.1101/cshperspect.a003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madden K., Snyder M. Cell polarity and morphogenesis in budding yeast. Annu. Rev. Microbiol. 1998;52:687–744. doi: 10.1146/annurev.micro.52.1.687. [DOI] [PubMed] [Google Scholar]
- 18.Thomson T.M., Benjamin K.R., Brent R. Scaffold number in yeast signaling system sets tradeoff between system output and dynamic range. Proc. Natl. Acad. Sci. USA. 2011;108:20265–20270. doi: 10.1073/pnas.1004042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu R.C., Pesce C.G., Brent R. Negative feedback that improves information transmission in yeast signalling. Nature. 2008;456:755–761. doi: 10.1038/nature07513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kranz J.E., Satterberg B., Elion E.A. The MAP kinase Fus3 associates with and phosphorylates the upstream signaling component Ste5. Genes Dev. 1994;8:313–327. doi: 10.1101/gad.8.3.313. [DOI] [PubMed] [Google Scholar]
- 21.Malleshaiah M.K., Shahrezaei V., Michnick S.W. The scaffold protein Ste5 directly controls a switch-like mating decision in yeast. Nature. 2010;465:101–105. doi: 10.1038/nature08946. [DOI] [PubMed] [Google Scholar]
- 22.Flotho A., Simpson D.M., Elion E.A. Localized feedback phosphorylation of Ste5p scaffold by associated MAPK cascade. J. Biol. Chem. 2004;279:47391–47401. doi: 10.1074/jbc.M405681200. [DOI] [PubMed] [Google Scholar]
- 23.Bhattacharyya R.P., Reményi A., Lim W.A. The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science. 2006;311:822–826. doi: 10.1126/science.1120941. [DOI] [PubMed] [Google Scholar]
- 24.Strickfaden S.C., Winters M.J., Pryciak P.M. A mechanism for cell-cycle regulation of MAP kinase signaling in a yeast differentiation pathway. Cell. 2007;128:519–531. doi: 10.1016/j.cell.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garrenton L.S., Braunwarth A., Thorner J. Nucleus-specific and cell cycle-regulated degradation of mitogen-activated protein kinase scaffold protein Ste5 contributes to the control of signaling competence. Mol. Cell. Biol. 2009;29:582–601. doi: 10.1128/MCB.01019-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi S., Pryciak P.M. Membrane localization of scaffold proteins promotes graded signaling in the yeast MAP kinase cascade. Curr. Biol. 2008;18:1184–1191. doi: 10.1016/j.cub.2008.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stauffer T.P., Ahn S., Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol. 1998;8:343–346. doi: 10.1016/s0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- 28.Dundr M., McNally J.G., Misteli T. Quantitation of GFP-fusion proteins in single living cells. J. Struct. Biol. 2002;140:92–99. doi: 10.1016/s1047-8477(02)00521-x. [DOI] [PubMed] [Google Scholar]
- 29.Hirschberg K., Miller C.M., Lippincott-Schwartz J. Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J. Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kofahl B., Klipp E. Modelling the dynamics of the yeast pheromone pathway. Yeast. 2004;21:831–850. doi: 10.1002/yea.1122. [DOI] [PubMed] [Google Scholar]
- 31.Schaber J., Kofahl B., Klipp E. A modelling approach to quantify dynamic crosstalk between the pheromone and the starvation pathway in baker’s yeast. FEBS J. 2006;273:3520–3533. doi: 10.1111/j.1742-4658.2006.05359.x. [DOI] [PubMed] [Google Scholar]
- 32.Shao D., Zheng W., Tang C. Dynamic studies of scaffold-dependent mating pathway in yeast. Biophys. J. 2006;91:3986–4001. doi: 10.1529/biophysj.106.081661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colman-Lerner A., Gordon A., Brent R. Regulated cell-to-cell variation in a cell-fate decision system. Nature. 2005;437:699–706. doi: 10.1038/nature03998. [DOI] [PubMed] [Google Scholar]
- 34.Fink G.R., Guthrie C. Academic Press; New York: 1991. Guide to Yeast Genetics and Molecular Biology. [Google Scholar]
- 35.Gordon A., Colman-Lerner A., Brent R. Single-cell quantification of molecules and rates using open-source microscope-based cytometry. Nat. Methods. 2007;4:175–181. doi: 10.1038/nmeth1008. [DOI] [PubMed] [Google Scholar]
- 36.Bush A., Chernomoretz A., Colman-Lerner A. Using Cell-ID 1.4 with R for microscope-based cytometry. Curr. Protoc. Mol. Biol. 2012;Chapter 14 doi: 10.1002/0471142727.mb1418s100. Unit 14.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cedersund G., Roll J. Systems biology: model based evaluation and comparison of potential explanations for given biological data. FEBS J. 2009;276:903–922. doi: 10.1111/j.1742-4658.2008.06845.x. [DOI] [PubMed] [Google Scholar]
- 38.Poritz M.A., Malmstrom S., Kamb A. Graded mode of transcriptional induction in yeast pheromone signalling revealed by single-cell analysis. Yeast. 2001;18:1331–1338. doi: 10.1002/yea.777. [DOI] [PubMed] [Google Scholar]
- 39.Yi T.M., Kitano H., Simon M.I. A quantitative characterization of the yeast heterotrimeric G protein cycle. Proc. Natl. Acad. Sci. USA. 2003;100:10764–10769. doi: 10.1073/pnas.1834247100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bishop A.C., Ubersax J.A., Shokat K.M. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- 41.Chapman S.A., Asthagiri A.R. Quantitative effect of scaffold abundance on signal propagation. Mol. Syst. Biol. 2009;5:313. doi: 10.1038/msb.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doi K., Gartner A., Matsumoto K. MSG5, a novel protein phosphatase promotes adaptation to pheromone response in S. cerevisiae. EMBO J. 1994;13:61–70. doi: 10.1002/j.1460-2075.1994.tb06235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elion E.A., Satterberg B., Kranz J.E. FUS3 phosphorylates multiple components of the mating signal transduction cascade: evidence for STE12 and FAR1. Mol. Biol. Cell. 1993;4:495–510. doi: 10.1091/mbc.4.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Drogen F., Stucke V.M., Peter M. MAP kinase dynamics in response to pheromones in budding yeast. Nat. Cell Biol. 2001;3:1051–1059. doi: 10.1038/ncb1201-1051. [DOI] [PubMed] [Google Scholar]
- 45.Motulsky H.J.M., Mahan L.C. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol. Pharmacol. 1984;25:1–9. [PubMed] [Google Scholar]
- 46.Eils R., Athale C. Computational imaging in cell biology. J. Cell Biol. 2003;161:477–481. doi: 10.1083/jcb.200302097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerlich D., Ellenberg J. 4D imaging to assay complex dynamics in live specimens. Nat. Cell Biol. 2003;(Suppl):S14–S19. [PubMed] [Google Scholar]
- 48.Gerlich D., Beaudouin J., Eils R. Four-dimensional imaging and quantitative reconstruction to analyse complex spatiotemporal processes in live cells. Nat. Cell Biol. 2001;3:852–855. doi: 10.1038/ncb0901-852. [DOI] [PubMed] [Google Scholar]
- 49.Cannell M.B., McMorland A., Soeller C. Image enhancement by deconvolution. In: Pawley J.B., editor. Handbook of Biological Confocal Microscopy. Springer; New York: 2006. pp. 488–500. [Google Scholar]
- 50.Holmes T.J., Biggs D., Abu-Tarif A. Blind deconvolution. In: Pawley J.B., editor. Handbook of Biological Confocal Microscopy. Springer; New York: 2006. pp. 468–487. [Google Scholar]
- 51.Stolze K., Holmes S.C., Gait M.J. Novel spermine-amino acid conjugates and basic tripeptides enhance cleavage of the hairpin ribozyme at low magnesium ion concentration. Bioorg. Med. Chem. Lett. 2001;11:3007–3010. doi: 10.1016/s0960-894x(01)00608-4. [DOI] [PubMed] [Google Scholar]
- 52.Zhang B., Zerubia J., Olivo-Marin J.C. Gaussian approximations of fluorescence microscope point-spread function models. Appl. Opt. 2007;46:1819–1829. doi: 10.1364/ao.46.001819. [DOI] [PubMed] [Google Scholar]
- 53.Uchida M., Sun Y., Larabell C.A. Quantitative analysis of yeast internal architecture using soft X-ray tomography. Yeast. 2011;28:227–236. doi: 10.1002/yea.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raue A., Kreutz C., Timmer J. Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood. Bioinformatics. 2009;25:1923–1929. doi: 10.1093/bioinformatics/btp358. [DOI] [PubMed] [Google Scholar]
- 55.Ozisik M.N. John Wiley & Sons; New York: 1993. Heat Conduction. [Google Scholar]
- 56.Loew L.M. The Virtual Cell Project. Novartis Found. Symp. 2002;247:151–160. discussion 160–151, 198–206, 244–152. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.