

Abstract
The consumption of high-fat/calorie diets in modern societies is likely a major contributor to the obesity epidemic, which can increase the prevalence of cancer, cardiovascular disease, and neurological impairment. Obesity may precipitate decline via inflammatory and oxidative signaling, and one factor linking inflammation to oxidative stress is the proinflammatory, pro-oxidant enzyme NADPH oxidase. To reveal the role of NADPH oxidase in the metabolic and neurological consequences of obesity, the effects of high-fat diet were compared in wild-type C57Bl/6 (WT) mice and in mice deficient in the NAPDH oxidase subunit NOX2 (NOX2KO). While diet-induced weight gains in WT and NOX2KO mice were similar, NOX2KO mice had smaller visceral adipose deposits, attenuated visceral adipocyte hypertrophy, and diminished visceral adipose macrophage infiltration. Moreover, the detrimental effects of HFD on markers of adipocyte function and injury were attenuated in NOX2KO mice; NOX2KO mice had improved glucose regulation, and evaluation of NOX2 expression identified macrophages as the primary population of NOX2-positive cells in visceral adipose. Finally, brain injury was assessed using markers of cerebrovascular integrity, synaptic density, and reactive gliosis, and data show that high-fat diet disrupted marker expression in WT but not NOX2KO mice. Collectively, these data indicate that NOX2 is a significant contributor to the pathogenic effects of high-fat diet and reinforce a key role for visceral adipose inflammation in metabolic and neurological decline. Development of NOX-based therapies could accordingly preserve metabolic and neurological function in the context of metabolic syndrome.
Keywords: brain injury, metabolic syndrome, NADPH oxidase, obesity
diet-induced obesity may be the primary cause of metabolic syndrome, which is associated with dramatically enhanced risk for many diseases, including type 2 diabetes, cardiovascular disease, stroke, and cancer (reviewed in Ref. 38). In recent years, diet-induced obesity has also been linked to brain pathology, cognitive dysfunction, and Alzheimer's disease (reviewed in Ref. 65). For example, studies have reported deficits in learning, memory, and executive function in obese compared with nonobese patients (28, 29, 93), and regression studies have demonstrated that increased body weight is associated with decreased brain volume (94). Additionally, clinical obesity is associated with reductions in focal gray matter volume and enlarged white matter, particularly in the frontal lobe (69). While it is not fully understood how obesity destabilizes health, obesity is closely associated with a pattern of chronic inflammation thought to originate in adipose tissue (85), resulting in enhanced cytokine production, increased acute-phase reactants, and other inflammatory mediators (14, 20, 42). Activation of these inflammatory markers correlates tightly with insulin resistance (75), cardiovascular disease (77), and cognitive impairment (27, 89).
While the molecular link(s) between excess adiposity and inflammation has not yet been identified, recent reports have implicated activation of the proinflammatory enzyme NADPH oxidase in the detrimental effects of diet-induced obesity. NADPH oxidase is a superoxide-producing complex consisting of membrane (gp91phox and p22phox) and cytosolic (p47phox, p67phox, and p40phox) components which assemble at the plasma membrane to form the active oxidase (8, 25). NADPH oxidase is expressed in many types of immune cells including macrophages, dendritic cells, and neutrophils, as well as in adipocytes, endothelial cells, and neurons. With regard to the participation of NADPH oxidase in obesity, studies from our laboratory show that NADPH oxidase activity/expression is increased in the brains of mice given a high-fat diet (18), a scenario also observed in rats (97). Obesity-associated alterations in both endothelial cells (22, 87) and leukocytes (74) have been linked to NADPH oxidase. Once activated in the brain, NADPH oxidase is thought to trigger inflammatory processes that can contribute to CNS disease (15, 51, 84). For example, data from our laboratory and others' show the critical role that NADPH oxidase plays in directing brain inflammation, particularly in the release of proinflammatory cytokines, including TNFα, IL-6, and IL-1β (23, 78, 90), all of which could underlie neurological impairment (2, 6, 52, 73, 91).
Depite such data supporting a role for NADPH oxidase in the adverse effects of obesity, the effects of NADPH oxidase subunit deletion have not yet been tested in models of diet-induced obesity. To address this issue, this study employed a mouse model of chronic granulomatous disease (CGD), an inherited deficiency in NADPH oxidase based on deletion of the catalytic gp91phox subunit (also known as NOX2). The effects of high-fat diet were thus compared in wild-type C57Bl/6 (WT) mice and NOX2-deficient (NOX2KO) mice to determine whether deletion of NOX2 would offer significant protection from the detrimental neurological and/or physiological consequences of diet-induced obesity.
MATERIALS AND METHODS
Animal treatments.
The Institutional Animal Care and Use Committee at the Pennington Biomedical Research Center approved all experimental protocols, which were compliant with NIH guidelines on the use of experimental animals. Four-month-old male C57Bl/6 (WT) and B6.129S-Cybbtm1Din/J (NOX2KO) mice were purchased from Jackson Laboratories (Bar Harbor, ME) and were housed in standard caging with a 12:12-h light-dark cycle and ad libitum access to food and water. C57Bl/6 mice were housed in standard conventional rooms, whereas NOX2KO mice were housed under sterile conditions to accommodate the CGD phenotype, which is characterized by recurrent bacterial and fungal infections leading to excessive inflammation (62). Both WT and NOX2KO mice were separated into groups given either a high-fat diet (HFD) or a low-fat control diet (CD) for 14 wk. The HFD was composed of 60% fat (pork lard), and the control diet was composed of 10% fat. Both diets were open source, were purchased from Research Diets (New Brunswick, NJ; HFD: D12492; CD: D12450B), and were provided in pelleted form. Data were compiled from two separate experiments, each composed of both WT and NOX2KO mice given CD or HFD, with 9–10 total animals in each group.
Body weight, food intake, and body composition [measured using a Bruker minispec LF90 time domain Nuclear Magnetic Resonance (NMR) analyzer; Bruker Optics, Billerica MA] were measured twice a month. Fasting blood glucose was measured in tail blood using a glucometer (Ascensia Elite, Bayer, Mishawaka, IN), and glucose tolerance was measured using a modified oral glucose tolerance test (OGTT). Briefly, mice were fasted for 4 h, baseline glucose was measured, and then mice were immediately administered glucose (2 g/kg) via oral gavage. Blood glucose was measured at 15, 30, 60, and 120 min, and area under the curve (AUC) was recorded as an index of glucose disposal. To measure circulating nonesterified fatty acids (NEFA) in the context of hyperglycemia, additional blood samples (∼50 μl) were collected at 0 and 60 min by lancing the submandibular vein (31), and NEFA were analyzed as described below. All mice were humanely euthanatized via isoflurane inhalation and cardiac puncture after a brief (6 h) fast, and blood, brain (anterior 1/3 of cerebral cortex), and visceral (epididymal) and subcutaneous (inguinal) adipose tissue depots were collected.
Clinical chemistry.
Whole blood was collected by cardiac puncture of terminally anesthetized mice and was allowed to clot at 4°C overnight and then centrifuged at 3,000 g for 30 min. Serum was collected and either analyzed immediately or aliquoted and stored at −80°C. Levels of total cholesterol, HDL-cholesterol, LDL-cholesterol, triglycerides, and NEFA in sera were measured colorimetrically using commercially available kits (Wako Chemicals, Richmond, VA). Insulin levels were evaluated by ELISA in accordance with the manufacturer's assay protocol (Crystal Chem, Downers Grove IL).
Histological and biochemical analyses of adipose tissue.
The inguinal and epididymal adipose depots were collected for histological and biochemical analyses. For histology, the tissues were drop-fixed in 10% neutral buffered formalin for 2–3 days, after which they were processed for paraffin embedding. Sections (5 μm) were then cut, collected, stained with hematoxylin and eosin, and digitized. Adipocyte size was measured by an investigator blinded to the experimental grouping in ×40 microscope fields by counting the total number of adipocytes within predefined area grids and then dividing the area by the total number of adipocytes within the grids to calculate average adipocyte size. For each sample, three replicate tissue sections were analyzed, with three fields counted in each section, for a total of nine fields averaged per sample.
For Western blot, adipose tissue samples were homogenized in RIPA buffer (G biosciences, St. Louis, MO) and then cleared by centrifugation at 5,000 g for 10 min at 4°C. Samples were denatured in SDS, and equivalent amounts of protein were electrophoretically separated in polyacrylamide gels and blotted onto nitrocellulose. Blots prepared from adipose tissue were processed using anti-Iba-1 (1:500, Wako Chemicals, Richmond, VA), anti-PPARγ1/2 (1:1,000, Abcam, Cambridge, MA), anti-adiponectin (1:1,000, Abcam), anti-GADD153/CHOP (1:5,000, Abcam), anti-GRP78 (1:500, Novus Biologicals), and anti-tubulin (1:1,000, Wako Chemicals). After incubation with primary antibodies, blots were washed and exposed to horseradish peroxidase-conjugated secondary antibodies and visualized using a chemiluminescence system (Amersham Biosciences, Pittsburgh, PA). Blot images were scanned and densitometrically analyzed for quantification. To ensure accurate quantification across multiple blots, samples from all groups (HFD and CD in both WT and NOX2KO) were included in each individual blot. Data were calculated as a ratio of expression over tubulin expression, which was included as an internal loading control. Protein expression in HFD mice was then calculated and presented as percent expression relative to CD mice of the same genotype.
For immunocytochemical analyses of adipose depots, tissue sections were processed using anti-Iba-1 (1:100, Wako Chemicals) and anti-NOX2 (1:100, Santa Cruz Biotechnology). Sections were incubated with biotinylated or peroxidase-linked secondary antibodies and then visualized using diaminobenzidine (for Iba1) or NOVAred (for NOX2) as chromagens following the manufacturer's instructions (Vector Laboratories, Burlingame, CA). To document nonspecific staining, the primary antibodies were omitted from the staining protocol. To visualize the cellular distribution of NADPH oxidase subunit expression, sections were first labeled for NOX2 and then double-labeled using Iba-1 as a macrophage cell marker.
Measures of brain injury by western blot.
Tissue samples generated from frontal cortices were homogenized and processed for Western blot with chemiluminescence, as described in previous reports (16, 76). Blots were processed using the following primary antisera: anti-claudin-5 (1:400, Abcam), anti-ZO-1 (1:100, Abcam), anti-occludin (1:8,000, Abcam), anti-matrix metalloproteinase-2 (MMP2; 1:1,000, Abcam), anti-MMP9 (1:1,000, Abcam), anti-synapsin 1 (1:10,000, Thermo Fisher Scientific, Pittsburg, PA), anti-phospho(Ser553)-synapsin 1 (1:10,000, Abcam), anti-synapse-associated protein-97 (SAP97; 1:2,500, Abcam), anti-glial fibrillary acidic protein (GFAP; 1:5,000, Abcam); anti-Iba-1 (1:500, Wako Chemicals), and anti-tubulin (1:1,000, Wako Chemicals). To ensure accurate quantification across multiple blots, samples from all groups were included in each individual blot. Data were first calculated as a ratio of expression over tubulin expression, which was included as an internal loading control, and then protein expression in HFD mice was calculated and presented as percent expression relative to CD mice of the same genotype.
Statistical analyses.
All data are shown as means ± SE. Body weight and composition data, adipocyte size, and all metabolic data were all analyzed with two-way analyses of variance (ANOVA) followed by planned Bonferroni posttests to determine the differential effects of HFD in WT compared with NOX2KO mice. Additionally, planned comparisons of WT with NOX2KO mice under both CD and HFD conditions were carried out using one-way ANOVA. Protein expression values generated by Western blot (ratios of expression over tubulin) were normalized to percent CD for each genotype to reconcile data from multiple blots and were analyzed by two-tailed, unpaired t-tests to determine if statistically significant differences existed between HFD and CD groups within each genotype. Statistical significance for all analyses was accepted at P < 0.05 (*P < 0.05, **P < 0.01, and ***P < 0.001).
RESULTS
Effects of HFD on body weight and composition in WT and NOX2KO mice.
Four-month-old male WT and NOX2KO mice were fed either HFD or CD for 14 wk as described in materials and methods. During that time, body weights progressively diverged such that, by the end of the diet exposure period, the HFD-fed mice weighed significantly more than CD mice, although all mice gained weight (Fig. 1A). Statistically, ANOVA for genotype X diet revealed a significant main effect of diet on body weight (F(1,35) = 82.82, P < 0.0001). The effect of genotype on body weight was also significant (F(1,35) = 2.27, P = 0.030), but the interaction was not. Planned comparisons of NOX2KO and WT mice revealed that, although there were no significant differences between body weights in mice given CD, HFD-fed NOX2KO mice weighed significantly more than HFD-fed WT mice after 4 and 6 wk of diet consumption (Fig. 1A). Analysis of food intake during this period did not reveal any differences between WT and NOX2KO mice with regard to diet (CD or HFD) ingestion (data not shown), indicating that NOX2 is not involved in feeding behavior and that the increased body weight in NOX2KO mice was not caused by increased food intake. In addition to body weight, total body fat was measured using NMR as described in materials and methods. Similar to what was observed for body weight, the amount of body fat (expressed as %total body weight) increased in all mice over the 14-wk feeding trial, but mice given HFD had the greatest amounts of body fat (Fig. 1B). Statistically, ANOVA to measure the effects of genotype and diet on percent body fat revealed a significant main effect of diet (F(1,33) = 53.68, P < 0.0001). The effect of genotype on body fat was also significant (F(1,33) = 4.41, P = 0.033), with a significant interaction between diet and genotype (F(1,33) = 8.63, P = 0.0038). Planned comparisons of NOX2KO and WT mice revealed no genotype-based differences in percent body fat in mice given CD but did reveal increased body fat in WT compared with NOX2KO mice after 11 and 14 wk of HFD (Fig. 1B). NMR-based measures of lean body mass (expressed as %total body weight) revealed decreases in all mice over the 14-wk feeding trial, with mice given HFD having the least amount of lean body mass by the end of the feeding trial (Fig. 1C). Statistically, two-way ANOVA revealed a significant main effect of diet on lean body mass (F(1,35) = 52.10, P < 0.0001). The effect of genotype on body weight was also significant (F(1,35) = 9.27, P = 0.0038), but the interaction was not. Planned comparisons of NOX2KO and WT mice revealed no differences in percent lean mass in mice given CD but did reveal decreased lean mass in WT mice compared with NOX2KO mice after 11 and 14 wk of HFD (Fig. 1C).
Fig. 1.

Effects of high-fat diet (HFD) on body weight and composition in WT and NAPDH oxidase subunit 2 knockout (NOX2KO) mice. Four-month old male C57Bl/6 (WT) mice and B6.129S-Cybbtm1Din/J (NOX2KO) mice were placed for 14 wk on HFD or the nutritionally matched low-fat control diet (CD) with 10 mice in each group. A: body weight in WT and NOX2KO mice over time following administration of CD or HFD. #Significant (P < 0.05) increase in body weight noted in NOX2KO vs. WT mice after 4 and 6 wk of diet. B: body fat as %total body weight in WT and NOX2KO mice over time following administration of CD or HFD. Significant (#P < 0.05, ##P < 0.01, respectively) increases in body fat in WT vs. NOX2KO mice after 11 and 14 wk of HFD. C: lean body mass as %total body weight in WT and NOX2KO mice over time following administration of CD or HFD. Significant (#P < 0.05, ##P < 0.01, respectively) decrease in lean body mass in WT vs. NOX2KO mice after 11 and 14 wk of HFD.
After the mice were euthanized, the subcutaneous inguinal and visceral epididymal adipose depots were collected for analysis. Comparison of the wet weights of the individual fat pads revealed that HFD significantly increased the size of the subcutaneous inguinal fat pad. Specifically, ANOVA for the effects of genotype X diet on the weight of the inguinal fat pad revealed a significant main effect of diet (F(1,35) = 78.19, P < 0.0001), but no effect of genotype and no interaction (Fig. 2A), whereas post hoc tests confirmed that HFD significantly increased the size of the inguinal fat pad in both WT and NOX2KO mice. Planned comparisons of NOX2KO and WT mice revealed no differences in the weight of the inguinal adipose depot in mice given CD; likewise, the inguinal adipose depot in HFD-fed WT mice was not statistically different in size from the inguinal depot isolated from HFD-fed NOX2KO mice (Fig. 2A). Visceral adipose was likewise analyzed, and ANOVA for the effects of genotype X diet on the weight of the epididymal fat pad showed no main effect of either diet or genotype but did reveal a significant interaction (F(1,35) = 10.51, P = 0.0401; Fig. 2B). Post hoc tests showed that HFD significantly increased epididymal adipose depot weight in WT mice but not in NOX2KO mice, whereas planned comparisons of NOX2KO and WT mice revealed that the epididymal adipose depot in HFD-fed WT mice was significantly heavier than that isolated from HFD-fed NOX2KO mice (Fig. 2B).
Fig. 2.

Effects of HFD on adipose depot weights in WT and NOX2KO mice. Subcutaneous inguinal and visceral epididymal fat pads were collected from WT and NOX2KO mice at the end of the 14-wk feeding trial and weighed. Data were collected from 10 mice in each group except for the NOX2KO/HFD group, which had 9 samples. A: inguinal fat depot weight in WT and NOX2KO mice following administration of CD or HFD. ***Significant (P < 0.001) increase in weight of inguinal fat depot in both WT and NOX2KO mice on HFD vs. CD. B: epididymal fat depots in WT and NOX2KO mice following administration of CD or HFD. *Significant (P < 0.05) increase in weight of epididymal fat depot in WT mice given HFD vs. WT mice on CD. #Significant (P < 0.05) increase in weight of epididymal fat depot in WT vs. NOX2KO mice on HFD.
Diet-induced adipocyte hypertrophy and inflammation in WT and NOX2KO mice.
As obesity and metabolic dysfunction are frequently associated with adipocyte enlargement or hypertrophy (12, 39), the size of individual adipocytes within subcutaneous inguinal and visceral epididymal adipose depots was evaluated in tissue sections as described in materials and methods. Such measurements revealed that HFD significantly increased the overall size of subcutaneous inguinal adipocytes (Fig. 3A). Specifically, ANOVA for the effects of genotype and diet on inguinal adipocyte size revealed a significant main effect of diet (F(1,33) = 47.79, P < 0.0001) but no effect of genotype and no interaction. Planned comparisons revealed no differences in the size of inguinal adipocytes in CD-fed NOX2KO and CD-fed WT mice and likewise showed that HFD increased subcutaneous adipocyte size similarly in both WT and NOX2KO mice (Fig. 3A). With respect to visceral adipocytes, two-way ANOVA on the effects of genotype and diet on epididymal adipocyte size revealed a significant main effect of diet (F(1,34) = 38.40, P < 0.0001). Although there was no significant effect of genotype on visceral adipocyte size, there was a significant interaction of diet and genotype (F(1,34) = 15.18, P < = 0.0015), and post hoc tests showed that HFD increased epididymal adipocyte size only in WT mice (Fig. 3B). Planned comparisons revealed that HFD-fed WT mice had significantly larger epididymal adipocytes than HFD-fed NOX2KO mice (Fig. 3B), with no differences in visceral adipocyte size between CD-fed mice. Representative images of hematoxylin and eosin-stained tissue sections prepared from visceral epididymal adipose depots likewise revealed that larger adipocytes in HFD-fed WT mice compared with HFD-fed NOX2KO mice or mice given CD (Fig. 3C).
Fig. 3.

Effects of HFD on adipocyte hypertrophy in WT and NOX2KO mice. Subcutaneous inguinal and visceral epididymal fat pads were collected from WT and NOX2KO mice at the end of the 14-wk feeding trial and processed for histological analyses of adipocyte size as described in materials and methods. Data were collected from 10 mice in each group except for the NOX2KO/HFD group, which had 9 samples. A: size of inguinal adipocytes in WT and NOX2KO mice after administration of CD or HFD. **Significant (P < 0.01) increase in adipocyte size in WT mice on HFD vs. WT mice on CD; ***significant (P < 0.001) increase in NOX2KO mice on HFD vs. NOX2KO mice on CD. B: size of epididymal adipocytes in WT and NOX2KO mice after administration of CD or HFD. ***Significant (P < 0.001) increase in adipocyte size in WT mice on HFD vs. WT mice on CD; ###significant decrease in adipocyte size in HFD-fed NOX2KO mice vs. HFD-fed WT mice. (C) Representative images of H&E stained adipocytes from which adipocyte size measures were based.
Obesity is frequently accompanied by chronic low-grade inflammation (14, 42), and evidence suggests that macrophages may mediate inflammatory pathways initiated within adipose tissues (95). Thus, macrophage infiltration into inguinal and epididymal adipose depots was evaluated by measuring expression of Iba-1, a calcium-binding protein specifically expressed in macrophages that is upregulated with activation (40, 53, 96) and can be used in Western blot and immunohistological analyses in paraffin-embedded tissues (1, 92). Evaluation of Western blots indicated clear increases in Iba-1 expression in WT, but not NOX2KO, adipose following HFD (Fig. 4). Quantification and two-tailed, unpaired t-test analyses of Iba-1 expression over multiple blots likewise revealed that HFD significantly increased Iba-1 expression in subcutaneous inguinal fat depots collected from both WT (t(18) = 4.43, P = 0.0003) and NOX2KO mice (t(18) = 3.17, P = 0.0053; Fig. 4A). Conversely, HFD increased visceral epididymal Iba-1 expression only in WT (t(18) = 2.75, P = 0.0132) but not NOX2KO mice (Fig. 4A). Representative images of hematoxylin and eosin-stained tissue sections prepared from visceral epididymal adipose depots likewise confirmed increased macrophage infiltration into adipose isolated from HFD-fed WT mice but not from HFD-fed NOX2KO mice or mice given CD (Fig. 4C).
Fig. 4.

Effects of HFD on macrophage infiltration into adipose depots in WT and NOX2KO mice. Iba-1 expression was used to measure macrophage infiltration/activation in inguinal and epididymal fat. Data were collected from 10 mice in each group except for the NOX2KO/HFD group, which had 9 samples. A: HFD-induced Iba-1 expression in inguinal and epididymal adipose tissue in WT and NOX2KO mice as percentage of expression in CD mice. *Significant (P < 0.05) HFD-induced increase in Iba-1 expression in inguinal and epididymal fat in WT mice and inguinal fat in NOX2KO mice. B: representative Western blot images showing Iba-1 (17 kDa) expression in epididymal fat taken from WT and NOX2KO mice given CD or HFD. C: representative images of Iba-1 immunostaining showing macrophage activation and infiltration into epididymal fat taken from WT and NOX2KO mice given CD or HFD.
Diet-induced alterations to visceral adipocyte physiology in WT and NOX2KO mice.
To determine whether the hypertrophy and inflammation noted in visceral adipose were accompanied by derangement in adipocyte physiology, markers of adipocyte function/injury in visceral adipose of WT and NOX2KO mice were examined by Western blot. Specifically, the overall expressions of PPARγ and adiponectin were evaluated, as they are both markers of mature functional adipocytes and also because loss of these factors contributes to obesity-induced metabolic decline (5, 82). Quantification and analyses of PPARγ expression revealed that HFD decreased PPARγ relative to levels in CD-fed mice only in WT (t(18) = 3.72, P = 0.0040) mice but not in NOX2KO mice (Fig. 5A). Conversely, adiponectin expression was decreased by HFD in visceral fat depots collected from both WT (t(18) = 10.21, P < 0.0001) and NOX2KO mice (t(18) = 5.2, P < 0.0001; Fig. 5B), but data also showed that the effect of HFD was significantly greater in WT mice than in NOX2KO mice (t(18) = 2.79, P = 0.0122; Fig. 5B).
Fig. 5.

Effects of HFD on markers of adipocyte function and injury in WT and NOX2KO mice. Expression of (A) PPARγ, (B) adiponectin, (C) GADD153/CHOP, and (D) GRP78 were evaluated in tissue homogenates prepared from epididymal adipose depots. Data were collected from 10 mice in each group except for the NOX2KO/HFD group, which had 9 samples, and depict means ± SE expression in HFD mice presented as percentage values in CD mice (100% line on graph). Significant (*P < 0.05, **P < 0.01, ***P < 0.001, respectively) changes in expression in HFD mice vs. CD mice; #significant (P < 0.05) decreases in adiponectin expression in WT-HFD adipose vs. changes in expression in NOX2KO-HFD mice.
Adipocyte injury was estimated by evaluating the expressions of GADD153/CHOP and GRP78, both of which are known to be increased in the context of obesity and thought to reflect endoplasmic reticulum (ER) stress in adipocytes caused by chronic and/or excessive inflammation (66, 67). Evaluation and statistical analysis of GADD153/CHOP blots revealed that HFD significantly increased GADD153 expression only in WT mice (t(18) = 3.77, P = 0.0014) but not in NOX2KO mice (Fig. 5C). Likewise, expression of GRP78 was also significantly increased by HFD in WT mice (t(18) = 2.5, P = 0.0217; Fig. 5D) but not in NOX2KO mice (Fig. 5D).
Histological pattern of NOX2 expression in visceral adipose of WT mice.
Although the above data indicate that NOX2 expression participates in HFD-induced adiposopathy, these data do not give any indication as to the cell type(s) mediating this effect. This issue is noteworthy because, while NOX2 expression has been detected in white adipose depots in both rodents (79) and humans (19), NOX2 expression in differentiated 3T3-L1 adipocyte cells appears to quite low (36). Indeed, cell culture data indicate that NADPH oxidase-based signaling in adipocyte cell lines may be mediated by NOX4 rather than NOX2 (57, 81), raising the possibility that adipose NOX2 expression may be restricted to macrophages, which are well known to be NOX2 positive (32). To thus better understand how NOX2 regulates adipose function, the expression of NOX2 was evaluated immunohistochemically in visceral epididymal tissue isolated from CD- and HFD-fed WT mice as described in materials and methods. Initial experiments verified the specificity of the selected NOX2 antibody by showing that it was unable to recognize an epitope in NOX2KO adipose tissues (Fig. 6A). Qualitative evaluation of NOX2 expression in the visceral adipose sections demonstrated prominent NOX2 immunoreactivity in cells with morphology typical of macrophages (Fig. 6B). To more accurately confirm the cell type-specific pattern of NOX2 expression, sections from CD- and HFD-fed WT mice were double-labeled for NOX2 and the macrophage markers Iba-1. Evaluation of tissue double-labeled sections showed extensive colocalization of NOX2 and Iba-1 staining (Fig. 6B), indicating that macrophages are the predominant cell type expressing NOX2 in the visceral adipose depot. However, an occasional and less intense pattern of NOX2 immunoreactivity that was not associated with Iba-1-positive cells could be observed (Fig. 6B, arrows), perhaps arising from preadipocytes or other stromovascular cells.
Fig. 6.
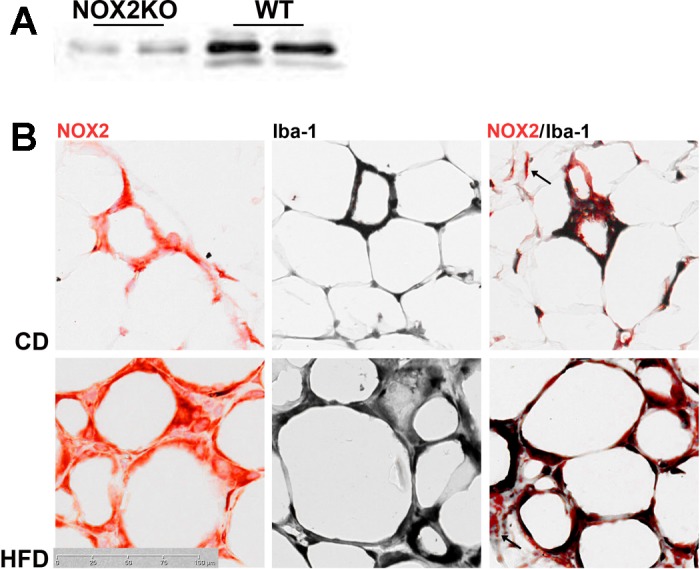
Expression of NOX2 in visceral adipose of WT mice. A: representative images of NOX2 Western blot showing that the antibody used for immunostaining did not recognize epitopes in NOX2KO tissue. B: representative images of NOX2 immunoreactivity (left, red), Iba-1 immunoreactivity (middle, black), and NOX2/Iba-1 double-labeled sections (right) of epididymal fat taken from WT mice given CD or HFD. Images reflect the predominant expression of NOX2 in Iba-1-positive macrophages, although NOX2-positive, Iba-1-negative cells can be observed (arrows in right), perhaps arising from preadipocytes or other stromovascular cells.
Diet-induced metabolic dysfunction in WT and NOX2KO mice.
To determine the extent of HFD-induced metabolic syndrome in WT and NOX2KO mice, data were collected, and for presentation purposes are thematically divided into syndromes separately describing insulin resistance and hyperlipidemia. To document insulin sensitivity and glycemic control, studies focused on regulation of fasting glucose and glucose tolerance. At the end of the 14-wk diet regimen, mice were fasted, and blood glucose and serum insulin were measured as described in materials and methods. Two-way ANOVA on the effects of genotype and diet on fasting blood glucose revealed significant main effects of diet (F(1,35) = 14.44, P = 0.0014) and genotype (F(1,35) = 35.67, P < 0.0001), with a statistically significant interaction between diet and genotype (F(1,35) = 6.72, P = 0.0239; Fig. 7A). Post hoc tests showed that HFD increased glucose levels in WT mice but not in NOX2KO mice, while planned comparisons revealed that glucose levels in HFD-fed WT mice were significantly higher than levels in HFD-fed NOX2KO mice (Fig. 7A). Conversely, two-way ANOVA on the effects of genotype and diet on fasting insulin revealed a significant effect of diet (F(1,35) = 14.44, P = 0.0014) but no effect of genotype and no interaction (Fig. 7B). Glucose tolerance was measured by OGTT as described in materials and methods, and statistical analyses showed significant main effects of diet (F(1,35) = 42.96, P < 0.0001) and genotype (F(1,35) = 5.83, P = 0.0401) on glucose tolerance but no significant interaction between diet and genotype (Fig. 7C). Post hoc tests showed that HFD impaired glucose tolerance (i.e., increased the area under the curve) in both WT and NOX2KO mice, but planned comparisons revealed that HFD-fed WT mice had increased area under the curve compared with HFD-fed NOX2KO mice (Fig. 7C). Finally, in light of evidence of adipocyte hypertrophy and inflammation in HFD-fed WT mice, experiments were designed to reveal alterations in adipocyte glucose tolerance. Specifically, the adipose lipolytic response to glucose loading was evaluated by quantifying serum levels of NEFA at both fasting and 60 min post-glucose conditions, as described in materials and methods. Fasting NEFA levels (measured as meq/dl) were not significantly affected by genotype or diet (WT-CD, 1.139 ± 0.054; WT-HFD, 1.1070 ± 0.044; NOX2KO-CD, 1.140 ± 0.027; NOX2KO-HFD, 1.060 ± 0.068). However, while glucose administration decreased NEFA in CD-fed mice, post-glucose NEFA levels in HFD-fed WT were much higher than in other groups of mice (Fig. 7D). Specifically, ANOVA analyses of post-glucose NEFA levels showed a significant main effect of diet (F(1,33) = 42.96, P < 0.0001) and of genotype (F(1,33) = 5.83, P = 0.0401) on post-glucose NEFA but no interaction (Fig. 7D). Post hoc tests revealed that HFD increased post-glucose levels of NEFA in both WT and NOX2KO mice, but planned comparisons showed that post-glucose NEFA were significantly higher in HFD-fed WT mice than in HFD-fed NOX2KO mice (Fig. 7D).
Fig. 7.

Effects of HFD on glucose regulation and tolerance in WT and NOX2KO mice. Mice were fasted for 4 h, after which an OGTT was performed to measure glucose regulation and insulin resistance. Data were collected from 10 mice in each group except for the NOX2KO/HFD group, which had 9 samples. A: fasting blood glucose levels measured in WT and NOX2KO mice after administration of HFD or CD. ***Significant (P < 0.001) increase in fasting blood glucose level in HFD-fed WT vs. CD-fed WT mice; ###significant decrease in fasting glucose in HFD-fed NOX2KO vs. HFD-fed WT mice. B: fasting insulin levels measured in WT and NOX2KO mice after administration of HFD or CD. ***Significant (P < 0.001) increase in fasting insulin in WT mice on HFD vs. WT mice on CD, and similarly an increase in NOX2KO mice on HFD vs. NOX2KO mice on CD. C: oral glucose tolerance, as indicated by area under the curve (AUC). ***Significantly (P < 0.001) larger AUC in WT mice on HFD vs. WT mice on CD; #significant decrease in glucose AUC in HFD-fed NOX2KO vs. HFD-fed WT mice. *Significantly (P < 0.05) larger AUC in NOX2KO mice on HFD vs. NOX2KO mice on CD. D: NEFA levels in mice 60 min after glucose (2 mg/kg) gavage. Significant (*P < 0.05 and **P < 0.001, respectively) increases in serum NEFA in HFD mice; ##significant decrease in NEFA following glucose load in HFD-fed NOX2KO vs. HFD-fed WT mice.
Studies next assessed a panel of bioactive serum lipids in WT and NOX2KO mice, measured under fasted conditions as described in materials and methods. Data showed a significant effect of diet (F(1,34) = 44.49, P < 0.0001) on total cholesterol, but no effect of genotype, no interaction, and no significant differences between WT and NOX2KO mice (Table 1). Likewise, there were significant effects of diet on LDL-cholesterol (F(1,34) = 26.17, P = 0.0013) and on HDL-cholesterol (F(1,34) = 30.13, P = 0.0004) but no effects of genotype and no interactions. Additionally, post hoc tests showed that HFD increased LDL- and HDL-cholesterol species in WT but not in NOX2KO mice (Table 1). Finally, there were no differences in levels of fasting triglycerides or NEFA in either WT or NOX2KO mice given either CD or HFD (Table 1).
Table 1.
Serum lipids in WT and NOX2KO mice following CD or HFD administration
| WT |
NOX2KO |
|||
|---|---|---|---|---|
| CD | HFD | CD | HFD | |
| Total cholesterol, mg/dl | 151.4 ± 8.8 | 211.5 ± 11.7*** | 159.0 ± 7.1 | 204.7 ± 11.0** |
| LDL-cholesterol, mg/dl | 28.3 ± 1.1 | 36.5 ± 1.1* | 29.9 ± 2.1 | 35.2 ± 3.0 |
| HDL-cholesterol, mg/dl | 80.0 ± 2.8 | 100.3 ± 4.6** | 88.1 ± 4.0 | 100.9 ± 5.0 |
| Triglycerides, mg/dl | 29.2 ± 1.5 | 24.2 ± 3.7 | 25.2 ± 2.3 | 23.3 ± 2.7 |
| NEFA, mEq/l | 1.13 ± 0.05 | 1.07 ± 0.04 | 1.14 ± 0.03 | 1.07 ± 0.07 |
Values are means ± SE of data collected from 9–20 animals. Male wild-type (WT; C57BL/6) and NAPDH oxidase subunit 2-deficient (NOX2KO) mice were administered control diet (CD) or high-fat diet (HFD) for 14 wk, and then serum lipids were measured under fasted conditions as described in materials and methods. Data were analyzed by 2-way ANOVA followed by planned Bonferroni posttests to determine effects of HFD in WT vs. NOX2KO mice. Significant differences (*P < 0.05,**P < 0.01, and ***P < 0.001, respectively) noted in mice given HFD vs. CD mice.
Effects of HFD on brain injury in WT and NOX2KO mice.
Experiments were next designed to determine the extent of brain injury caused by HFD in both strains of mice. Analyses were thematically split into evaluations of cerebrovascular integrity, synaptic density, and reactive gliosis measured in the anterior neocortex, a CNS site of increased NOX activity/expression following HFD (18). Cerebrovascular and blood-brain barrier (BBB) integrity were evaluated by measuring the expressions of the essential tight junction proteins claudin-5, ZO-1, and occludin, as well as the matrix metalloproteinases MMP2 and MMP9, via Western blot, as described in materials and methods. These specific signals were chosen because decreased expression of tight junction proteins is well known to accompany many neurological disorders including multiple sclerosis, stroke, Alzheimer's disease, Parkinson's disease, and epilepsy (reviewed in Ref. 13), while MMP are well known to degrade basement membranes and connective tissue of the BBB during inflammatory responses, contributing to both loss of BBB integrity and cerebral hypoperfusion (63). Western blot data indicated cerebrovascular injury in WT but not NOX2KO mice following HFD (Fig. 8). Specifically, two-tailed unpaired t-tests reveal significant, HFD-induced decreases in claudin-5 (t(18) = 2.75, P = 0.0191) and occludin expression (t(18) = 6.53, P < 0.0001) in WT but not NOX2KO, mice (Fig. 8A). Likewise, MMP2 expression was increased (t(18) = 3.21, P = 0.0048) by HFD in WT but not NOX2KO mice (Fig. 8A), while ZO1 and MMP9 were not affected by diet in either strain of mice (Fig. 8A).
Fig. 8.

Effects of HFD on markers of brain injury in WT and NOX2KO mice. A: markers of cerebrovascular integrity (expression of tight junction proteins claudin-5, ZO-1, and occludin and matrix metalloproteinases MMP2 and MMP9). B: synaptic density [expression of postsynaptic marker protein synapse-associated protein-97 (SAP97), presynaptic protein synapsin 1, and phosphorylated synapsin 1] were evaluated in tissue homogenates prepared from the frontal cortex. C: reactive gliosis (expression of GFAP, Iba-1, iNOS, COX2). Data were collected from 10 mice in each group except for the NOX2KO/HFD group, which had 9 samples, and depict means ± SE expression in HFD mice presented as %values in CD mice (100% line) on graph. Significant (*P < 0.05, **P < 0.01, ***P < 0.001, respectively) changes in expression in WT HFD vs. WT CD mice.
Evaluations of synaptic density were based on expression of the postsynaptic marker synapse-associated protein-97 (SAP97) and total and phosphorylated forms of the presynaptic protein synapsin 1 (SYN1). Quantification of total SYN1 expression revealed no differences in expression between groups (Fig. 8B), but levels of phosphorylated SYN1 (t(18) = 2.86, P = 0.0104) and SAP97 expression (t(18) = 2.34, P = 0.0309) in were significantly decreased by HFD in WT but not NOX2KO mice (Fig. 8B).
To determine whether HFD consumption affected inflammatory gliosis in mice, the expression of astrocyte and microglial markers, as well as the proinflammatory/pro-oxidant enzymes inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX2), were evaluated in cortical homogenates by use of Western blot. The intermediate filament protein glial fibrillary acidic protein (GFAP) was used to evaluate astrocyte hypertrophy (64), and data showed significant increases in GFAP caused by HFD in WT (t(18) = 7.58, P < 0.0001; Fig. 8C) but not NOX2KO mice. Microglial reactivity was evaluated by measuring expression of Iba-1, and blots likewise revealed significant increases in Iba-1 expression following HFD on WT mice (t(18) = 2.62, P = 0.0174; Fig. 8C) but not in NOX2KO mice. iNOS and COX2 were not affected by diet (Fig. 8C).
While NOX2KO mice have been used in numerous studies of brain injury, including models of aging (71), stroke (21), Alzheimer's disease (72), and traumatic injury (26), systematic, side-by-side comparisons of markers of cerebrovascular integrity, synaptic density, and reactive gliosis in WT and NOX2KO mice have not been reported. This is an important issue, as not only is NADPH oxidase thought to contribute to brain injury, but data also show that NADPH oxidase participates in synaptic plasticity and memory formation (47, 48, 88), raising the possibility that compensatory changes in CNS physiology caused by genetic deletion of NOX2 could confound comparisons between WT and NOX2KO animals. Thus, to determine whether NOX2 deletion results in significant alterations in the murine CNS, tissue homogenates were prepared from the anterior cerebral cortex of WT and NOX2KO mice maintained on CD and were evaluated for markers of cerebrovascular integrity, synaptic density, and reactive gliosis, as described in materials and methods. Data show that claudin-5 (t(18) = 2.42, P = 0.0264) and occludin expression (t(18) = 4.27, P + 0.0005) were significantly decreased in NOX2KO mice compared with WT mice (Table 2). Conversely, the expressions of all other cerebrovascular, synaptic, and glial/inflammatory markers were not significantly altered in NOX2KO mice compared with WT mice (Table 2).
Table 2.
Effects of NOX2KO on baseline values for markers of brain injury
| %WT | |
|---|---|
| Markers of cerebrovascular injury | |
| Claudin-5 | 79.6 ± 5.4* |
| Occludin | 71.3 ± 6.3*** |
| ZO-1 | 121.1 ± 12.6 |
| MMP2 | 111.7 ± 7.4 |
| MMP9 | 126.4 ± 17.8 |
| Markers of synaptic injury | |
| Phosphorylated synapsin | 96.0 ± 4.3 |
| Total synapsin | 109.7 ± 7.6 |
| SAP97 | 94.8 ± 3.2 |
| Markers of reactive gliosis | |
| GFAP | 116.7 ± 13.8 |
| Iba-1 | 108.7 ± 6.9 |
| iNOS | 83.1 ± 9.2 |
| COX2 | 79.1 ± 9.5 |
Data show means ± SE expression in NOX2KO mice presented as %WT and were analyzed by 2-tailed, unpaired t-tests. Tissue homogenates were prepared from the anterior cerebral cortex of male WT (C57BL/6) and NOX2KO mice maintained on CD were evaluated for markers of cerebrovascular integrity, synaptic density, and reactive gliosis as described in materials and methods. MMP, matrix metalloproteinase; SAP97, synapse-associated protein-97; GFAP, glial fibrillary acidic protein; COX2, cyclooxygenase 2. Significant differences (*P < 0.05 and **P < 0.01, respectively) noted in NOX2KO mice vs. WT mice.
DISCUSSION
Data in this manuscript strongly support a key role for NOX2 in the detrimental effects of diet-induced obesity. Specifically, data show that while both WT and NOX2KO mice became obese following HFD administration, NOX2KO mice had attenuated adipose pathology and preserved adipose function, particularly in visceral adipose deposits. Additionally, glucose tolerance was normalized in NOX2KO mice compared with WT mice following HFD, and HFD-induced brain injury was prevented in NOX2KO mice. Overall, these data are consistent with previous studies demonstrating increased NADPH oxidase activity/expression in models of obesity (18, 22, 97) and are also in agreement with the growing body of literature describing the sensitivity of the brain to obesity-induced metabolic dysfunction (reviewed in Refs. 17 and 60). Moreover, these data significantly extend previous studies with the demonstration that NOX2 is a specific and powerful mediator of pathogenic effects of diet-induced obesity that extend from adipocytes to brain cells. These detrimental effects could arise from NOX2-positive visceral adipose macrophages, whose action precipitates loss of adipocyte function and the development of ER stress within adipose tissues, triggering pathways that may ultimately result in loss of metabolic and neurological function. Collectively, these data raise the possibility that NOX2-based therapies could be used clinically to preserve both metabolic and neurological function in the context of obesity.
These data support a key role for NOX2 in adipose responses to HFD. Visceral adipose deposits from HFD-fed NOX2KO mice were smaller, with decreased hypertrophy and macrophage infiltration, compared with WT mice given HFD. These data also show that NOX2 deletion attenuates the development of ER stress in adipose tissues and also preserves the expression of mature adipocyte markers, suggesting that sustained expression/activity of NOX2 leads to an overall destabilization of adipocyte physiology through ER stress. These data are in agreement with existing literature on the role of free radicals in general, and NADPH oxidase specifically, in adipocyte physiology. For example, many studies have established that HFDs increase adipose NADPH oxidase (24, 33, 58) and that NADPH oxidase regulates adipocyte chemokine expression (33, 36), which could explain our observations of decreased macrophage infiltration into NOX2-deficient adipose. Data also support an important role for NADPH oxidase in long-term adipocyte responses to insulin, such as proliferation and differentiation (81); however, this paper is the first to provide evidence for a specific role for NOX2 in these pathways. The human genome contains five NOX members, NOX1 through NOX5 (34, 50), and while NOX2 expression is found in white adipose deposits in both rodents (79) and humans (19), NOX2 expression in differentiated 3T3-L1 adipocytes appears to be quite low (36), raising questions as to which adipose-resident cells express NOX2. Indeed, cell culture data indicate that many of the free radical-based pathways involved in adipocyte insulin sensitivity and proliferation may be mediated by NOX4 rather than NOX2 (57, 81). However, in contrast to the protective effects of NOX2 deletion reported here, NOX4-deficient mice have recently been shown to have enhanced susceptibility to diet-induced obesity, with accelerated insulin resistance, enhanced adipocyte hypertrophy, and increased adipose tissue hypoxia, inflammation and apoptosis (54). These findings thus suggest differential roles for the NADPH oxidase subunits in adipose tissue, with NOX4 involved in maintaining physiological events such as insulin receptor signaling and adipocyte proliferation/differentiation, and NOX2 driving sustained inflammatory changes in response to stimuli like fatty acids or oxidized lipoproteins.
These data also suggest that NOX2 might play a particularly deleterious role specifically in visceral adipose. Subcutaneous and visceral adipocytes derive from different progenitor cells that exhibit a different gene expression pattern and thus may respond quite differently to the effects of diet or NADPH oxidase inhibition. Indeed, visceral fat tissue is strongly associated with insulin resistance, diabetes mellitus, dyslipidemia, hypertension, atherosclerosis, hepatic steatosis, and overall mortality, whereas subcutaneous fat seems to have intrinsic beneficial metabolic properties (35). While the reasons for this are not yet clear, several studies have documented that visceral adipose is distinguished by increased inflammation. For example, the number of macrophages has been estimated at two- to fourfold higher in visceral compared with subcutaneous fat irrespective of adiposity levels (37); likewise, the expression of proinflamamtory cytokines is elevated in visceral compared with subcutaneous fat (7). This relatively enhanced level of inflammation in the visceral fat has been repeatedly and directly linked to obesity-related insulin resistance and type 2 diabetes (44, 86). For example, the cytokine TNFα has been demonstrated to mediate obesity-induced insulin resistance (reviewed in Ref. 43), while the chemokine monocyte chemotactic protein-1 (MCP-1) has also been shown to impair adipocyte insulin sensitivity (80). Increased proinflammatory cytokines can induce insulin resistance by several mechanisms, including via suppressor of cytokine signaling-3 (SOCS3) expression (30) and/or the activation of numerous intracellular serine kinases such as c-Jun NH2-terminal kinase (JNK) and inhibitor of κB kinase (IKK) (41). Finally, it is important to note that data from our laboratories and others' have firmly established the critical role that NADPH oxidase plays in directing macrophage inflammation, particularly the release of proinflammatory cytokines including TNFα, IL-6, and IL-1β (23, 78, 90). Thus, the data in this paper raise the possibility that inhibition of NOX2 just within visceral adipose may be sufficient to prevent the pattern of macrophage accumulation and inflammation that precipitates metabolic and neurological decline. This finding is both highly clinically significant and easily translated into new therapies.
While obesity is known to predispose individuals to a myriad of diseases (reviewed in Ref. 38), the brain may be one of the more critical sites in light of the increasing costs associated with cognitive impairment and dementia. These data indicate that NOX2 may be a key aspect of diet-induced brain injury, in agreement with a large body of literature supporting a role for NADPH oxidase in neurodegenerative pathways (15, 51, 84). For example, the same NOX2KO mice are protected from postischemic neuroinflammation and inflammatory cytokine-mediated brain damage (21) and also from microglia-mediated injury in mouse models of traumatic brain injury (26), indicating that NOX2 mediates neurotoxic brain inflammation. In further support of this potential mechanism, data from our laboratories and others' have established the critical role that NOX plays in the release of proinflammatory cytokines including TNFα, IL-6, and IL-1β (23, 78, 90), all of which could underlie neurological impairment (2, 6, 52, 73, 91). Although cytokine expression was not documented in this study, data show that HFD-induced reactive gliosis was prevented in NOX2KO mice, and previous studies from our laboratory show a tight association of reactive gliosis with both CNS cytokine release and cognitive impairment following HFD (76). Interestingly, however, NADPH oxidase also participates in physiological events including neuronal signaling and memory formation (47, 48, 88). Cognitive function was not measured in this study, as data would have been very difficult to interpret, since both humans and mice with similar NOX2 mutations have been shown to have some degree of cognitive dysfunction (46, 70). In this regard, the lack of significant differences in basal expression of synaptic markers suggests that cognitive deficits associated with NOX2KO likely reflect alterations in synaptic signaling rather than decreases in synaptic number, which is in keeping with current thoughts on the role of NADPH oxidase in cognition (3, 49). Collectively, these data raise the possibility that neurological function would be best preserved if the proinflammatory, detrimental consequences of glial NOX activation could be inhibited while preserving physiological neuronal NOX signaling. Indeed, genetically engineered mice with LoxP sites flanking the NOX2 gene are currently under development, and these mice may significantly advance understating of the complex and nuanced role of NOX2 in the brain. It should also be pointed out that commercially available NOX2KO mice have been available as homozygous breeders for many years, raising the possibility of subtle genetic drift between commercially available NOX2KO mice and the parental C57Bl/6 strain, which could potentially contribute to reported differences between strains. Indeed, it is possible that genetic drift and/or compensatory changes in the expression of other NADPH oxidase subunits could be partially responsible for the alterations in basal claudin-5 and occludin expression noted in NOX2KOmice (see Table 2). The use of newly developed, tissue-specific NOX2 knockout mice could thus also circumvent any artifacts introduced by subtle genetic drift and/or compensatory changes in the NOX2KO and C57Bl/6 strains of mice.
One key site whereby aberrant and/or sustained NADPH oxidase activation could undermine brain function is in the cerebrovascular compartment. Indeed, data in this paper reveal that HFD-induced perturbations to cerebrovascular integrity (decreased tight junction protein/increased MMP expression) are blunted in NOX2KO mice. While direct cerebrovascular function was not measured in this study, numerous reports have confirmed that alterations in these markers faithfully reflect physiological impairments in BBB function (56, 59). In addition to loss of BBB integrity, there is increasing evidence that hypoperfusion and/or loss of neurovascular coupling may be a key pathway whereby oxidative stress connects vascular-related diseases to cognitive impairment (45, 55). Indeed, evidence suggests that diabetes-induced cognitive decline may be induced via disruption of neurovascular coupling, with physiological impairment arising from both cerebrovascular elements and also glial cells (61, 83). In this light, it is important to note that NADPH oxidase is a major player in both endothelial and glial physiology. Overall, therefore, these data collectively support the hypothesis that excessive or sustained NADPH oxidase activation could disrupt brain homeostasis through any of several different cerebrovascular mechanisms, raising the possibility that targeted NOX2 inhibition could be a viable therapeutic strategy to preserve neurological function in the context of pathogenic obesity.
While NOX2KO mice were protected from many of the metabolic and neurological effects of HFD, it is clear that both strains of mice became obese in response to HFD, and indeed, NOX2KO mice were actually heavier than WT mice at times. Thus, these data reiterate that obesity per se is not sufficient to precipitate metabolic or neurological decline. This scenario is also reflected in clinical settings, but it remains unclear why increased adiposity appears to cause disease in some people but not in others (10). Recently devised theories posit that obesity in susceptible individuals is uniquely associated with pathological dysfunction in adipocytes (sick fat or adiposopathy) and that these abnormalities in fat function, rather than increases in fat mass, precipitate physiological decline (4, 10, 12). Adiposopathy is generally defined anatomically by adipocyte hypertrophy and physiologically by impaired fatty acid regulation (elevated release, particularly under high-glucose conditions), disrupted adipokine secretion, and increased inflammation (4, 9, 11, 39). Data in this paper suggest that NOX2 may mediate, at least in part, the pathological processes of adiposopathy. As obesity remains increasingly prevalent and seemingly resistant to clinical remediation (68), these data suggest that new therapies to preserve health in the presence of obesity could be based on manipulation of NOX2.
GRANTS
This work was supported by the National Institutes of Health (AG-05119) and used PBRC Core facilities (Animal Phenotyping and Imaging) funded by the NIH (P20 RR-021945 and P30 DK-072476).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.K.P., L.G.F., and S.G. performed experiments; J.K.P. and A.J.B.-K. analyzed data; J.K.P. and A.J.B.-K. interpreted results of experiments; J.K.P. drafted manuscript; J.K.P. and A.J.B.-K. edited and revised manuscript; J.K.P., L.G.F., S.G., J.N.K., and A.J.B.-K. approved final version of manuscript; J.N.K. and A.J.B.-K. conception and design of research; A.J.B.-K. prepared figures.
REFERENCES
- 1.Ahmed Z, Shaw G, Sharma VP, Yang C, McGowan E, Dickson DW. Actin-binding proteins coronin-1a and IBA-1 are effective microglial markers for immunohistochemistry. J Histochem Cytochem 55: 687–700, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole G, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383–421, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ali SS, Young JW, Wallace CK, Gresack J, Jeste DV, Geyer MA, Dugan LL, Risbrough VB. Initial evidence linking synaptic superoxide production with poor short-term memory in aged mice. Brain Res 1368: 65–70, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Appachi S, Kelly KR, Schauer PR, Kirwan JP, Hazen S, Gupta M, Kashyap SR. Reduced cardiovascular risk following bariatric surgeries is related to a partial recovery from “adiposopathy”. Obes Surg 12: 1928–1936, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aprahamian TR, Sam F. Adiponectin in cardiovascular inflammation and obesity. Int J Inflam 2011: 376909, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arvin B, Neville LF, Barone FC, Feuerstein GZ. The role of inflammation and cytokines in brain injury. Neurosci Biobehav Rev 20: 445–452, 1996 [DOI] [PubMed] [Google Scholar]
- 7.Atzmon G, Yang XM, Muzumdar R, Ma XH, Gabriely I, Barzilai N. Differential gene expression between visceral and subcutaneous fat depots. Horm Metab Res 34: 622–628, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Babior BM. The respiratory burst oxidase and the molecular basis of chronic granulomatous disease. Am J Hematol 37: 263–266, 1991 [DOI] [PubMed] [Google Scholar]
- 9.Bays H. Adiposopathy: role of adipocyte factors in a new paradigm. Expert Rev Cardiovasc Ther 3: 187–189, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Bays H, Blonde L, Rosenson R. Adiposopathy: how do diet, exercise and weight loss drug therapies improve metabolic disease in overweight patients? Expert Rev Cardiovasc Ther 4: 871–895, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Bays HE. Adiposopathy is “sick fat” a cardiovascular disease? J Am Coll Cardiol 57: 2461–2473, 2011 [DOI] [PubMed] [Google Scholar]
- 12.Bays HE, González-Campoy JM, Bray GA, Kitabchi AE, Bergman DA, Schorr AB, Rodbard HW, Henry RR. Pathogenic potential of adipose tissue and metabolic consequences of adipocyte hypertrophy and increased visceral adiposity. Expert Rev Cardiovasc Ther 6: 343–368, 2008 [DOI] [PubMed] [Google Scholar]
- 13.Bednarczyk J, Lukasiuk K. Tight junctions in neurological diseases. Acta Neurobiol Exp (Warsaw) 71: 393–408, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res 96: 939–949, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Block ML. NADPH oxidase as a therapeutic target in Alzheimer's disease. BMC Neurosci 9, Suppl 2: S8, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruce-Keller AJ, Gupta S, Knight AG, Beckett TL, McMullen JM, Davis PR, Murphy MP, Van Eldik LJ, St Clair D, Keller JN. Cognitive impairment in humanized APP×PS1 mice is linked to Aβ(1–42) and NOX activation. Neurobiol Dis 44: 317–326, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruce-Keller AJ, Keller JN, Morrison CD. Obesity and vulnerability of the CNS. Biochim Biophys Acta 1792: 395–400, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruce-Keller AJ, White CL, Gupta S, Knight AG, Pistell PJ, Ingram DK, Morrison CD, Keller JN. NOX activity in brain aging: Exacerbation by high-fat diet. Free Radic Biol Med 49: 22–30, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Rotellar F, Valentí V, Silva C, Gil MJ, Fernández-Real JM, Salvador J, Frühbeck G. Increased levels of calprotectin in obesity are related to macrophage content: impact on inflammation and effect of weight loss. Mol Med 17: 1157–1167, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chandalia M, Abate N. Metabolic complications of obesity: inflated or inflamed? J Diabetes Complications 21: 128–136, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Chen H, Kim GS, Okami N, Narasimhan P, Chan PH. NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiol Dis 42: 341–348, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chinen I, Shimabukuro M, Yamakawa K, Higa N, Matsuzaki T, Noguchi K, Ueda S, Sakanashi M, Takasu N. Vascular lipotoxicity: endothelial dysfunction via fatty-acid-induced reactive oxygen species overproduction in obese Zucker diabetic fatty rats. Endocrinology 148: 160–165, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Clark RA, Valente AJ. Nuclear factor kappa B activation by NADPH oxidases. Mech Ageing Dev 125: 799–810, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Coate KC, Huggins KW. Consumption of a high glycemic index diet increases abdominal adiposity but does not influence adipose tissue pro-oxidant and antioxidant gene expression in C57BL/6 mice. Nutr Res 30: 141–150, 2010 [DOI] [PubMed] [Google Scholar]
- 25.DeLeo FR, Quinn MT. Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J Leukoc Biol 60: 677–691, 1996 [DOI] [PubMed] [Google Scholar]
- 26.Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K, Song D, Tsunawaki S, Shioda S, Aruga T. Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation 7: 41, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dziedzic T. Systemic inflammatory markers and risk of dementia. Am J Alzheimers Dis Other Demen 21: 258–262, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elias MF, Elias PK, Sullivan LM, Wolf PA, D'Agostino RB. Lower cognitive function in the presence of obesity and hypertension: the Framingham Heart Study. Int J Obes Relat Metab Disord 27: 260–268, 2003 [DOI] [PubMed] [Google Scholar]
- 29.Elias MF, Elias PK, Sullivan LM, Wolf PA, D'Agostino RB. Obesity, diabetes and cognitive deficit: the Framingham Heart Study. Neurobiol Aging 26: 11–16, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Emanuelli B, Peraldi P, Filloux C, Chavey C, Freidinger K, Hilton DJ, Hotamisligil GS, Van Obberghen E. SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-alpha in the adipose tissue of obese mice. J Biol Chem 276: 47944–47949, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Fernández I, Peña A, Del Teso N, Pérez V, Rodríguez-Cuesta J. Clinical biochemistry parameters in C57BL/6J mice after blood collection from the submandibular vein and retroorbital plexus. J Am Assoc Lab Anim Sci 49: 202–206, 2010 [PMC free article] [PubMed] [Google Scholar]
- 32.Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med 166: S4–S8, 2002 [DOI] [PubMed] [Google Scholar]
- 33.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114: 1752–1761, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geiszt M, Leto TL. The Nox family of NAD(P)H oxidases: host defense and beyond. J Biol Chem 279: 51715–51718, 2004 [DOI] [PubMed] [Google Scholar]
- 35.Gil A, Olza J, Gil-Campos M, Gomez-Llorente C, Aguilera CM. Is adipose tissue metabolically different at different sites? Int J Pediatr Obes 6, Suppl 1 : 13–20, 2011 [DOI] [PubMed] [Google Scholar]
- 36.Han CY, Umemoto T, Omer M, Den Hartigh LJ, Chiba T, LeBoeuf R, Buller CL, Sweet IR, Pennathur S, Abel ED, Chait A. NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J Biol Chem 287: 10379–10393, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harman-Boehm I, Blüher M, Redel H, Sion-Vardy N, Ovadia S, Avinoach E, Shai I, Klöting N, Stumvoll M, Bashan N. Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesity. J Clin Endocrinol Metab 92: 2240–2247, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Haslam DW, James WP. Obesity. Lancet Neurol 366: 1197–1209, 2005 [DOI] [PubMed] [Google Scholar]
- 39.Heilbronn L, Smith SR, Ravussin E. Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus. Int J Obes Relat Metab Disord 28, Suppl 4 : S12–S21, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Hilton GD, Stoica BA, Byrnes KR, Faden AI. Roscovitine reduces neuronal loss, glial activation, and neurological deficits after brain trauma. J Cereb Blood Flow Metab 28: 1845–1859, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature 420: 333–336, 2002 [DOI] [PubMed] [Google Scholar]
- 42.Hotamisligil GS. Inflammation and metabolic disorders. Nature 444: 860–867, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Hotamisligil GS. Mechanisms of TNF-alpha-induced insulin resistance. Exp Clin Endocrinol Diabetes 107: 119–125, 1999 [DOI] [PubMed] [Google Scholar]
- 44.Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol 8: 923–934, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim HA, Miller AA, Drummond GR, Thrift AG, Arumugam TV, Phan TG, Srikanth VK, Sobey CG. Vascular cognitive impairment and Alzheimer's disease: role of cerebral hypoperfusion and oxidative stress. Naunyn Schmiedebergs Arch Pharmacol 385: 953–959, 2012 [DOI] [PubMed] [Google Scholar]
- 46.Kishida KT, Hoeffer CA, Hu D, Pao M, Holland SM, Klann E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol Cell Biol 26: 5908–5920, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid Redox Signal 9: 233–244, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kishida KT, Pao M, Holland SM, Klann E. NADPH oxidase is required for NMDA receptor-dependent activation of ERK in hippocampal area CA1. J Neurochem 94: 299–306, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knapp LT, Klann E. Role of reactive oxygen species in hippocampal long-term potentiation: contributory or inhibitory?. J Neurosci Res 70: 1–7, 2002 [DOI] [PubMed] [Google Scholar]
- 50.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4: 181–189, 2004 [DOI] [PubMed] [Google Scholar]
- 51.Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med 43: 332–347, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lane TE, Buchmeier MJ, Watry DD, Fox HS. Expression of inflammatory cytokines and inducible nitric oxide synthase in brains of SIV-infected rhesus monkeys: applications to HIV-induced central nervous system disease. Mol Med 2: 27–37, 1996 [PMC free article] [PubMed] [Google Scholar]
- 53.Lee CH, Hwang IK, Lee IS, Yoo KY, Choi JH, Lee BH, Won MH. Differential immunoreactivity of microglial and astrocytic marker protein in the hippocampus of the seizure resistant and sensitive gerbils. J Vet Med Sci 70: 1405–1409, 2008 [DOI] [PubMed] [Google Scholar]
- 54.Li Y, Mouche S, Sajic T, Veyrat-Durebex C, Supale R, Pierroz D, Ferrari S, Negro F, Hasler U, Feraille E, Moll S, Meda P, Deffert C, Montet X, Krause KH, Szanto I. Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. Int J Obes (Lond) 36: 1503–1513, 2012 [DOI] [PubMed] [Google Scholar]
- 55.Liu H, Zhang J. Cerebral hypoperfusion and cognitive impairment: the pathogenic role of vascular oxidative stress. Int J Neurosci 122: 494–499, 2012 [DOI] [PubMed] [Google Scholar]
- 56.Liu W, Chen Q, Liu J, Liu KJ. Normobaric hyperoxia protects the blood brain barrier through inhibiting Nox2 containing NADPH oxidase in ischemic stroke. Med Gas Res 1: 22, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth JD, Goldstein BJ. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol 24: 1844–1854, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsuzawa-Nagata N, Takamura T, Ando H, Nakamura S, Kurita S, Misu H, Ota T, Yokoyama M, Honda M, Miyamoto K, Kaneko S. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism 57: 1071–1077, 2008 [DOI] [PubMed] [Google Scholar]
- 59.McColl BW, Rose N, Robson FH, Rothwell NJ, Lawrence CB. Increased brain microvascular MMP-9 and incidence of haemorrhagic transformation in obese mice after experimental stroke. J Cereb Blood Flow Metab 30: 267–272, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 66: 1210–1215, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mogi M, Horiuchi M. Neurovascular coupling in cognitive impairment associated with diabetes mellitus. Circ J 75: 1042–1048, 2011 [DOI] [PubMed] [Google Scholar]
- 62.Morgenstern DE, Gifford MA, Li LL, Doerschuk CM, Dinauer MC. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus.. J Exp Med 185: 207–218, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakaji K, Ihara M, Takahashi C, Itohara S, Noda M, Takahashi R, Tomimoto H. Matrix metalloproteinase-2 plays a critical role in the pathogenesis of white matter lesions after chronic cerebral hypoperfusion in rodents. Stroke 37: 2816–2823, 2006 [DOI] [PubMed] [Google Scholar]
- 64.O'Callaghan JP, Sriram K. Glial fibrillary acidic protein and related glial proteins as biomarkers of neurotoxicity. Expert Opin Drug Saf 4: 433–442, 2005 [DOI] [PubMed] [Google Scholar]
- 65.Olufadi R, Byrne CD. Clinical and laboratory diagnosis of the metabolic syndrome. J Clin Pathol 61: 697–706, 2008 [DOI] [PubMed] [Google Scholar]
- 66.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11: 381–389, 2004 [DOI] [PubMed] [Google Scholar]
- 67.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457–461, 2004 [DOI] [PubMed] [Google Scholar]
- 68.Padwal R, Li SK, Lau DC. Long-term pharmacotherapy for obesity and overweight. Cochrane Database Syst Rev 3: CD004094, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pannacciulli N, Del Parigi A, Chen K, Le DS, Reiman EM, Tataranni PA. Brain abnormalities in human obesity: a voxel-based morphometric study. Neuroimage 31: 1419–1425, 2006 [DOI] [PubMed] [Google Scholar]
- 70.Pao M, Wiggs EA, Anastacio MM, Hyun J, DeCarlo ES, Miller JT, Anderson VL, Malech HL, Gallin JI, Holland SM. Cognitive function in patients with chronic granulomatous disease: a preliminary report. Psychosomatics 45: 230–234, 2004 [DOI] [PubMed] [Google Scholar]
- 71.Park L, Anrather J, Girouard H, Zhou P, Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab 27: 1908–1918, 2007 [DOI] [PubMed] [Google Scholar]
- 72.Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, Iadecola C. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci USA 105: 1347–1352, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parnet P, Kelley KW, Bluthé RM, Dantzer R. Expression and regulation of interleukin-1 receptors in the brain. Role in cytokines-induced sickness behavior. J Neuroimmunol 125: 5–14, 2002 [DOI] [PubMed] [Google Scholar]
- 74.Patel C, Ghanim H, Ravishankar S, Sia CL, Viswanathan P, Mohanty P, Dandona P. Prolonged reactive oxygen species generation and nuclear factor-kappaB activation after a high-fat, high-carbohydrate meal in the obese. J Clin Endocrinol Metab 92: 4476–4479, 2007 [DOI] [PubMed] [Google Scholar]
- 75.Pickup JC, Crook MA. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 41: 1241–1248, 1998 [DOI] [PubMed] [Google Scholar]
- 76.Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, Bruce-Keller AJ. Cognitive impairment following high-fat diet consumption is associated with brain inflammation. J Neuroimmunol 219: 25–32, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rader DJ. Inflammatory markers of coronary risk. N Engl J Med 343: 1179–1182, 2000 [DOI] [PubMed] [Google Scholar]
- 78.Robertson AK, Cross AR, Jones OT, Andrew PW. The use of diphenylene iodonium, an inhibitor of NADPH oxidase, to investigate the antimicrobial action of human monocyte derived macrophages. J Immunol Methods 133: 175–182, 1990 [DOI] [PubMed] [Google Scholar]
- 79.Sakurai T, Izawa T, Kizaki T, Ogasawara JE, Shirato K, Imaizumi K, Takahashi K, Ishida H, Ohno H. Exercise training decreases expression of inflammation-related adipokines through reduction of oxidative stress in rat white adipose tissue. Biochem Biophys Res Commun 379: 605–609, 2009 [DOI] [PubMed] [Google Scholar]
- 80.Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci USA 100: 7265–7270, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schröder K, Wandzioch K, Helmcke I, Brandes RP. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol 29: 239–245, 2009 [DOI] [PubMed] [Google Scholar]
- 82.Semple RK, Chatterjee VK, O'Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest 116: 581–589, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Serlin Y, Levy J, Shalev H. Vascular pathology and blood-brain barrier disruption in cognitive and psychiatric complications of type 2 diabetes mellitus. Cardiovasc Psychiatry Neurol 2011: 609202, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimoto S. Activation of NADPH oxidase in Alzheimer's disease brains. Biochem Biophys Res Commun 273: 5–9, 2000 [DOI] [PubMed] [Google Scholar]
- 85.Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology 132: 2169–2180, 2007 [DOI] [PubMed] [Google Scholar]
- 86.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 116: 1793–1801, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Silver AE, Beske SD, Christou DD, Donato AJ, Moreau KL, Eskurza I, Gates PE, Seals DR. Overweight and obese humans demonstrate increased vascular endothelial NAD(P)H oxidase-p47(phox) expression and evidence of endothelial oxidative stress. Circulation 115: 627–637, 2007 [DOI] [PubMed] [Google Scholar]
- 88.Tejada-Simon MV, Serrano F, Villasana LE, Kanterewicz BI, Wu GY, Quinn MT, Klann E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol Cell Neurosci 29: 97–106, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trollor JN, Smith E, Agars E, Kuan SA, Baune BT, Campbell L, Samaras K, Crawford J, Lux O, Kochan NA, Brodaty H, Sachdev P. The association between systemic inflammation and cognitive performance in the elderly: the Sydney Memory and Ageing Study. Age (Dordr) 34: 1295–1308, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Turchan-Cholewo J, Dimayuga VM, Gupta S, Gorospe RM, Keller JN, Bruce-Keller AJ. NADPH oxidase drives cytokine and neurotoxin release from microglia and macrophages in response to HIV-Tat. Antioxid Redox Signal 11: 193–204, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tyor WR, Glass JD, Friffin JW. Cytokine expression in the brain during AIDS. Ann Neurol 31: 349–360, 1992 [DOI] [PubMed] [Google Scholar]
- 92.Vega-Avelaira D, Moss A, Fitzgerald M. Age-related changes in the spinal cord microglial and astrocytic response profile to nerve injury. Brain Behav Immun 21: 617–623, 2007 [DOI] [PubMed] [Google Scholar]
- 93.Waldstein SR, Katzel LI. Interactive relations of central versus total obesity and blood pressure to cognitive function. Int J Obes (Lond) 30: 201–207, 2006 [DOI] [PubMed] [Google Scholar]
- 94.Ward MA, Carlsson CM, Trivedi MA, Sager MA, Johnson SC. The effect of body mass index on global brain volume in middle-aged adults: a cross sectional study. BMC Neurol: 23, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AWJ. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zecca L, Wilms H, Geick S, Claasen JH, Brandenburg LO, Holzknecht C, Panizza ML, Zucca FA, Deuschl G, Sievers J, Lucius R. Human neuromelanin induces neuroinflammation and neurodegeneration in the rat substantia nigra: implications for Parkinson's disease. Acta Neuropathol 116: 47–55, 2008 [DOI] [PubMed] [Google Scholar]
- 97.Zhang X, Dong F, Ren J, Driscoll MJ, Culver B. High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp Neurol 191: 318–325, 2005 [DOI] [PubMed] [Google Scholar]