

Abstract
The assembly of proteins into multidomain complexes is critical for their function. In eukaryotic nonmuscle cells, regulation of the homodimeric actin cross-linking protein α-actinin-4 (ACTN4) during cell migration involves signaling receptors with intrinsic tyrosine kinase activity, yet the underlying molecular mechanisms are poorly understood. As a first step to address the latter, we validate here an atomic model for the ACTN4 end region, which corresponds to a ternary complex between the N-terminal actin-binding domain (ABD) and an adjacent helical neck region of one monomer, and the C-terminal calmodulin-like domain of the opposite antiparallel monomer. Mutagenesis experiments designed to disrupt this ternary complex confirm that its formation reduces binding to F-actin. Molecular dynamics simulations show that the phosphomimic mutation Y265E increases actin binding by breaking several interactions that tether the two calponin homology domains into a closed ABD conformation. Simulations also show a disorder-to-order transition in the double phosphomimic mutant Y4E/Y31E of the 45-residue ACTN4 N-terminal region, which can inhibit actin binding by latching both calponin homology domains more tightly. Collectively, these studies provide a starting point for understanding the role of external cues in regulating ACTN4, with different phenotypes resulting from changes in the multidomain assembly of the protein.
Introduction
The α-actinin (ACTN) family of actin-binding proteins plays a critical role in the tightly regulated remodeling of the actin cytoskeleton (1,2). Generally considered responsible for actin filament (F-actin) cross-linking, the ACTNs are a highly conserved family of proteins with four mammalian isoforms: ACTN2 and ACTN3 are specific to muscle cells, whereas ACTN1 and ACTN4 are expressed in nonmuscle cells. In addition to F-actin crosslinking, ACTNs may bridge the cytoskeletal network to the cell membrane with ACTN4, in particular, essential for maintaining normal cell motility (3–5).
The sheer size of the ACTNs (Fig. 1) has so far only allowed us to resolve parts of their structures that involve pairwise interactions between domains. For instance, formation of antiparallel homodimers is essential for ACTN function and is facilitated through the sturdy spectrin repeats, with a Kd of dimer formation at the picomolar level (6,7). Experimental evidence has also shown a possible interaction between the second globular head of the CaM-like domain (CaM2) and a region referred to as the neck (residues 271–300, numbering based on human ACTN4 sequence) (8). Finally, crystal structures of the actin-binding domain (ABD) for three of the ACTN isoforms show both of its component calponin homology (CH) domains linked together in what is referred to as a closed ABD conformation (9–11). Interestingly, a recent low-resolution cryo-electron microscopy (cryo-EM) structure of the complex between the ABD and F-actin strongly suggests that prior opening of the ABD via separation of its two CH domains is required for actin binding to take place (12). The putative actin-binding site (ABS) is helix G of CH1, which binds at the hydrophobic cleft between actin subdomains 1 and 3, a purported hot spot for actin-binding proteins (13).
Figure 1.
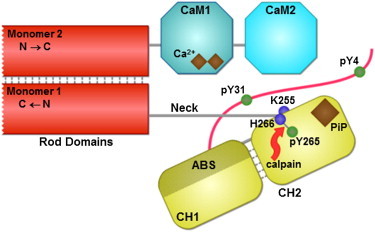
Domain architecture of ACTN4. Human ACTN4 (911 residues) forms antiparallel homodimers through binding of the spectrin rod domains. Monomer 1 shows an unstructured region; an ABD comprised of two CH domains (CH1 contains the ABS and CH2 has a putative PiP-binding site); and a neck region that connects CH2 to the first spectrin repeat of the rod domain. Monomer 2 shows the rod domain and C-terminal CaM-like domain (only CaM1 contains putative binding sites for calcium ions). Also shown are a target for m-calpain proteolysis between Y265 and H266, and residue K255 that has been implicated as a disease mutation site. The tyrosine residues Y4, Y31, and Y265 are phosphorylatable.
Multiple mechanisms exist to regulate the binding of ACTNs to F-actin. These include tyrosine phosphorylation (14,15); binding of calcium ions in the Ca2+-sensitive nonmuscle isoforms (16,17); limited proteolysis by ubiquitous intracellular enzymes calpain-1 and calpain-2 (18,19); and binding of phosphatidylinositol intermediaries (e.g., PiP2 and PiP3) to a putative binding site on the ABD (20,21). Even though much biological insight has been gained from structural studies, the majority of human proteins are composed of multiple interacting domains (22) whose assemblies and interactions, especially in response to various external cues, still remain to be deciphered. For instance, no molecular mechanism has been put forward yet to rationalize how the interactions between actin-binding proteins and F-actin are differentially regulated. In the case of nonmuscle ACTNs, their dynamic control by extracellular signals during cell migration provides a special impetus to understand the structural changes they undergo as a result of various covalent and noncovalent modifications.
As a first step to understanding how external cues lead to regulation of actin binding in human ACTN4, we validate here a structural model for the end regions of this homodimer, which we found involves a multidomain complex between the ABD, the neck region, and CaM2. Three independent sets of mutants that were designed to disrupt this complex confirm the structure of this module. Molecular dynamics (MD) simulations of our model provide a possible mechanism that can explain why phosphorylation at residue Y265 in the CH2 domain leads to enhanced actin binding (15). Simulations and virtual docking also show a disorder-to-order transition in a double phosphomimic mutant Y4E/Y31E of the 45-residue N-terminal region, which can account for the observed decrease in actin binding upon double phosphorylation at Y4/Y31 (15). Our structural model of the ACTN4 end region thus offers, to our knowledge, a first glimpse of how multi-domain interactions can be used by highly regulated systems to trigger the coherent signaling expected in the inner working of cells.
Materials and Methods
Structural modeling of the neck-CaM2 complex
Using the best representative conformer (model 1) from the solution structure of the complex between titin and ACTN2 CaM2 (PDB ID 1H8B) (23), two homology models for the ACTN4 neck-CaM2 complex were built by doing in silico mutations (with fixed backbones) in PyMOL Molecular Graphics System, version 1.5.0.4 (Schrödinger, LLC) to match the ACTN4 neck and CaM2 sequences. The first model was based on a previous multiple sequence alignment (8) between titin and ACTN2 neck (which shares 83% and 97% sequence identity and similarity, respectively, with ACTN4 neck). The second model has the neck shifted by one helical turn relative to the first model such that only hydrophobic residues comprise the neck surface at the binding interface. To assess the stability of these homology models, a 50-ns MD simulation was performed for each as described below.
Structural modeling of the ACTN4 homodimer end region
In a recent low-resolution cryo-EM structure of chicken ACTN1 (PDB ID 1SJJ) (25), one end of the homodimer has a closed ABD conformation with the CH2 domain in close proximity to CaM2. By structurally aligning this closed ABD end with the second neck-CaM2 homology model mentioned previously, using CaM2 as the reference for alignment, we came up with a structural model for a possible ternary complex involving ABD-neck-CaM2. To test if this model is feasible, we attempted to add the other ACTN domains. First, we added the spectrin rod domains by connecting the helical neck from this model to the first spectrin repeat (SR1) from one of the monomers in the human ACTN2 rod dimer crystal structure (PDB ID 1HCI) (26). Next, by structurally aligning CaM2 from this model and the calcium-free solution structure of calmodulin (PDB ID 1CFC) (27), we found that the first globular head (CaM1) from the solution structure can be easily connected to the fourth spectrin repeat (SR4) from the other monomer in the rod dimer, thereby allowing us to add CaM1 to this model. In silico mutations were then done in PyMOL to match the ACTN4 sequence for both the rod dimer and CaM1. Based on sequence alignments, an additional insertion of five residues had to be made in the loop connecting the two EF-hand motifs of CaM1. We also replaced the ABD from the cryo-EM structure with the crystal structure for a human ACTN4 ABD mutant (PDB ID 2R0O) (11), with K255E back-mutated in silico to Lys. The different domains were joined together in Chimera (28), followed by energy minimization via steepest descent for 1000 steps to fix the peptide bonds between the connected domains. From this full wild-type (WT) atomic model (containing ABD, neck, spectrin rods, CaM1, and CaM2), an in silico mutation was done to make a separate full model for the Y265E mutant. For both full models, a 100-ns MD simulation was performed as described below.
MD simulations
Each initial protein conformation was solvated with TIP3P water molecules (29) in an octahedral water box with 15.0 Å between the edge of the protein and the edge of the water box. Counterions were added to neutralize the total system charge. A cutoff of 8.0 Å was used for calculating short-range Lennard-Jones and electrostatic interactions, and long-range electrostatic contributions were computed using the particle-mesh Ewald approach (30). A time step of 2 fs was used and the length of all bonds involving hydrogen were kept constant using the SHAKE algorithm (or SETTLE for water) (31,32). Initial energy minimization was done using the steepest descent algorithm for 1000 steps. This was followed by equilibration under NVT at 300 K for 50 ps using Langevin dynamics with a collision frequency of 1.0 ps−1, and then equilibration under NPT at 1 atm for another 50 ps using a pressure time constant of 2 ps. All heavy atoms had position restraints up to this point. Production runs were finally done under NVT conditions with no position restraints. The Amber ff12SB force field was used in all runs. Simulations were GPU-accelerated using version 12 of the Amber MD software suite (33,34). The resulting trajectories were visualized in VMD (35). Changes in solvent accessible surface area (ΔSASA) were computed in VMD and changes in relative binding free energy upon complex formation were estimated using FastContact (36).
Langevin dynamics simulations
Simulations of WT and mutant Y4E/Y31E sequences of the 45-residue N-terminal were performed using Langevin dynamics under implicit solvent conditions. Both constructs were initially set in fully extended conformations. The Amber96 force field was used (37), with GBSA implicit solvation implemented using the OBC algorithm (38). Initial energy minimization was done using the steepest descent algorithm for 1000 steps. Production runs were then done for a total simulation time of 100 ns with a time step of 2 fs. All runs were GPU-accelerated using the Zephyr GUI of OpenMM (39). The resulting trajectories were visualized in VMD, where for each trajectory a residue helicity plot over sequential 100-ps windows was computed.
Virtual docking
A helical model of the N-terminal region comprising residues 31–41 and with the Y31E mutation was constructed in PyMOL. This model was used as ligand for docking to the crystal structure of a human ACTN4 ABD mutant (PDB ID 2R0O), with K255E back-mutated to Lys. Virtual docking was done using ClusPro (40,41), with top-ranked docked models filtered based on balanced contributions between electrostatic and hydrophobic contacts.
Mutagenesis of ACTN4 and construction of expression plasmid
Mutagenesis of ACTN4 was performed by site-directed polymerase chain reaction (PCR). After purification by agarose gel electrophoresis and digestion with appropriate restricted enzymes, WT and mutant ACTN4 PCR fragments were cloned into bacterial expression vector pET-28a to construct His-tagged proteins. Positive plasmid was further transformed into a BL21 strain and then induced with 1 mM isopropyl 1-thio-D-galactopyranoside for 4 h at 37°C to express target proteins. Cell pellet was lysed in nickel column-binding buffer in the presence of a protease inhibitors mixture set V (catalog No. 539137; EMD Calbiochem, Billerica MA). The soluble ACTN4 was purified with HisTrapTM HP (catalog No. 17-5247-01; GE Healthcare). Fractions containing proteins of interest were pooled and completely dialyzed against actin-binding buffer in the absence of ATP for 24 h at 4°C with three changes of buffer.
F-actin sedimentation assays
F-actin sedimentation assays were performed as described previously (15).
Results
Structural model of the neck-CaM2 complex
An outstanding question about the antiparallel homodimer is whether there are intermolecular interactions of note between the two monomers, besides those involving the spectrin repeats. That this is possible was shown by Young and Gautel (8), and it is further supported by homology to known complexes. CaM2 of muscle-specific ACTN isoforms can bind to titin, where the interaction site on the latter has been localized to the N-terminal Z-repeat motifs (42–45). These Z-repeats were shown to gain helical secondary structure upon complex formation with CaM2 (46,47), which was confirmed in a solution structure of this complex (PDB ID 1H8B) (23). Multiple sequence alignment of ACTN2 with several Z-repeat variants led to a putative binding site within the neck region, which was confirmed to bind CaM2 with nanomolar affinity (8).
Using the 1H8B solution structure, we built a homology model for the ACTN4 neck-CaM2 complex such that the binding interface of the neck is predominantly nonpolar (Fig. 2 A). This model has hydrophobic residues covering the three helical turns of the neck surface that face the hydrophobic pocket of CaM2, similar to what is seen in the solution structure (Fig. 2 B, top alignment). Note that our homology model is shifted by about one helical turn compared to a model based on the alignment proposed in (8) (Fig. 2 B, bottom alignment), which has a polar NQ sequence buried on the third helical turn. The 30 NMR models in PDB 1H8B have an average change in solvent accessible surface area (ΔSASA) of 1326 ± 88 Å2 upon forming the titin-CaM2 complex, very similar to our neck-CaM2 homology model ΔSASA of 1318 Å2 (see Table 1). We estimate the relative binding free energy for the contacts in our complex model at −19.7 kcal/mol using FastContact (36). We similarly estimate the binding free energy of the titin-CaM2 complex at −23.3 ± 5.0 kcal/mol (error based on estimates from each NMR model); this indicates that our neck-CaM2 model is predicted to form a tight complex similar to titin-CaM2, consistent with experimental results obtained from isothermal calorimetry (8). Fig. 2 C shows that, in addition to the hydrophobic interactions within the binding interface, polar contacts also contribute in stabilizing our neck-CaM2 model. MD simulations also confirm the stability of our homology model relative to a model based on the alignment in (8) (Fig. 2 D).
Figure 2.
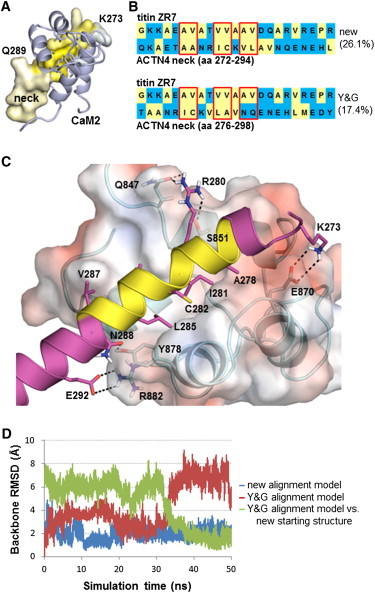
Neck-CaM2 complex homology model. (A) The helical neck is shown in surface representation with coloring based on side-chain hydrophobicity using the Eisenberg scale (52). Colors go from white to yellow, in the direction of increased side-chain hydrophobicity. CaM2 is shown in blue. (B) Comparison of our pairwise sequence alignment between titin repeat ZR7 and the neck region of ACTN4 (residues 272–294) with that suggested by Young and Gautel (residues 276–298) (8). Red boxes denote matching of neck residues with those in titin ZR7 that are in contact with CaM2 based on the solution structure in PDB 1H8B (23). Numbers in parentheses give the percent sequence similarity for the global alignment as computed by the EMBOSS (53) stretcher algorithm (using default parameters) at the EMBL-EBI website (http://www.ebi.ac.uk/Tools/psa/emboss_stretcher/). (C) Surface representation is of CaM2, colored according to electrostatic potential using APBS (54). The neck is shown in magenta, except for the three helical turns in yellow that contribute hydrophobic residues to the binding interface. Also shown are polar contacts that stabilize neck-CaM2, including K273-E870, R280-Q847, R280-S851, N288-Y878, and E292-R882. (D) Backbone root mean-square deviation (RMSD) plots of the neck region from 50-ns explicit solvent MD simulations of the neck-CaM2 complex. Blue curve shows that the simulation of the homology model based on our alignment was stable throughout the trajectory; the model based on the Y&G alignment (magenta curve) shows a large increase in backbone RMSD at around 34 ns into the simulation when the neck region twisted to a position similar to our homology model (green curve; RMSD relative to our model).
Table 1.
Comparison of ΔSASA and relative binding ΔG for complexes between titin & CaM2, neck & CaM2, and ABD & neck-CaM2
The ABD, neck region, and CaM2 form a novel ternary complex
Given the presence of multiple domains within the ACTN molecule, revealing the molecular basis of its interactions with F-actin requires understanding if and how its individual domains couple to each other. In a published cryo-EM structure (PDB ID 1SJJ) (25), one end of the antiparallel dimer has a closed ABD interacting with CaM2, whereas the other end has an open ABD. Although a stable complex between the neck and CaM2 was shown to form in vitro (8), this was not resolved in the cryo-EM structure. However, due to the lack of symmetry at both ends of the ACTN dimer and shortcomings during freezing that could weaken hydrophobic contacts (48) responsible for the neck-CaM2 complex, alternative interactions not seen in the cryo-EM (such as between neck-CaM2) can still be possible. To rationalize this data, we show that the closed ABD conformation from the cryo-EM structure is fully consistent with our homology model of the neck-CaM2 complex in Fig. 2.
Using CaM2 as the reference for structural alignment between the closed ABD end of the cryo-EM and our neck-CaM2 model, Fig. 3 A shows that, without any further rearrangement, the helical neck fits in between CaM2 and the closed ABD (CH2 in particular), with a relative binding free energy of −15.1 kcal/mol between the neck and CH2 as estimated with FastContact. Compared to the mainly hydrophobic interface that stabilizes the neck-CaM2 complex (Fig. 2, A and C), the neck-CH2 interface is more polar in nature (Fig. 3 B). In addition, CaM2 and CH2 have an estimated relative binding energy of −9.6 kcal/mol, giving a total relative binding energy of −24.7 kcal/mol between neck-CaM2 and CH2 (see Table 1 for computed ΔSASA). Our structural model altogether reveals interactions that are fully consistent with a ternary complex between the ABD, neck, and CaM2. This complex is shown in the full model in the center of Fig. 4, with the neck attached to the first spectrin repeat (SR1) and with the CaM-like domain modeled after calcium-free calmodulin in PDB ID 1CFC (27). It is noteworthy that the connectivity between CaM1 and the fourth spectrin repeat (SR4), both from the opposite monomer than the ABD and neck in the dimer, is fully preserved. Thus, in the absence of calcium the neck-CaM2 interaction is easily accessible in full-length ACTN4.
Figure 3.
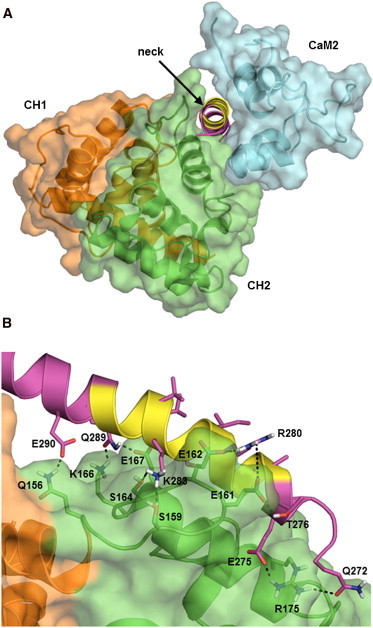
The ACTN4 neck fits in between the ABD and CaM2. (A) Surface representation shows the ABD-CaM2 complex from the closed ABD end of cryo-EM structure PDB 1SJJ (25). View of the neck (shown in cartoon only) is through its helical axis. (B) Interactions between the neck and CH2 of the ABD are predominantly polar in nature. Residues that form polar interactions are shown in sticks. Several nonpolar residues are also shown extending from three helical turns and away from CH2. These residues are part of the binding interface with CaM2.
Figure 4.
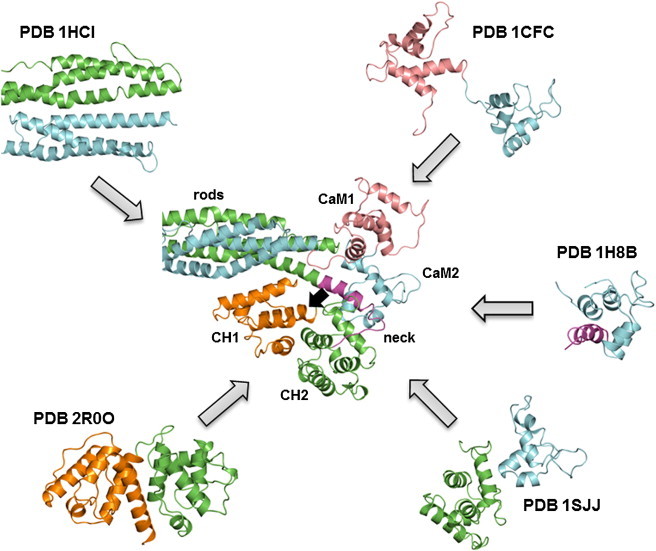
Atomic model of six domains of ACTN4 linked by a novel ternary complex between ABD, neck, and CaM2 resolved from known pairwise interactions. The full model (shown in the center) was built from homology models to published structures (PDB IDs of known pairwise interactions shown surrounding the central model). Monomer 1 containing ABD is colored green, except for CH1 (orange) and the neck region (magenta). Monomer 2 containing the CaM-like domain is colored light blue, with CaM1 (pink) positioned on top of the ABD-neck-CaM2 ternary complex. The black arrow points to the ABS on CH1, whose access to F-actin is partially blocked by the rod domains in the closed ABD structure. For clarity, the disordered N-terminal is not shown.
It is important to note that our full model of the ACTN4 end region (center of Fig. 4) involves the superposition of individually resolved pairwise interactions (periphery of Fig. 4, shown with corresponding PDB IDs). With the ABD in its closed conformation, the ABS on CH1 (black arrow in Fig. 4) is blocked by the nearby spectrin domains, thereby requiring the ABD to be in an open conformation to bind F-actin. This is fully consistent with the cryo-EM structure of F-actin decorated with human ACTN2 ABD (PDB ID 3LUE) (12), where only CH1 was found at the binding interface. Further supporting the viability of our model, CH1 is not involved in interactions with either the neck or CaM2, which allows the ABD to open without affecting the structure of the core complex. Interestingly, by superimposing CH2 from the open ABD of the cryo-EM structure onto our model in Fig. 4, CH1 is positioned at the tip of the dimer (Fig. 5). This predicted configuration appears optimal for binding, because the ABS is fully exposed to bind F-actin.
Figure 5.
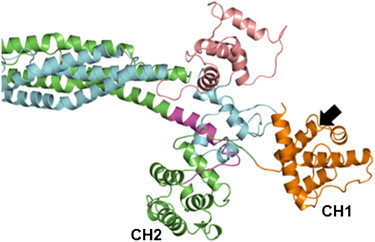
Atomic model of open ABD in ACTN4. Same reference and color scheme as in Fig. 4. Except for the unbinding of CH1 and CH2, none of the other domains need to move to access the open ABD state. Note that this state is in an optimal position to bind F-actin because the other domains do not hinder the ABS (pointed to by black arrow) on CH1.
Experimental validation of the ternary complex
The ternary complex predicts that CH2 makes stable interactions with both the neck and CaM2. The model in the center of Fig. 4 reveals three critical interfaces, and key interactions are highlighted in Figs. 6–8: a), a cluster of polar residues (Q174, N184, and Q186) of CH2 with R868/R869 of CaM2; b), E162 and S164 in the loop between CH1 and CH2 with R280 and K283 of the neck and Q847 of CaM2; and c), H189 of CH2 and D874 of CaM2. All these residues are highly conserved among the nonmuscle ACTN isoforms (Fig. S1 in the Supporting Material) and have a direct role on the stability of CH2 binding to neck-CaM2. Disrupting these contacts should tend to free the ABD from the ternary complex, unblocking the ABS and allowing easier access to the F-actin. Gel-based assays using mutant constructs for each of these sets of residues validate this prediction.
Figure 6.
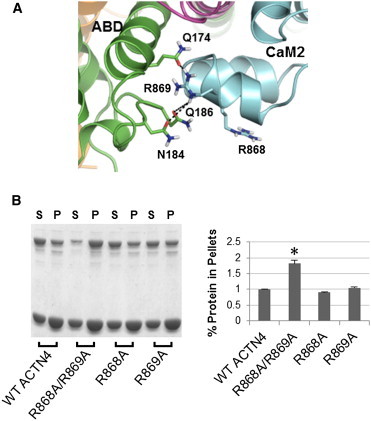
Experimental validation of residues R868/R869 that stabilize the ternary complex. (A) ABD-CaM2 interface: R869 of CaM2 makes HBs (dashed lines) with polar side chains from CH2. R868 can make these same H-bonds in the R869A mutant by turning toward the polar cluster. (B) Representative image (left) and quantitative results (right) of Coomassie Blue G-250 stained polyacrylamide gels for WT, R868A/R869A, R868A, and R869A constructs of full-length human ACTN4. Image analysis and quantitation of three experiments are shown in the plot, with the percent of ACTN4 that cosedimented with actin for each construct normalized relative to WT. Student’s t-test was performed to compare each mutant with WT. Asterisks denote p < 0.05 between the labeled mutant and WT. Error bars indicate ± SD.
Figure 7.
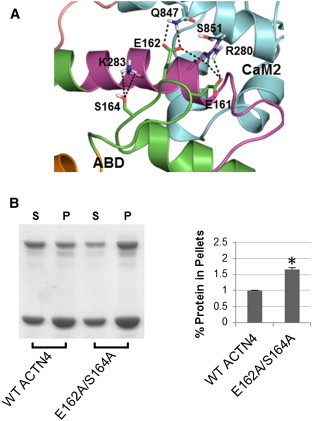
Experimental validation of residues E162/S164 that stabilize the ternary complex. (A) ABD-neck-CaM2 interface: E161, E162, and S164 of the ABD interact with R280 and K283 of the neck and Q847 of CaM2 to keep the core ternary complex stable. (B) Representative image (left) and quantitative results of three experiments (right) of Coomassie Blue G-250 stained polyacrylamide gels (as in Fig. 6B) for WT and E162A/S164A constructs of full-length human ACTN4.
Figure 8.
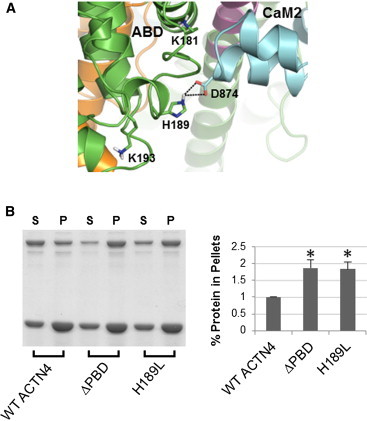
Experimental validation of residue H189 that stabilizes the ternary complex. (A) ABD-CaM2 interface: H189 of ABD interacts with D874 of CaM2. The other two residues (K181 and K193) of the putative PiP-binding site on CH2 are also shown, with neither of them able to make contacts with CaM2. (B) Representative image (left) and quantitative results of three experiments (right) of Coomassie Blue G-250 stained polyacrylamide gels (as in Fig. 6B) for WT, H189L, and ΔPBD (see text) constructs of full-length human ACTN4.
Fig. 6 A shows that R869 makes hydrogen bonds (HBs) with CH2. However, we found that single alanine mutations of either R869 or R868 have little effect on the binding profile (Fig. 6 B), and only the double mutant R868A/R869A increases actin binding to ∼187% relative to WT. The origin of this redundancy is apparent from the structure because either arginine can reach to form HBs with CH2. This type of robustness is not uncommon in regulatory proteins. For instance, single mutations of R469 or R470 in the NPR-C receptor show no effect but the double mutant has a huge impact on PLC-β activity (49).
We have also validated interactions with CH2 that include both the neck region and CaM2. The double mutant E162A/S164A increases actin binding to around 166% of WT by freeing the loop between CH1 and CH2 from the neck-CaM2 complex (Fig. 7, A and B). Finally, actin-binding assays for the H189L mutation in CH2 also show increased actin binding to around 187% of WT (Fig. 8 A and B). Interestingly, H189 is part of a trio of residues (20) purported to act as the binding site for phosphoinositides, which when mutated to hydrophobic residues (ΔPBD mutant) binds the same amount of F-actin as our single mutant H189L. Thus, our data suggest a mechanism by which phosphoinositides disrupts H189 contacts with CaM2. We should emphasize that our structural models and data show that neck-CaM2 forms a stable complex even if mutations in R868/R869, E162/S164, and H189 disengage ABD from the ternary core structure.
Phosphorylation at Y265 eases ABD opening
Increased actin binding was previously observed upon phosphorylation of CH2 residue Y265 (15). Crystal structures of the ABD reveal that the hydroxyl group of Y265 makes HBs with the backbone N-H and C-O groups of S159 and the adjacent H266 interacts with the backbone oxygen atom of A154 (Fig. 9 A). These bonds stabilize the closed ABD form, and constrain the loop between CH1 and CH2 that contacts the neck. A 100-ns MD simulation of our full WT model shows that these noncovalent bonds are highly stable, whereas the negatively charged moiety of the phosphomimic mutation Y265E immediately disrupts them (Fig. 9 B). We note that the open ABD structure shown in Fig. 5 does not necessarily require breaking the loop-neck interactions involving residues E162 and S164, but only the E161-R280 contact in Fig. 7 A. These results indicate that phosphorylation at Y265 can regulate actin binding by breaking contacts with the loop connecting CH1 and CH2, thereby disengaging the CH domains and allowing the ABD to adopt an open conformation as in Fig. 5. To confirm this prediction, we performed actin-binding assays on the ACTN4 1-300 fragment, which removes any influence of other ACTN4 domains, for both WT and Y265E. The mutant was almost all bound to F-actin (Fig. 9 C), demonstrating that the phosphomimic allows the ABD to be in a predominantly open state.
Figure 9.
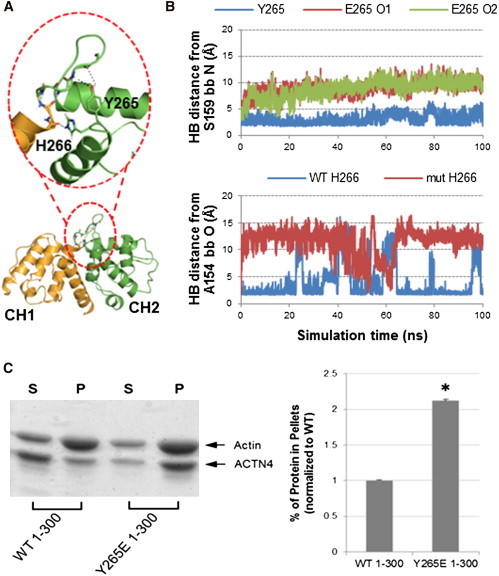
ACTN4 regulation by phosphorylation at Y265. (A) Crystal structure of CH1 (orange) and CH2 (green) from ACTN4 ABD (PDB 2R0O) (11). Zoomed area highlights HBs of Y265 and H266 with the backbone of the loop connecting the two CH domains. (B) HB distance plots between the S159 backbone and the side chain at residue 265 (top) and between the A154 backbone and H266 side chain (bottom) from 100-ns explicit solvent MD simulations. For the WT simulation (blue curves), these HBs are kept mostly stable throughout the trajectory. For the Y265E simulation (magenta and green curves), the HB to the S159 backbone is not formed with either carboxylate oxygen of Glu, whereas the HB involving H266 is broken for most of the trajectory. (C) Phosphorylation mimic at Y265 increases actin binding for the 1-300 construct. Representative image (left) and quantitative results of three experiments (right) of Coomassie Blue G-250-stained polyacrylamide gels (as in Fig. 6B) for WT and Y265E constructs of human ACTN4 1-300.
Dual phosphorylation of Y4/Y31 may induce a disorder-to-order transition in the N-terminal
The only region not included in our structural model is the 45-residue unstructured N-terminal before the ABD. Dual phosphorylation of residues Y4/Y31 in this region was previously found to inhibit actin binding (15). We have gained some insight into the molecular basis of ACTN4 regulation through Y4/Y31 phosphorylation by running Langevin dynamics simulations in implicit solvent for both WT and Y4E/Y31E N-terminal sequences. Starting from extended structures, the WT sequence folded into a globule with no regular secondary structure (Fig. 10 A), whereas the double phosphomimic mutant Y4E/Y31E formed helical structures (Fig. 10 B). Secondary structure predictions further confirm the observed higher helicity for Y4E/Y31E (Fig. S2). These results suggest that dual phosphorylation may trigger a disorder-to-order transition in the N-terminal region, most noticeable around residue 31. Docking experiments of the helical region between residues 31 to 41 from Y4E/Y31E N-terminal to the ABD strongly suggest that the helix can bind to a hydrophobic crevice (comprising residues L169, L170, L171, V185, F188, W192, M258, and V261) on the surface of CH2. This interaction will disrupt the binding of ABD and CaM2 in the ternary complex (Fig. 10 C), which taken alone would lead to a predicted increase in actin binding similar to that seen for the three sets of artificial mutations (R868A/R869A, E162A/S164A, and H189L). However, this interaction would also lead to tighter latching between CH1 (to which the N-terminal is covalently attached) and CH2, thereby further stabilizing the closed ABD state and leading to the observed decrease in actin binding.
Figure 10.
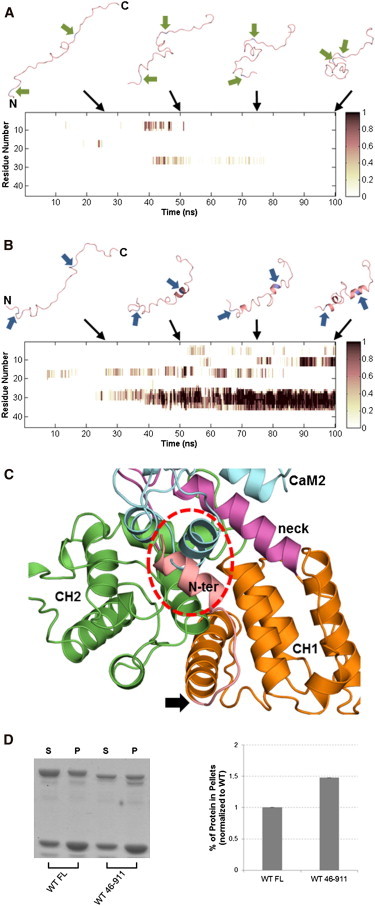
ACTN4 regulation by the N-terminal region. Helicity plots over 100-ns implicit solvent Langevin dynamics simulations of WT (A) and Y4E/Y31E (B) constructs of the 45-residue N-terminal region. Time points were averaged per residue over sequential 100-ps windows. Darker shades signify higher helical content. Above each plot are representative snapshots at 25, 50, 75, and 100 ns into each trajectory. The termini of both constructs are labeled for the 25-ns snapshots. The two arrows adjacent to each peptide (A) point to WT Y4 and Y31 residues, or (B) point to the phosphomimic mutations Y4E and Y31E. Both simulations were started with constructs in fully extended conformations. (C) Top-ranked docked model of the helical motif around Y31E and ACTN4 ABD superimposed with the core ternary complex. The circled area highlights a clash between the N-terminal helix and CaM2. Arrow points to where the N-terminal connects to CH1, indicating that this is a feasible docked model. (D) Removal of the N-terminal enhances binding to F-actin. Representative image (left) and quantitative results (right) of Coomassie Blue G-250-stained polyacrylamide gel for full-length (FL) and 46-911 truncation mutant constructs of human ACTN4. Image analysis and quantitation are shown in the plot, with percent protein in pellets for the mutant normalized relative to WT.
Interestingly, we have found that deletion of the N-terminal increases actin binding by 140% relative to full-length WT (Fig. 10 D). These preliminary results suggest that the presence of the WT N-terminal already inhibits actin binding even before it is phosphorylated at Y4/Y31, possibly by latching CH1 and CH2 together (with even stronger latching upon dual phosphorylation). More detailed structural experiments are required to fully validate the interactions involving the unstructured N-terminal.
Discussion
We have validated a structural model of the end region of the ACTN4 homodimer, with the core structure involving a ternary complex that assembles the ABD and neck region from one monomer and CaM2 from the opposite monomer. All pairwise complexes used in building this model are based on experimentally resolved structures (periphery of Fig. 4), such as ABD (PDB ID 2R0O) (11), rod domains (PDB ID 1HCI) (26), CaM2 (PDB ID 1H8B) (23), and a cryo-EM structure (PDB ID 1SJJ) (25) of open/closed ABD and the ABD-CaM2 interaction. Mutagenesis experiments on three different contact areas of the ABD and the neck-CaM2 complex show the same phenotype of increased ACTN4 binding to F-actin, strongly suggesting that the formation of the ternary complex negatively regulates actin cross-linking by ACTN4.
Our model of the ternary complex shows that CaM2 and CH2 form a stable complex, consistent with the one end of the cryo-EM structure (PDB ID 1SJJ) that has a closed ABD conformation. Although the cryo-EM did not show a helical neck as part of the CH2-CaM2 complex, experimental evidence from others (8) suggests that the neck and CaM2 interact in a similar fashion as the titin Z-repeat and CaM2 in muscle cells. Our homology model for the neck-CaM2 complex is fully consistent with these experiments, as well as the CH2-CaM2 contacts seen in the cryo-EM.
It is interesting that in the predicted ternary complex, we find that: i), closed ABD keeps the ABS on CH1 blocked by the spectrin rods and inaccessible to actin; ii), CH1 does not contribute to interactions that stabilize the ternary complex; iii), disruption of the ternary complex does not need to happen for the ABD to open (i.e., CH1 can pivot around the CH2-neck-CaM2 complex), so ABD opening only requires breaking of the CH1-CH2 interface, as suggested by a recent cryo-EM structure involving human ACTN3 ABD and F-actin (PDB ID 3LUE) (12); and iv), opening of the ABD in this model, based on the open ABD conformation in the PDB 1SJJ cryo-EM, positions CH1 at the tip of the homodimer, an ideal position for binding F-actin.
Further analysis of the atomic model presented here should provide insight into understanding the molecular mechanisms by which external cues (e.g., tyrosine phosphorylation, binding of calcium ions, binding of phosphoinositides, and calpain proteolysis) lead to the regulation of actin binding by ACTN4. In fact, MD simulations on our model provide credible evidence as to the roles of phosphorylation in the regulation of ACTN4. We found through MD simulations that the Y265E mutation (a mimic for phosphorylation at Y265) breaks several noncovalent interactions involving the mutated Y265 and the adjacent H266 that contribute to tethering the CH1 and CH2 domains (Fig. 9 B). Loss of these interactions, which are stable in the WT simulation, should then allow for ease of ABD opening, although the timescale of our simulations are not enough to capture this large-scale structural transition. For the N-terminal, we found that the double phosphomimic mutation Y4E/Y31E stabilizes helical regions around the mutated residues (Fig. 10 B). This result, when taken in conjunction with our virtual docking results (Fig. 10 C), suggests that the mechanism by which double phosphorylation at Y4/Y31 inhibits actin binding is by latching more tightly the two CH domains to hinder ABD opening.
Conclusions
F-actin cross-linking is an important structural determinant of cell shape and morphogenesis. Many actin-binding proteins are known to participate in this process, but very little is known about how their activities are regulated. In this work, we propose a detailed structural model for the ACTN4 homodimer end region, based on a core structure involving a ternary complex between the ABD, neck region, and CaM2. This model allows us to suggest mechanistic insights that could be explored with more detailed experiments. Our findings provide perhaps the first, to our knowledge, glimpse on how effectors can modulate the interactions within the multidomain structure of ACTN4, thereby allowing the protein to efficiently switch between bound and unbound signals. More generally, the high similarity between ACTNs and other actin-binding proteins (50,51) suggests that the assembly of multidomains is a general mechanism for regulating protein signaling.
Acknowledgments
This work was supported by NIH grant R01 GM97082 to CJC and R01 GM69668 to AW. TT was supported by T32 Training Grant 5T32EB009403-02.
We are grateful to Dr. David Koes for valuable comments and suggestions on the manuscript.
All atomic structures of ACTN4 will be made available for download at http://smoothdock.ccbb.pitt.edu.
Supporting Material
References
- 1.Otey C.A., Carpen O. Alpha-actinin revisited: a fresh look at an old player. Cell Motil. Cytoskeleton. 2004;58:104–111. doi: 10.1002/cm.20007. [DOI] [PubMed] [Google Scholar]
- 2.Sjöblom B., Salmazo A., Djinović-Carugo K. Alpha-actinin structure and regulation. Cell. Mol. Life Sci. 2008;65:2688–2701. doi: 10.1007/s00018-008-8080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dandapani S.V., Sugimoto H., Pollak M.R. Alpha-actinin-4 is required for normal podocyte adhesion. J. Biol. Chem. 2007;282:467–477. doi: 10.1074/jbc.M605024200. [DOI] [PubMed] [Google Scholar]
- 4.Shao H., Wang J.H., Wells A. α-actinin-4 is essential for maintaining the spreading, motility and contractility of fibroblasts. PLoS ONE. 2010;5:e13921. doi: 10.1371/journal.pone.0013921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanchanawong P., Shtengel G., Waterman C.M. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468:580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flood G., Kahana E., Critchley D.R. Association of structural repeats in the alpha-actinin rod domain. Alignment of inter-subunit interactions. J. Mol. Biol. 1995;252:227–234. doi: 10.1006/jmbi.1995.0490. [DOI] [PubMed] [Google Scholar]
- 7.Flood G., Rowe A.J., Gratzer W.B. Further analysis of the role of spectrin repeat motifs in alpha-actinin dimer formation. Eur. Biophys. J. 1997;25:431–435. doi: 10.1007/s002490050057. [DOI] [PubMed] [Google Scholar]
- 8.Young P., Gautel M. The interaction of titin and alpha-actinin is controlled by a phospholipid-regulated intramolecular pseudoligand mechanism. EMBO J. 2000;19:6331–6340. doi: 10.1093/emboj/19.23.6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franzot G., Sjöblom B., Djinović Carugo K. The crystal structure of the actin binding domain from alpha-actinin in its closed conformation: structural insight into phospholipid regulation of alpha-actinin. J. Mol. Biol. 2005;348:151–165. doi: 10.1016/j.jmb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 10.Borrego-Diaz E., Kerff F., Dominguez R. Crystal structure of the actin-binding domain of alpha-actinin 1: evaluating two competing actin-binding models. J. Struct. Biol. 2006;155:230–238. doi: 10.1016/j.jsb.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 11.Lee S.H., Weins A., Dominguez R. Crystal structure of the actin-binding domain of alpha-actinin-4 Lys255Glu mutant implicated in focal segmental glomerulosclerosis. J. Mol. Biol. 2008;376:317–324. doi: 10.1016/j.jmb.2007.11.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galkin V.E., Orlova A., Egelman E.H. Opening of tandem calponin homology domains regulates their affinity for F-actin. Nat. Struct. Mol. Biol. 2010;17:614–616. doi: 10.1038/nsmb.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dominguez R. Actin-binding proteins—a unifying hypothesis. Trends Biochem. Sci. 2004;29:572–578. doi: 10.1016/j.tibs.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 14.Izaguirre G., Aguirre L., Haimovich B. The cytoskeletal/non-muscle isoform of alpha-actinin is phosphorylated on its actin-binding domain by the focal adhesion kinase. J. Biol. Chem. 2001;276:28676–28685. doi: 10.1074/jbc.M101678200. [DOI] [PubMed] [Google Scholar]
- 15.Shao H., Wu C., Wells A. Phosphorylation of alpha-actinin 4 upon epidermal growth factor exposure regulates its interaction with actin. J. Biol. Chem. 2010;285:2591–2600. doi: 10.1074/jbc.M109.035790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witke W., Hofmann A., Noegel A.A. The Ca(2+)-binding domains in non-muscle type alpha-actinin: biochemical and genetic analysis. J. Cell Biol. 1993;121:599–606. doi: 10.1083/jcb.121.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imamura M., Sakurai T., Masaki T. Molecular cloning of low-Ca(2+)-sensitive-type non-muscle alpha-actinin. Eur. J. Biochem. 1994;223:395–401. doi: 10.1111/j.1432-1033.1994.tb19006.x. [DOI] [PubMed] [Google Scholar]
- 18.Selliah N., Brooks W.H., Roszman T.L. Proteolytic cleavage of alpha-actinin by calpain in T cells stimulated with anti-CD3 monoclonal antibody. J. Immunol. 1996;156:3215–3221. [PubMed] [Google Scholar]
- 19.Sprague C.R., Fraley T.S., Greenwood J.A. Phosphoinositide binding to the substrate regulates susceptibility to proteolysis by calpain. J. Biol. Chem. 2008;283:9217–9223. doi: 10.1074/jbc.M707436200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukami K., Sawada N., Takenawa T. Identification of a phosphatidylinositol 4,5-bisphosphate-binding site in chicken skeletal muscle alpha-actinin. J. Biol. Chem. 1996;271:2646–2650. doi: 10.1074/jbc.271.5.2646. [DOI] [PubMed] [Google Scholar]
- 21.Fraley T.S., Tran T.C., Greenwood J.A. Phosphoinositide binding inhibits alpha-actinin bundling activity. J. Biol. Chem. 2003;278:24039–24045. doi: 10.1074/jbc.M213288200. [DOI] [PubMed] [Google Scholar]
- 22.Ekman D., Björklund Å.K., Elofsson A. Multi-domain proteins in the three kingdoms of life: orphan domains and other unassigned regions. J. Mol. Biol. 2005;348:231–243. doi: 10.1016/j.jmb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Atkinson R.A., Joseph C., Pastore A. Ca2+-independent binding of an EF-hand domain to a novel motif in the alpha-actinin-titin complex. Nat. Struct. Biol. 2001;8:853–857. doi: 10.1038/nsb1001-853. [DOI] [PubMed] [Google Scholar]
- 24.Reference deleted in proof.
- 25.Liu J., Taylor D.W., Taylor K.A. A 3-D reconstruction of smooth muscle alpha-actinin by CryoEm reveals two different conformations at the actin-binding region. J. Mol. Biol. 2004;338:115–125. doi: 10.1016/j.jmb.2004.02.034. [DOI] [PubMed] [Google Scholar]
- 26.Ylänne J., Scheffzek K., Saraste M. Crystal structure of the alpha-actinin rod reveals an extensive torsional twist. Structure. 2001;9:597–604. doi: 10.1016/s0969-2126(01)00619-0. [DOI] [PubMed] [Google Scholar]
- 27.Kuboniwa H., Tjandra N., Bax A. Solution structure of calcium-free calmodulin. Nat. Struct. Biol. 1995;2:768–776. doi: 10.1038/nsb0995-768. [DOI] [PubMed] [Google Scholar]
- 28.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 29.Jorgensen W.L. Quantum and statistical mechanical studies of liquids. 10. Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J. Am. Chem. Soc. 1981;103:335–340. [Google Scholar]
- 30.Darden T., York D., Pedersen L. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 31.Ryckaert J.-P., Ciccotti G., Berendsen H. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 32.Miyamoto S., Kollman P.A. SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 33.Case D.A., Darden T.A., Kollman P.A. University of California; San Francisco: 2012. AMBER 12. [Google Scholar]
- 34.Götz A.W., Williamson M.J., Walker R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012;8:1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 36.Champ P.C., Camacho C.J. FastContact: a free energy scoring tool for protein-protein complex structures. Nucleic Acids Res. 2007;35(Web Server issue):W556–W560. doi: 10.1093/nar/gkm326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cornell W.D., Cieplak P., Kollman P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 38.Onufriev A., Bashford D., Case D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 39.Friedrichs M.S., Eastman P., Pande V.S. Accelerating molecular dynamic simulation on graphics processing units. J. Comput. Chem. 2009;30:864–872. doi: 10.1002/jcc.21209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Comeau S.R., Gatchell D.W., Camacho C.J. ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004;32(Web Server issue):W96–W99. doi: 10.1093/nar/gkh354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Comeau S.R., Gatchell D.W., Camacho C.J. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 42.Ohtsuka H., Yajima H., Kimura S. Binding of the N-terminal 63 kDa portion of connectin/titin to alpha-actinin as revealed by the yeast two-hybrid system. FEBS Lett. 1997;401:65–67. doi: 10.1016/s0014-5793(96)01432-9. [DOI] [PubMed] [Google Scholar]
- 43.Ohtsuka H., Yajima H., Kimura S. The N-terminal Z repeat 5 of connectin/titin binds to the C-terminal region of alpha-actinin. Biochem. Biophys. Res. Commun. 1997;235:1–3. doi: 10.1006/bbrc.1997.6534. [DOI] [PubMed] [Google Scholar]
- 44.Sorimachi H., Freiburg A., Labeit S. Tissue-specific expression and alpha-actinin binding properties of the Z-disc titin: implications for the nature of vertebrate Z-discs. J. Mol. Biol. 1997;270:688–695. doi: 10.1006/jmbi.1997.1145. [DOI] [PubMed] [Google Scholar]
- 45.Young P., Ferguson C., Gautel M. Molecular structure of the sarcomeric Z-disk: two types of titin interactions lead to an asymmetrical sorting of alpha-actinin. EMBO J. 1998;17:1614–1624. doi: 10.1093/emboj/17.6.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Atkinson R.A., Joseph C., Pastore A. Binding of alpha-actinin to titin: implications for Z-disk assembly. Biochemistry. 2000;39:5255–5264. doi: 10.1021/bi991891u. [DOI] [PubMed] [Google Scholar]
- 47.Joseph C., Stier G., Pastore A. A structural characterization of the interactions between titin Z-repeats and the alpha-actinin C-terminal domain. Biochemistry. 2001;40:4957–4965. doi: 10.1021/bi002739r. [DOI] [PubMed] [Google Scholar]
- 48.Privalov P.L. Cold denaturation of proteins. Crit. Rev. Biochem. Mol. Biol. 1990;25:281–305. doi: 10.3109/10409239009090612. [DOI] [PubMed] [Google Scholar]
- 49.Zhou H., Murthy K.S. Identification of the G protein-activating sequence of the single-transmembrane natriuretic peptide receptor C (NPR-C) Am. J. Physiol. Cell Physiol. 2003;284:C1255–C1261. doi: 10.1152/ajpcell.00520.2002. [DOI] [PubMed] [Google Scholar]
- 50.Gimona M., Djinovic-Carugo K., Winder S.J. Functional plasticity of CH domains. FEBS Lett. 2002;513:98–106. doi: 10.1016/s0014-5793(01)03240-9. [DOI] [PubMed] [Google Scholar]
- 51.Korenbaum E., Rivero F. Calponin homology domains at a glance. J. Cell Sci. 2002;115:3543–3545. doi: 10.1242/jcs.00003. [DOI] [PubMed] [Google Scholar]
- 52.Eisenberg D., Schwarz E., Wall R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 1984;179:125–142. doi: 10.1016/0022-2836(84)90309-7. [DOI] [PubMed] [Google Scholar]
- 53.Rice P., Longden I., Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 54.Baker N.A., Sept D., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.UniProt Consortium Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40(Database issue):D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sievers F., Wilm A., Higgins D.G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Combet C., Blanchet C., Deléage G. NPS@: network protein sequence analysis. Trends Biochem. Sci. 2000;25:147–150. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.