

Abstract
Oxidative stress is acknowledged to play a role in kidney disease progression. Genetic variants that affect the capacity to handle oxidative stress may therefore influence the outcome of kidney disease. We examined whether genetic variants of the GSTM1 gene, a member of a superfamily of glutathione S-transferases, influence the course of kidney disease progression in participants of the African American Study of Kidney Disease (AASK) trial. Groups with and without the common GSTM1 null allele, GSTM1(0), differed significantly in the time to a glomerular filtration rate (GFR) event or dialysis (P = 0.04) and in the time to GFR event, dialysis, or death (P = 0.02). The hazard ratios (HR) for the time to a GFR event or dialysis in those with two or one null allele relative to those possessing none were 1.88 [95% confidence interval (CI), 1.07 to 3.30, P = 0.03] and 1.68 (95% CI, 1.00 to 2.84, P < 0.05), respectively. For the time to GFR event, dialysis, or death, the HR for two null alleles was 2.06 (95% CI, 1.20 to 3.55, P = 0.01) and for one null allele 1.70 (95% CI, 1.02 to 2.81, P = 0.04). We demonstrated that GSTM1 directly regulates intracellular levels of 4-hydroxynonenal (4-HNE) in vascular smooth muscle cells. Furthermore, we showed that renal 4-HNE levels and GSTM1 are both increased after reduction of renal mass (RRM) in the mouse. We conclude that GSTM1 is normally upregulated in chronic kidney disease (CKD) in a protective response to increased oxidative stress. A genetic variant that results in loss of GSTM1 activity may be deleterious in CKD.
Keywords: glutathione S-transferase-μ1, gene variant, oxidative stress, kidney disease, AASK
oxidative stress is thought to play an important role in the development of many chronic diseases including cancer and atherosclerotic vascular disease. Nevertheless, even within a population with shared environmental stressors, the risk of developing these diseases varies. It thus seems likely that these varying risks are attributable to genetic differences in pathways capable of mitigating the effects of this oxidative stress. One important class of enzymes that has evolved to combat the damaging effects of reactive chemical species is the glutathione S-transferases. In particular, the μ class isoform 1 (GSTM1) has emerged as a potential modifier of multiple chronic diseases in humans.
GSTM1 belongs to a super family of glutathione S-transferases that participate in the metabolism of xenobiotics and electrophilic species. In humans, a state of complete GSTM1 enzyme deficiency exists in those who are homozygous for the GSTM1 null allele, GSTM1(0), which carries a 20-kb deletion in the GSTM1 gene. The prevalence of this polymorphism varies with race, but is as high as 50% in Caucasian and Asian populations, and is ∼ 27% in African Americans (11). The significance of this polymorphism was first appreciated in the oncology literature where subjects carrying the GSTM1(0) allele were found to be at higher risks of common malignancies (4, 8). Subsequently, human studies of cardiovascular disease (CVD) demonstrated that subjects who are homozygous for the GSTM1(0) allele have an increased risk of atherosclerosis (35), and coronary heart disease (34). The prevailing explanation for these findings pertains to a reduced ability to handle oxidative stress and the resultant cellular damage.
Our recent work supports such an explanation and also further implicates GSTM1 as a potential modifier of vascular injury, especially in the kidney. We previously characterized a mouse model expressing the major pathological features of human arteriolar nephrosclerosis, including medial hypertrophy and hyperplasia of the renal interlobular arteries and arterioles (21). Using microarray analysis, we identified Gstm1 as a strong candidate gene that modifies susceptibility to vascular injury. Compared with the resistant strain [129S6/SvEv (129)], the susceptible strain [C57BL/6 (B6)] has a 50% reduction in Gstm1 expression and higher levels of reactive oxygen species (ROS) in both kidneys and vascular smooth muscle cells (VSMCs) and has VSMCs that proliferate and migrate at a faster rate (38). The difference in Gstm1 gene expression can be explained by a difference in gene copy number. We found that the 129 strain has twice the Gstm1 gene copy number compared with the B6 strain, confirming a published report by Henrichsen et al. (15). We established cause and effect in vitro by demonstrating that small interfering (si) RNA knockdown of Gstm1 increases VSMC proliferation in a dose-dependent manner and increases VSMC migration rates and ROS levels (38).
We next queried whether GSTM1 polymorphisms may modify the course of human hypertensive nephrosclerosis. We hypothesized that patients with the GSTM1(0) allele will have an accelerated progression of kidney disease. To test this hypothesis, we examined the association between the GSTM1(0) allele and clinical outcomes in a well-characterized cohort with hypertensive nephrosclerosis (HN), the African American Study of Kidney Disease and Hypertension (AASK).
MATERIALS AND METHODS
Setting and Participants
AASK was a multicenter randomized clinical trial designed to test the effectiveness of three antihypertensive medications and two levels of blood pressure control on the progression of hypertensive kidney disease in a 3 × 2 factorial design (12). Details on the study design and conduct are available elsewhere (9, 37). Briefly, The AASK trial originally included 1,094 African Americans aged 18–70 yr with clinical diagnosis of HN and an I-iothalamate glomerular filtration rate (GFR) between 20 and 65 ml·min−1·1.73 m−2 and with no other identified causes of renal insufficiency. Between February 1995 and September 1998, participants were randomly assigned to one of two mean arterial pressure (MAP) goals, 102–107 mmHg (usual; n = 554) or 92 mmHg or less (lower; n = 540) and to initial treatment with either a β-blocker (metoprolol, 50–200 mg/d; n = 441), an angiotensin-converting enzyme inhibitor (ramipril 2.5–10 mg/d; n = 436), or a dihydropyridine calcium channel blocker (amlodipine, 5–10 mg/d; n = 217). Open-label agents were added to achieve the assigned blood pressure goals. The amlodipine arm was halted in September 2000 based on the recommendation of the Data and Safety Monitoring Board (patients were switched to open-label medications), while the other two arms were followed up to September 2001 as planned. Beginning in 2002, 994 patients were eligible (still alive and not lost to follow-up) for the AASK Genomics Study. Of these, 850 patients consented to provide DNA samples. In this study, we were provided 731 available DNA samples for genotyping by the AASK Ancillary Study Committee. The Institutional Review Board had approved use of the DNA samples and clinical data from the AASK trial.
DNA Preparation and Genotyping
Genomic DNA was extracted according to previously published methods (3). All primers and real-time PCR reactions were performed using the sequences and method described by Girault et al. (13). As the precise amount of genomic sample DNA added to each genotyping reaction is difficult to assess, the human ALBUMIN gene was used as a reference disomic gene to normalize the input of genomic DNA. As the ALBUMIN and GSTM1 PCR efficiencies are similar (between 90 and 100%) (13), the GSTM1 gene dose may be expressed as the N-fold difference in the target gene relative to ALBUMIN, designated NGST = 2(Ct ALB − Ct GST), where Ct is the threshold cycle when the PCR product first appears. The resulting NGST value allows classification into three GSTM1 genotypes: two null alleles (0/0; homozygous null), one null allele (1/0; heterozygous), or no null alleles (1/1; homozygously active) (13).
Outcome Measures
Clinical outcome measures were chosen a priori and were identical to those used in the original AASK trial as these were thought to reflect important clinical endpoints. The primary analysis focused on the relationship between the time to occurrence of clinically important events and the GSTM1 genotype. Events of interest were a GFR event defined as a 50% reduction or absolute 25 ml·min−1·1.73 m−2 decline in measured GFR, initiating dialysis, a composite of a GFR event or dialysis, and a composite of a GFR event, dialysis, or death. The time to the doubling of the baseline urine protein-to-creatinine ratio (UP/C) was also analyzed.
In secondary analyses, differences in the occurrence of cardiovascular events (CVE; cardiovascular deaths and hospitalizations for myocardial infarctions, stroke, heart failure, or revascularization procedures) among the genotype groups were also explored.
Population Admixture
African Americans represent an admixed population with genetic contributions from both African and European biogeographical origins. To adjust for the potential population admixture in the AASK study and ensure that AASK participants were comparable in genetic background, our analyses were corrected for population stratification using a multidimensional scaling (MDS) approach. Specifically, individual genotypes of a panel of 126 bi-allelic markers were used to construct an identity-by-state (IBS) distance matrix (10), and the two most significant MDS components (C1 and C2) were extracted and used as covariates in adjusted analyses. Thus the observed associations would not be simply due to an artifact of differential admixture between the GSTM1 genotype groups.
Statistical Analysis
Participants' genotypes were represented as a categorical variable with three levels: 0/0, 1/0, and 1/1. Differences between genotype groups were tested using χ2 for categorical patient characteristics and using one-way ANOVA for continuous measures.
Survival probabilities for time-to-event outcomes were estimated using the Kaplan-Meier method, and survival differences between genotype groups were assessed by a log-rank test. The effect of genotype on clinical outcomes was analyzed with the Cox proportional hazards model with adjustment for the same set of baseline covariates used in the AASK trial: age, gender, log-transformed UP/C, MAP at baseline, and history of CVD, mean baseline GFR, antihypertensive drug group, and the MDS components C1 and C2 described above. In the Cox regression analyses, the 1/1 genotype group was considered as the reference group. The assumption of proportional hazards was assessed using time-dependent covariates. Statistical interaction (effect modification) between the genotype and the following variables were also explored: trial drug, trial blood pressure group, mean baseline GFR, and log-transformed UP/C. Participants were administratively censored for loss to follow-up or else at the end of the study or September 2000 for the participants randomized to amlodipine. The time at risk for all outcomes was the time that a participant was in the AASK trial. When clinical events not included in each composite outcome analysis occurred, participants were censored as in the original trial (37). Logistic regression was used to explore differences in CVE among the three genotype groups, adjusted for the same covariates as in the Cox regression analysis.
All analyses were performed using SAS version 9.2 (Cary, NC). Two-sided P values and 95% CIs are reported. The statistical methods used in this study parallel the analyses of clinical outcomes in the original trial (37). The sponsors had no role in the data analysis or preparation of the manuscript.
Animal Studies
Primary VSMC culture.
Briefly, VSMCs from aortas of 3- to 4-wk-old wild-type C57BL/6 (Jackson Laboratory, Bar Harbor, ME) and 129S6 (Taconic) mice were isolated by enzymatic digestion using collagenase (1.5 mg/ml, Sigma) while suspended in DMEM (GIBCO Laboratories) containing, l-glutamine, HEPES, penicillin, and streptomycin, as previously described (38). Cells were washed and grown in DMEM supplemented with 10% heat-inactivated calf serum, penicillin (100 U/ml), streptomycin (100 μg/ml) in 75-cm2 Corning tissue culture flasks at 37°C in a humidified environment of 5% CO2 and air.
RNA interference and cell transfection.
High-performance purity grade (>90% pure) siRNAs against Gstm1 (Gstm1 siRNA) was obtained from Ambion. siRNA, with a nonsilencing oligonucleotide sequence (nonsilencing siRNA) that does not recognize any known homology to mammalian genes, was used as a negative control (control siRNA), as previously described (38). VSMCs were seeded at a density of 5 × 104 cells/well in six-well plates and grown in DMEM containing 10% FCS. One day after seeding, cells are transfected with 100 pmol of control siRNA, or 100 pmol of Gstm1 siRNA using Lipofectomine NAiFect Transfection Reagent (Qiagen) according to the manufacturer's instructions. Seventy-two hours posttransfection, the cells were then analyzed by Western blotting.
Overexpression of GSTM1.
Overexpression of GSTM1 in VSMCs was accomplished using a wild-type murine Gstm1 expression vector (kindly provided by Dr. Eui-Ju Choi) (6) and TransFectin reagent (Bio-Rad) as per the manufacturer's suggestions.
Western blotting.
The method used has been previously described (23). VSMCs were lysed in RIPA buffer with protease inhibitors. Rabbit anti-GSTM1 antibody (generous gift of Dr. John Hayes) (14) was used at 1:2,000 dilution. Rabbit anti-nuclear factor-erythroid 2 (NRF2; Santa Cruz Biotechnology) and rabbit anti-4-hydroxynonenal (4-HNE; Abcam) antibodies were used at 1:1,000 dilution.
Renal ablation model of chronic kidney disease.
All mice were bred and maintained on a 12:12-h light-dark cycle with free access to standard chow and water in the animal facility at the University of Virginia. Only male mice were used in our studies. Experiments were carried out in accordance with local and the National Institutes of Health guidelines, and the animal protocol was approved by the University of Virginia Institutional Animal Care and Use Committee. Reduction of renal mass (RRM) model was performed as previously reported (28) using C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME). Under general anesthesia, (1.5% isoflurane), the right kidney was removed, and the blood supply of the upper half of the left kidney was interrupted by ligation of the upper branch of the two main branches of the left renal artery (for subtotal nephrectomy). Western blot analyses for NRF2, GSTM1, and 4-HNE protein adducts in the remnant kidney were performed 4 wk after surgery.
RESULTS
Baseline Characteristics of AASK Trial Participants
Of the 731 DNA samples obtained, 9 were duplicates. Of the remaining 722 unique samples, 692 were successfully genotyped (95.8%). The baseline patient characteristics were similar between those with DNA samples and those without DNA samples (not shown). Of 692 participants, 186 (27%) were homozygous for the null allele, 361 had one null allele (51%), and 145 (22%) had no null allele of the GSTM1 gene. These frequencies closely match those reported by Garte et al. (11) in a large (15,000) population survey of metabolic gene polymorphisms. Baseline characteristics were not significantly different among the three genotype groups except for a history of CVD (P = 0.04, Table 1). The homozygous null group has a higher prevalence of CVD, consistent with previous observations (34, 35). Considering the trial interventions, the distribution of the genotypes among the drug and blood pressure group assignments were not significantly different (Table 1). The continuous baseline measures including mean GFR were not significantly different among the three genotype groups (P = 0.41), nor were the two leading MDS components (P = 0.423 and 0.347, respectively).
Table 1.
Baseline patient characteristics by genotype group
| Patient Characteristics or Measures at Baseline | Homozygous Null (0/0), n = 186 | Heterozygous (1/0), n = 361 | Homozygous Active (1/1), n = 145 |
|---|---|---|---|
| Age, yr | 54.9 (0.8) | 54.0 (0.6) | 53.9 (0.9) |
| Sex, % male | 58.1 | 62.1 | 56.6 |
| History of CVD, % | 58.6* | 47.5* | 48.9* |
| MAP, mmHg | 113.6 (1.2) | 113.2 (0.9) | 116.7 (1.4) |
| GFR, ml·min−1·1.73 −2 | 46.8 (0.9) | 47.1 (0.7) | 48.7 (1.2) |
| Serum creatinine, mg/dl | 1.96 (0.05) | 1.98 (0.04) | 1.92 (0.05) |
| Urine protein/creatinine ratio | 0.30 (0.48) | 0.32 (0.52) | 0.26 (0.47) |
| Trial antihypertensive drug | |||
| Ramipril, % | 27.8 | 50.0 | 22.2 |
| Metoprolol, % | 27.4 | 52.6 | 20.0 |
| Amlodipine, % | 24.1 | 55.6 | 20.3 |
| Trial blood pressure goal | |||
| MAP 92 mmHg, % | 25.2 | 55.0 | 19.8 |
| MAP 102-107 mmHg, % | 28.7 | 49.1 | 22.2 |
Values are shown as mean (SD) for continuous measures and percentage (%) for categorical measures. CVD, cardiovascular disease; MAP, mean arterial pressure; GFR, glomerular filtration rate. The differences among genotype groups were tested with 1-way ANOVA for continuous measures and χ2 for categorical measures. The only significant difference at baseline is a history of CVD (
P = 0.04).
Time-to-Event Analysis
GSTM1(0) is associated with a worse composite outcome.
Table 2 shows the Cox regression results for the composite outcome of a GFR event, dialysis, or death. In these 692 participants, the baseline GFR and UP/C were significantly associated with increased risks of the composite outcome. These results are similar to the findings from the original trial (37), reassuring that this smaller cohort is representative of the whole trial cohort. The GSTM1 genotype groups differed significantly in the time to the composite outcome of a GFR event, dialysis, or death (Fig. 1, log-rank, P = 0.015). At 5 yr, the number of participants reaching this composite endpoint were 51/186 (27.4%) in the 0/0 group, 80/361(22.1%) in the 1/0 group, and 20/145 (13.8%) in the 1/1 group. The adjusted hazard ratio (HR) for the 0/0 group is 2.03 (95% CI, 1.17–3.53, P = 0.012) and for 1/0 group 1.71 (95% CI, 1.03–2.85, P = 0.039) relative to the 1/1 group. Additionally, for the time to the composite outcome of a GFR event or dialysis, the genotype groups did differ significantly (Fig. 2, P = 0.041). At 5 yr, the number of participants reaching this composite endpoint were 45/186 (24.1%) in the 0/0 group, 77/361(21.3%) in the 1/0 group, and 19/145 (13.1%) in the 1/1 group. The adjusted HR for 0/0 or 1/0 are 1.82 (95% CI, 1.03–3.23, P = 0.041) and 1.70 (95% CI, 1.00–2.87, P = 0.049), respectively. Considering time to dialysis only, the genotype groups did not differ significantly (P = 0.154). The adjusted HR for 0/0 or 1/0 are 2.42 (95% CI, 1.10–5.32, P = 0.028) and 1.53 (95% CI, 0.74–3.17, P = 0.255), respectively. Finally, the time to a GFR event only was not significantly different among groups (P = 0.096). The adjusted HR for 0/0 or 1/0 individuals were 1.65 (95% CI, 0.86–3.17, P = 0.132) and 1.80 (95% CI, 0.99–3.25, P = 0.052), respectively. The groups did not differ significantly in the time to doubling of the UP/C (log-rank, P = 0.94). The two leading MDS components were not significantly associated with any of the outcomes of interest, including a GFR event, dialysis, or the composites of a GFR event or dialysis, or that of a GFR event, dialysis, or death.
Table 2.
Results of Cox regression for time to the composite outcome of GFR event, dialysis, or death
| Variable | Hazard Ratio | 95% CI | P Value |
|---|---|---|---|
| Age, yr | 1.00 | (0.99, 1.02) | 0.819 |
| Gender (female vs. male) | 0.96 | (0.68, 1.36) | 0.827 |
| History of CVD | 0.76 | (0.54, 1.08) | 0.130 |
| MAP, mmHg | 1.00 | (0.99, 1.01) | 0.779 |
| Baseline GFR, ml·min−1·1.73 −2 | 0.98 | (0.96, 0.99) | <0.001 |
| Log-transformed UP/C | 1.86 | (1.63, 2.13) | <0.001 |
| MDS components | |||
| C1 | 0.02 | (0.00, 1.05) | 0.053 |
| C2 | 0.09 | (0.00, 5.32) | 0.244 |
| Trial Antihypertensive drug | |||
| Amlodipine (reference) | 1.00 | ||
| Ramipril | 0.63 | (0.39, 1.01) | 0.054 |
| Metoprolol | 0.74 | (0.46, 1.19) | 0.215 |
| Genotype group | |||
| 1/1 Group (reference) | 1.00 | ||
| 1/0 Group | 1.71 | (1.03, 2.85) | 0.039 |
| 0/0 Group | 2.03 | (1.17, 3.53) | 0.012 |
CI, confidence interval; UP/C, urine protein-to-creatinine ratio; MDS, multidimensional scaling.
Fig. 1.

Kaplan-Meier curve of composite outcome of glomerular filtration rate (GFR) event, dialysis, or death among genotype groups. A GFR event is defined as a 50% reduction or 25 ml·min−1·1.73 m−2 decline in measured GFR (iothalamate). The genotype groups are significantly different in the composite outcome of GFR event, dialysis, or death (log rank; P = 0.015).
Fig. 2.

Kaplan-Meier curve of composite outcome of GFR event or dialysis among genotype groups. GFR event is defined as in Fig. 1. The 3 genotype groups differed in time to a GFR event or dialysis (log-rank; P = 0.041).
No statistically significant interactions were detected between genotype groups and trial drug groups, trial blood pressure groups, baseline GFR, or proteinuria (P > 0.10 for all interactions). Thus only the main effect of genotype groups was considered in the final Cox regression analyses.
CVE
There were no significant differences in the observed CVE during the trial period (P = 0.27), with the event rate as 21/186 (11.3%), 41/361 (11.4%), and 15/145 (10.3%) for the 0/0, 1/0, and 1/1 groups, respectively. Similarly, logistic regression adjusted for baseline characteristics showed that no significant differences were found for the CVE risks among the three genotype groups.
GSTM1 is Part of the Nrf2 Stress-Response Pathway
How might the GSTM1 enzyme function to modify a hypertension-related kidney disease course? We previously reported that knockdown of GSTM1 expression by siRNA resulted in an increase in the reactive species (ROS) O2·− and H2O2 in VSMCs (38). The influence of GSTM1 on intracellular levels of ROS is likely due to its role in regulating the levels of endogenously generated reactive aldehydes that are products of lipid peroxidation. Among these reactive aldehydes, 4-HNE has been shown to induce mitochondrial generation of ROS in VSMCs (22). Enzyme kinetic assays have demonstrated that GSTM1 has activity against the reactive aldehydes acrolein and 4-HNE (2, 17, 25–26). However, the extent of the role of GSTM1 in regulating intracellular levels and activities of reactive aldehydes is not known. To determine whether loss of GSTM1 alters intracellular levels of 4-HNE, we performed Western blot analysis of isolated mouse primary VSMCs using an antibody that detects 4-HNE-modified proteins (4-HNE adducts). Treatment of primary VSMCs from the 129 mouse strain (that has high levels of GSTM1) (38) with Gstm1 siRNA to knock down GSTM1 expression resulted in significantly higher levels of 4-HNE adducts compared with control siRNA (Fig. 3A). Conversely, overexpression of GSTM1 in primary VSMCs from the B6 strain (that has low levels of GSTM1) (38) by a Gstm1 expression vector (Fig. 3B, left) significantly reduced 4-HNE adduct levels (Fig. 3B, right). These data support that GSTM1 regulates the biological levels of reactive aldehydes such as 4-HNE.
Fig. 3.
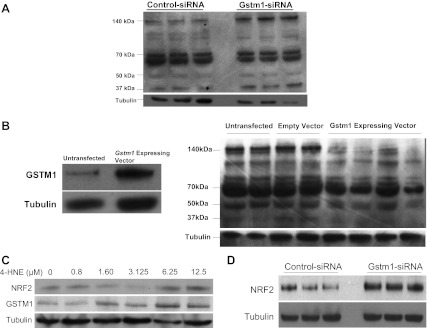
A: effect of knockdown of glutathione S-transferase-μ1 (GSTM1) on intracellular levels of 4-hydroxynonenal (4-HNE) protein adducts in vascular smooth muscle cells (VSMCs) from the 129S6/SvEv (129) mouse strain. Knockdown of GSTM1 with Gstm1-small interfering (si) RNA results in significantly increased levels of 4-HNE protein adducts compared with control-siRNA (P = 0.0008 by densitometry). Bands at molecular weights of 140, 70, 50, and 37 kDa correspond to proteins that have been modified by 4-HNE. Each lane represents separate cell treatment conditions. B, left: successful overexpression of GSTM1 in C57BL/6 VSMCs by a Gstm1-expressing vector. Right: effect of overexpression of GSTM1 on intracellular levels of 4-HNE protein adducts in VSMCs from the B6 mouse strain. Overexpression of GSTM1 results in significantly decreased levels of 4-HNE protein adducts compared with untransfected and empty vector conditions (P = 0.0002 by densitometry). Each lane represents separate cell treatment conditions. C: effect of 4-HNE on expression of Nrf2 and GSTM1. Exposure to 4-HNE results in increased VSMC expression of Nrf2 and GSTM1 proteins in a dose-dependent manner. D: effect of GSTM1 on expression of NRF2. Knockdown of Gstm1 by siRNA results in increased expression of NRF2 in VSMCs.
Intracellularly, exposure to reactive aldehydes results in the release of Nrf2 from the Nrf2-Keap complex and the escape of Nrf2 from Keap1-mediated ubiquitination (degradation) (18), leading to increased availability of Nrf2 in the cytoplasm to translocate to the nucleus to activate transcription of phase II oxidative stress response genes, including Gstm1 (7, 20, 33). Deletion of Nrf2 in the mouse results in decreased expression of Gstm1–Gstm4, and Gsta1 and Gsta2 (5), and increased sensitivity to hepatic, pulmonary, ovarian, and neurotoxic consequences of acute exposures to environmental agents and drugs (19). These data suggest that the Nrf2-Gstm1 pathway is upregulated as a cellular protective mechanism in response to stressful stimuli. In support, we found that exposure of cultured VSMCs to 4-HNE resulted in increased NRF2 and GSTM1 protein levels in a dose-dependent manner (Fig. 3C). To determine whether GSTM1 can influence the expression of NRF2, we next used siRNA to knock down Gstm1 in VSMCs and found that reduced expression of GSTM1 results in a significant increase in NRF2 levels compared with control (scrambled) siRNA (Fig. 3D) (P = 0.006 by densitometry, relative to tubulin). There was no difference in Nrf2 mRNA expression (data not shown). Thus, while Nrf2 regulates the transcription of Gstm1, Gstm1 in turn regulates the expression of Nrf2 post-transcriptionally. Our data suggest that the increased NRF2 protein levels may be related to accumulation of reactive aldehydes due to loss of GSTM1. Taken together, our data support that GSTM1 regulates intracellular levels of reactive aldehydes such as 4-HNE and can therefore, in turn, determine NRF2 levels through its capacity to metabolize reactive aldehydes. Loss of GSTM1 leads to increased accumulation of endogenously generated reactive aldehydes, such as 4-HNE, resulting in NRF2 escaping KEAP1-mediated ubiquitination and allowing NRF2 nuclear translocation to induce transcription of the phase II gene in response to increased electrophilic stress (18).
It is generally recognized that kidney failure is associated with a significant increase in lipid peroxidation (16, 24). Siems et al. (31) reported that serum levels of 4-HNE are 3- to 10-fold higher in chronic kidney disease (CKD) patients than in healthy subjects (31). To more directly examine the effect of CKD on 4-HNE levels, we used the murine ischemic RRM model of CKD that has been previously shown to cause a significant reduction in GFR (27), as well as inducing renal histopathological changes and albuminuria (28). Consistent with results from studies by Siems et al. (31) in humans with CKD, we found that renal 4-HNE adduct levels were significantly increased in the remnant kidney compared with prenephrectomy levels (Fig. 4A). In addition, Western blot analysis demonstrated that expression of both NRF2 and GSTM1 were significantly increased in the remnant kidney compared with prenephrectomy renal expression in the same mice (Fig. 4B).
Fig. 4.

A: comparison of renal 4-HNE protein adduct levels pre- and postnephrectomy. Reduction of renal mass (RRM) results in significantly higher levels of 4-HNE protein adducts in the remnant kidney compared with prenephrectomy state. Analysis was performed using kidney tissues from the same 3 mice, before and after RRM. B: effect of RRM on renal expression of nuclear factor-erythroid 2 (NRF2) and GSTM1 proteins. Induction of chronic kidney disease in the mouse results in significantly increased renal levels of Nrf2 and GSTM1 in the remnant kidney. Analysis was performed using kidney tissues from the same 3 mice, before and after RRM.
DISCUSSION
We have demonstrated that the GSTM1 genotype modifies the progression of kidney disease in hypertensive patients with CKD in the AASK Trial, and patients with the GSTM1(0) allele(s) progressed more rapidly. Previous evidence exists that GSTM1 polymorphisms may modify outcomes in renal disease. In a case-control study of 184 end-stage renal disease (ESRD) patients and 569 age- and sex-matched population controls in Northern India, Agrawal et al. (1) demonstrated that those individuals with GSTM1(0) alleles have increased odds of ESRD relative to those without null alleles [odds ratio (OR) for GSTM1(0) allele = 1.45, 95% CI = 1.03–2.02]. In another case-control study, Singh et al. (32) demonstrated that renal allograft participants who were homozygous for the GSTM1(0) allele had a 3.35-fold risk [95% CI = (1.27,8.84), P = 0.01] of rejection overall and a lower mean time to first rejection (log-rank, P = 0.002). These studies, by virtue of their design, yield weaker conclusions than our analysis but support the hypothesis that GSTM1 polymorphisms may modify important clinical outcomes in patients with renal disease.
Based on the laboratory data, we hypothesized that there may exist a genetic dosage effect of GSTM1 among 0/0, 1/0, and 1/1 groups. The estimated Kaplan-Meier survival of AASK data in Figs. 1 and 2 suggested such a genetic dosage effect of GSTM1 for the composite outcome of GFR event or dialysis and for that of GFR event, dialysis, or death. Accordingly, the Cox regression results for these two composite outcomes also seemed to suggest the similar trends, in that the adjusted HRs for 0/0 group were greater than the HRs for 1/0 group. We tested such dosing effect by switching 1/0 group as the reference group. Comparing to 1/0 group, the HR for 0/0 was 1.13 (P = 0.54) for the composite outcome of GFR event or dialysis and 1.26 (P = 0.23) for that of GFR event, dialysis, or death. Although the HRs were not statistically significant (which may be due to an insufficient number of events), their directions of higher risks were biologically plausible and would be consistent with the notion that the patients with the 0/0 genotype are at highest risks due to a complete absence of GSTM1 enzymatic activity.
There are limitations to our analysis. When components of the composite outcomes were analyzed separately (GFR event or dialysis), we did not attain statistical significance (though there was a noteworthy trend in the unadjusted analysis; data not shown), possibly due to the fact of too few events to show an effect of the GSTM1 genotype. The choice of the composite outcomes was made before this study and was based on the original trial and the rationale that the occurrence of any combination of the component events is clinically important. We also acknowledge that considering death in the composite outcome may lack specificity for an effect on renal disease progression. Nevertheless, its inclusion may identify broader effects of the GSTM1 null allele worthy of additional exploration.
Considering other limitations, the results of this study apply to African Americans with hypertension and CKD who were also enrolled in a randomized trial, and thus the generalizability may be somewhat limited. There may also be residual confounding from the trial interventions. Nevertheless, our analyses demonstrate that the distribution of the genotype (of primary interest) was not significantly different among the trial intervention groups at baseline, nor were there significant statistical interactions between the GSTM1 genotype and trial interventions in the outcome analysis. Finally, in the Cox regression analyses, adjusting for the antihypertensive drug group assignment or the blood pressure goal group did not change the overall conclusion that GSTM1 genotypes are significantly associated with the chosen clinical outcomes.
Regarding our genotyping methods, our primers are quite specific, and it seems unlikely that our PCR products would result from other unexpected amplifications of other GSTM genes or other GST classes of genes. Additionally, our analysis revealed that the MDS components did not differ significantly among the three genotype groups and that they were not significantly associated with any of the chosen outcomes. Our analysis further adjusted for potential bias from population stratification, and the significant associations between the null allele(s) and the outcomes of interest were not affected by inclusion of the MDS components as covariates. Thus our findings are reasonably robust with respect to the impact of the potential population admixture. Finally, it is possible that other GSTM1 variants that are in linkage disequilibrium with GSTM1(0) may be the actual risk alleles. Even if this were the case, no GSTM1 variant to date has been shown to correlate as strongly with GSTM1 enzyme activity as the null allele. Seidegard et al. (29) demonstrated that the null allele reduces GSTM1 activity in vitro in a dose-dependent manner such that those in the population that are homozygous null have absolutely no activity in vitro. Moreover, there is already a significant body of evidence implicating the GSTM1(0) allele in many common human diseases.
Our observation has generated additional clinical questions that warrant further study. First, does the null allele have similar effects in other racial groups and in kidney diseases not attributable to hypertension? Second, in those individuals with hypertension, does the GSTM1(0) allele predispose them to the development of hypertensive kidney disease? Finally, does the presence of the GSTM1(0) allele modify the effect of other genetic risk factors for CKD, such as APOL1?
What might be the biological effect by which GSTM1 influences kidney disease progression? We demonstrated in vitro that loss of GSTM1 results in increased levels of the reactive aldehyde 4-HNE. Our findings are consistent with the report that GSTM1(0) homozygous subjects have higher plasma levels of the reactive aldehyde malondialdehyde MDA than those with the active allele (30). In patients with renal failure, plasma 4-HNE and MDA levels are significantly elevated (36) and have been demonstrated to directly correlate with the degree of renal anemia (31). Determination of the association between GSTM1 genotypes and circulating levels of 4-HNE in normal and CKD states will be a focus of future studies.
We found that 4-HNE stimulated the expression of NRF2 and GSTM1 proteins in VSMCs in a dose-dependent manner. Using the RRM model of CKD in the mouse, we found that 4-HNE levels were significantly increased in the remnant kidney and this was associated with significantly increased mRNA (not shown) and protein levels of Nrf2 and Gstm1. Collectively, our data suggest that the Nrf2-Gstm1 pathway is normally upregulated in CKD/RRM in a protective response to increased oxidative stress, and that this may be in part mediated by increased levels of reactive aldehydes. Thus a genetic polymorphism of GSTM1 that results in the loss of the capacity to engage this protective response would lead to an environment of exaggerated oxidative stress with consequential acceleration of disease progression. Identification of individuals who are deficient in the GSTM1 enzyme has the potential to identify those at risk for accelerated adverse outcome.
GRANTS
This work was supported by the National Capital Area and Virginia Chapters of the National Kidney Foundation, National Institutes of Health Grant DK094907 to T. H. Le, and National Institutes of Health training grant T32 DK072922 to J. Chang.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.C., Q.Z., S.C., and A.G. performed experiments; J.C., J.Z.M., C.N., D.T.O., and T.H.L. analyzed data; J.C., J.Z.M., M.S.L., and T.H.L. interpreted results of experiments; J.C., J.Z.M., and T.H.L. prepared figures; J.C. and T.H.L. drafted manuscript; J.C., J.Z.M., and T.H.L. edited and revised manuscript; J.C., J.Z.M., Q.Z., S.C., A.G., D.T.O., M.S.L., and T.H.L. approved final version of manuscript; T.H.L. provided conception and design of research.
ACKNOWLEDGMENTS
We thank the AASK trial participants and Ancillary Studies Committee for granting access to the DNA and Trial data.
REFERENCES
- 1. Agrawal S, Tripathi G, Khan F, Sharma R, Baburaj VP. Relationship between GSTs gene polymorphism and susceptibility to end stage renal disease among North Indians. Ren Fail 29: 947–953, 2007 [DOI] [PubMed] [Google Scholar]
- 2. Berhane K, Widersten M, Engstrom A, Kozarich JW, Mannervik B. Detoxication of base propenals and other alpha, beta-unsaturated aldehyde products of radical reactions and lipid peroxidation by human glutathione transferases. Proc Natl Acad Sci USA 91: 1480–1484, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhatnagar V, O'Connor DT, Schork NJ, Salem RM, Nievergelt CM, Rana BK, Smith DW, Bakris GL, Middleton JP, Norris KC, Wright JT, Cheek D, Hiremath L, Contreras G, Appel LJ, Lipkowitz MS. Angiotensin-converting enzyme gene polymorphism predicts the time-course of blood pressure response to angiotensin converting enzyme inhibition in the AASK trial. J Hypertens 25: 2082–2092, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carlsten C, Sagoo GS, Frodsham AJ, Burke W, Higgins JP. Glutathione S-transferase M1 (GSTM1) polymorphisms and lung cancer: a literature-based systematic HuGE review and meta-analysis. Am J Epidemiol 167: 759–774, 2008 [DOI] [PubMed] [Google Scholar]
- 5. Chanas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR, Henderson CJ, Wolf CR, Moffat GJ, Itoh K, Yamamoto M, Hayes JD. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J 365: 405–416, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cho SG, Lee YH, Park HS, Ryoo K, Kang KW, Park J, Eom SJ, Kim MJ, Chang TS, Choi SY, Shim J, Kim Y, Dong MS, Lee MJ, Kim SG, Ichijo H, Choi EJ. Glutathione S-transferase mu modulates the stress-activated signals by suppressing apoptosis signal-regulating kinase 1. J Biol Chem 276: 12749–12755, 2001 [DOI] [PubMed] [Google Scholar]
- 7. Dickinson DA, Levonen AL, Moellering DR, Arnold EK, Zhang H, Darley-Usmar VM, Forman HJ. Human glutamate cysteine ligase gene regulation through the electrophile response element. Free Radic Biol Med 37: 1152–1159, 2004 [DOI] [PubMed] [Google Scholar]
- 8. Economopoulos KP, Sergentanis TN. GSTM1, GSTT1, GSTP1, GSTA1 and colorectal cancer risk: a comprehensive meta-analysis. Eur J Cancer 46: 1617–1631, 2010 [DOI] [PubMed] [Google Scholar]
- 9. Fogo A, Breyer JA, Smith MC, Cleveland WH, Agodoa L, Kirk KA, Glassock R. Accuracy of the diagnosis of hypertensive nephrosclerosis in African Americans: a report from the African American Study of Kidney Disease (AASK) Trial. AASK Pilot Study Investigators Kidney Int 51: 244–252, 1997 [DOI] [PubMed] [Google Scholar]
- 10. Fung MM, Chen Y, Lipkowitz MS, Salem RM, Bhatnagar V, Mahata M, Nievergelt CM, Rao F, Mahata SK, Schork NJ, Brophy VH, O'Connor DT. Adrenergic beta-1 receptor genetic variation predicts longitudinal rate of GFR decline in hypertensive nephrosclerosis. Nephrol Dial Transplant 24: 3677–3686, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garte S, Gaspari L, Alexandrie AK, Ambrosone C, Autrup H, Autrup JL, Baranova H, Bathum L, Benhamou S, Boffetta P, Bouchardy C, Breskvar K, Brockmoller J, Cascorbi I, Clapper ML, Coutelle C, Daly A, Dell'Omo M, Dolzan V, Dresler CM, Fryer A, Haugen A, Hein DW, Hildesheim A, Hirvonen A, Hsieh LL, Ingelman-Sundberg M, Kalina I, Kang D, Kihara M, Kiyohara C, Kremers P, Lazarus P, Le Marchand L, Lechner MC, van Lieshout EM, London S, Manni JJ, Maugard CM, Morita S, Nazar-Stewart V, Noda K, Oda Y, Parl FF, Pastorelli R, Persson I, Peters WH, Rannug A, Rebbeck T, Risch A, Roelandt L, Romkes M, Ryberg D, Salagovic J, Schoket B, Seidegard J, Shields PG, Sim E, Sinnet D, Strange RC, Stucker I, Sugimura H, To-Figueras J, Vineis P, Yu MC, Taioli E. Metabolic gene polymorphism frequencies in control populations. Cancer Epidemiol Biomarkers Prev 10: 1239–1248, 2001 [PubMed] [Google Scholar]
- 12. Gassman JJ, Greene T, Wright JT, Jr, Agodoa L, Bakris G, Beck GJ, Douglas J, Jamerson K, Lewis J, Kutner M, Randall OS, Wang SR. Design and statistical aspects of the African American Study of Kidney Disease and Hypertension (AASK). J Am Soc Nephrol 14: S154–S165, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Girault I, Lidereau R, Bieche I. Trimodal GSTT1 and GSTM1 genotyping assay by real-time PCR. Int J Biol Markers 20: 81–86, 2005 [PubMed] [Google Scholar]
- 14. Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol 45: 51–88, 2005 [DOI] [PubMed] [Google Scholar]
- 15. Henrichsen CN, Vinckenbosch N, Zollner S, Chaignat E, Pradervand S, Schutz F, Ruedi M, Kaessmann H, Reymond A. Segmental copy number variation shapes tissue transcriptomes. Nat Genet 41: 424, 2009 [DOI] [PubMed] [Google Scholar]
- 16. Himmelfarb J. Oxidative stress in hemodialysis. Contrib Nephrol 161: 132–137, 2008 [DOI] [PubMed] [Google Scholar]
- 17. Hubatsch I, Ridderstrom M, Mannervik B. Human glutathione transferase A4–4: an alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxynonenal and other genotoxic products of lipid peroxidation. Biochem J 330: 175–179, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Itoh K, Wakabayashi N, Katoh Y, Ishii T, O'Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8: 379–391, 2003 [DOI] [PubMed] [Google Scholar]
- 19. Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47: 89–116, 2007 [DOI] [PubMed] [Google Scholar]
- 20. Kwak MK, Kensler TW, Casero RA., Jr Induction of phase 2 enzymes by serum oxidized polyamines through activation of Nrf2: effect of the polyamine metabolite acrolein. Biochem Biophys Res Commun 305: 662–670, 2003 [DOI] [PubMed] [Google Scholar]
- 21. Le TH, Fogo AB, Salzler HR, Vinogradova T, Oliverio MI, Marchuk DA, Coffman TM. Modifier locus on mouse chromosome 3 for renal vascular pathology in AT1A receptor-deficiency. Hypertension 43: 445–451, 2004 [DOI] [PubMed] [Google Scholar]
- 22. Lee JY, Jung GY, Heo HJ, Yun MR, Park JY, Bae SS, Hong KW, Lee WS, Kim CD. 4-Hydroxynonenal induces vascular smooth muscle cell apoptosis through mitochondrial generation of reactive oxygen species. Toxicol Lett 166: 212–221, 2006 [DOI] [PubMed] [Google Scholar]
- 23. Malakauskas SM, Quan H, Fields TA, McCall SJ, Yu MJ, Kourany WM, Frey CW, Le TH. Aminoaciduria and altered renal expression of luminal amino acid transporters in mice lacking novel gene collectrin. Am J Physiol Renal Physiol 292: F533–F544, 2007 [DOI] [PubMed] [Google Scholar]
- 24. Martin CJ, Goeddeke-Merickel CM. Oxidative stress in chronic kidney disease. Nephrol Nurs J 32: 683–685, 2005 [PubMed] [Google Scholar]
- 25. Pal A, Hu X, Zimniak P, Singh SV. Catalytic efficiencies of allelic variants of human glutathione S-transferase Pi in the glutathione conjugation of alpha, beta-unsaturated aldehydes. Cancer Lett 154: 39–43, 2000 [DOI] [PubMed] [Google Scholar]
- 26. Paumi CM, Smitherman PK, Townsend AJ, Morrow CS. Glutathione S-transferases (GSTs) inhibit transcriptional activation by the peroxisomal proliferator-activated receptor gamma (PPAR gamma) ligand, 15-deoxy-delta 12,14 prostaglandin J2 (15-d-PGJ2). Biochemistry 43: 2345–2352, 2004 [DOI] [PubMed] [Google Scholar]
- 27. Qi Z, Whitt I, Mehta A, Jin J, Zhao M, Harris RC, Fogo AB, Breyer MD. Serial determination of glomerular filtration rate in conscious mice using FITC-inulin clearance. Am J Physiol Renal Physiol 286: F590–F596, 2004 [DOI] [PubMed] [Google Scholar]
- 28. Salzler HR, Griffiths R, Ruiz P, Chi L, Frey C, Marchuk DA, Rockman HA, Le TH. Hypertension and albuminuria in chronic kidney disease mapped to a mouse chromosome 11 locus. Kidney Int 72: 1226–1232, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seidegard J, Vorachek WR, Pero RW, Pearson WR. Hereditary differences in the expression of the human glutathione transferase active on trans-stilbene oxide are due to a gene deletion. Proc Natl Acad Sci USA 85: 7293–7297, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Siems W, Grune T. Intracellular metabolism of 4-hydroxynonenal. Mol Aspects Med 24: 167–175, 2003 [DOI] [PubMed] [Google Scholar]
- 31. Siems W, Quast S, Carluccio F, Wiswedel I, Hirsch D, Augustin W, Kraemer K, Hampl H, Sommerburg O. Oxidative stress in cardio renal anemia syndrome: correlations and therapeutic possibilities. Clin Nephrol 60, Suppl 1: S22–S30, 2003 [PubMed] [Google Scholar]
- 32. Singh R, Manchanda PK, Kesarwani P, Srivastava A, Mittal RD. Influence of genetic polymorphisms in GSTM1, GSTM3, GSTT1 and GSTP1 on allograft outcome in renal transplant recipients. Clin Transplant 23: 490–498, 2009 [DOI] [PubMed] [Google Scholar]
- 33. Tirumalai R, Rajesh Kumar T, Mai KH, Biswal S. Acrolein causes transcriptional induction of phase II genes by activation of Nrf2 in human lung type II epithelial (A549) cells. Toxicol Lett 132: 27–36, 2002 [DOI] [PubMed] [Google Scholar]
- 34. Wang J, Zou L, Huang S, Lu F, Lang X, Han L, Song Z, Xu Z. Genetic polymorphisms of glutathione S-transferase genes GSTM1, GSTT1 and risk of coronary heart disease. Mutagenesis 25: 365–369, 2010 [DOI] [PubMed] [Google Scholar]
- 35. Wang XL, Greco M, Sim AS, Duarte N, Wang J, Wilcken DE. Glutathione S-transferase mu1 deficiency, cigarette smoking and coronary artery disease. J Cardiovasc Risk 9: 25–31, 2002 [DOI] [PubMed] [Google Scholar]
- 36. Wiswedel I, Hirsch D, Carluccio F, Hampl H, Siems W. F2-isoprostanes as biomarkers of lipid peroxidation in patients with chronic renal failure. Biofactors 24: 201–208, 2005 [DOI] [PubMed] [Google Scholar]
- 37. Wright JT, Jr, Bakris G, Greene T, Agodoa LY, Appel LJ, Charleston J, Cheek D, Douglas-Baltimore JG, Gassman J, Glassock R, Hebert L, Jamerson K, Lewis J, Phillips RA, Toto RD, Middleton JP, Rostand SG. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: results from the AASK trial. JAMA 288: 2421–2431, 2002 [DOI] [PubMed] [Google Scholar]
- 38. Yang Y, Parsons KK, Chi L, Malakauskas SM, Le TH. Glutathione S-transferase-micro1 regulates vascular smooth muscle cell proliferation, migration, and oxidative stress. Hypertension 54: 1360–1368, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]