

Abstract
Many bacterial species are capable of assessing their local population densities through a cell-cell signaling mechanism termed quorum sensing (QS). This intercellular communication process is mediated by small molecule or peptide ligands and their cognate protein receptors. Numerous pathogens use QS to initiate virulence once they achieve a threshold cell number on a host. Consequently, approaches to intercept QS have attracted considerable attention as potential anti-infective therapies. Our interest in the development of small molecule tools to modulate QS pathways motivated us to evaluate triazole-containing analogs of natural N-acyl L-homoserine lactone (AHL) signals as non-native QS agonists and antagonists in Gram-negative bacteria. We synthesized 72 triazole derivatives of five broad structure types in high yields and purities using efficient Cu(I)-catalyzed azide-alkyne couplings. These compounds were evaluated for their ability to activate or inhibit two QS receptors from two prevalent pathogens – LasR from Pseudomonas aeruginosa and AbaR from Acinetobacter baumannii – using bacterial reporter strains. Several triazole derivatives were identified that were capable of strongly modulating the activity of LasR and AbaR. These compounds represent a new and synthetically accessible class of AHL analogs, and could find utility as chemical tools to study QS and its role in bacterial virulence.
Introduction
Bacteria can monitor their local population densities using diffusible, small molecule signals (or autoinducers) in a process called quorum sensing (QS).1–3 As the bacterial population grows and these signals reach a certain threshold concentration, the bacterial community will alter gene expression levels to initiate a range of behaviors that will benefit the group.4, 5 These collective processes are exceptionally diverse, and include luminescence, virulence factor production, swarming motility, sporulation, and biofilm production.6–8 In pathogens, QS allows the bacteria to only initiate virulence after reaching a sufficiently high cell density that will overwhelm the host.7, 9 Accordingly, QS is actively being pursued as a new target for the development of anti-infective strategies, particularly against the antibiotic-resistant and increasingly prevalent pathogens Pseudomonas aeruginosa and Staphylococcus aureus.10–13
A range of approaches to intercept bacterial QS has been examined over the past ~20 years, and chemical methods to block QS signals have received significant recent attention from the chemical biology and drug discovery communities.14–17 Most bacterial QS circuits are mediated by a low molecular weight ligand binding to its cognate receptor, and this binding event regulates the transcription of QS genes.18, 19 Productive ligand:receptor binding occurs only when a “quorum” of bacteria is reached (and the ligand concentration is sufficient). In view of this mechanism, one straightforward chemical approach to intercept QS is to block signal:receptor binding using non-native small molecules, and our labs and several others have explored the design and synthesis of such agents.14, 20–25 Compounds that antagonize or agonize QS receptors can thus result in QS inhibition or activation, respectively. Either class of compound could hold value as a chemical tool to study QS pathways and phenotypes. In particular, the QS inhibitors provide a pathway to the development of novel anti-virulence approaches.
Numerous synthetic QS modulators have been reported to date. The majority of these compounds are mimics of either natural QS signal molecules or natural products shown to modulate QS, such as the halogenated furanones produced by certain algae as natural antifoulants.26, 27 In Gram-negative bacteria, QS signal molecules are typically N-acyl L-homoserine lactones (AHLs), and considerable recent research has focused on the interception of AHL signalling via chemical means. Originally studied in the marine symbiont Vibrio fischeri, AHLs are generated by LuxI-type proteins (the AHL synthases) and are perceived by intracellular LuxR-type proteins (the AHL receptors).18, 28 Dependent on the synthase, the AHL products typically have acyl chains of 4–18 carbon atoms, and may have units of unsaturation and/or altered oxidation states at the 3-position. Once productive AHL:LuxR-type receptor binding occurs, this complex will most commonly dimerize, bind to specific QS-associated promoters, and activate transcription of QS target genes.
A range of non-native AHLs have been synthesized and examined as QS agonists and antagonists in many bacterial species.14, 20–22, 25, 29–44 These signal analogs are believed to act by competitively binding LuxR-type receptors. Several recent reviews provide comprehensive descriptions of the known structure-activity relationships (SARs) for LuxR-type receptor modulation by non-native AHLs;14, 24, 45, 46 we summarize the most salient SAR features here. The homoserine lactone (HL) head group of native AHLs has been replaced by a range of heterocycles and cycloalkanes, and several of these analogs are strong QS agonists with activities comparable to native ligands, while others are strong antagonists. Evaluation of the HL head group stereochemistry has demonstrated that AHLs containing the natural HL stereoisomer (i.e., L-HL) are most often markedly more potent than their D-HL analogs, whether agonists or antagonists. Beyond these head group modifications, the AHL acyl chain has been the focus of most other AHL derivatization studies, and our laboratories have examined these analogs extensively over the past decade.30, 32, 33, 35–37, 47–52 A larger proportion of these non-native AHLs display antagonistic as opposed to agonistic activities, suggesting that the structural requirements for activation of LuxR-type receptors by AHLs are more stringent. Indeed, most non-native, AHL-type LuxR-type agonists have acyl chains that closely mimic native AHL ligands. In turn, the most potent antagonists have largely contained moderately bulky, aromatic moieties on the acyl group. Such substituents have been postulated to promote hydrophobic interactions in the LuxR-type receptor ligand-binding pocket (assuming these non-native AHLs target the same site), which could act, in part, to inactivate the receptor. Lastly, a series of studies has probed replacement of the AHL amide bond with various isosteres, including sulfonamides, ureas, and sulfonylureas, and this replacement has largely yielded LuxR-type antagonists.24
Our laboratory has focused primarily on the development of solid-phase synthetic routes to non-native AHLs and AHL analogs, and these methods have allowed for the rapid construction of several focused AHL libraries.30, 33 We now seek to develop complementary solution-phase methods for AHL synthesis to expand our set of non-native AHLs for study. We have focused our attention on robust reactions that are tolerant of a range of functional groups and utilize readily available building blocks. In the current study, we report our development of solution-phase AHL synthesis methods based on Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reactions (Figure 1).53–55 The azide and alkyne building blocks were either commercially available or generated from simple precursors via Sonogashira cross-coupling and Cu-catalyzed aromatic azidation of aromatic halides. Combination of these building blocks with complementary reagents yielded four sets of novel triazole-containing AHL analogs (types I–IV, Figure 1A) and varied combinations of complementary building blocks yielded multivalent AHL analogs (type V, Figure 1B) in a minimal number of synthetic steps. Notably, the type I analogs had the acyl triazole positioned such that the 2-N atom of the triazole ring overlapped with the 3-oxo functionality of native AHLs (circled moieties, Fig. 1A); we reasoned that such overlap could possibly enhance AHL-receptor interactions, if these triazole analogs target the ligand-binding site of LuxR-type receptors. To our knowledge, triazole HLs of these structure types are yet to be evaluated for QS activity.56, 57 Screening of the triazole HLs for modulation of LuxR-type receptors in two prominent pathogens (P. aeruginosa and Acinetobacter baumannii) revealed several potent QS agonists and antagonists. These results suggest that these triazole HLs represent a new class of non-native AHL derivatives for use as QS modulators. Furthermore, the efficient synthetic routes described herein should be readily applicable for the future generation of expanded sets of triazole HLs to improve their potency and further elucidate their SAR.
Figure 1.

A. Structures of the type I IV triazole HLs and their corresponding building blocks. The 3-oxo group of the natural AHL (left) and the corresponding triazole 2-nitrogen in the type I analog are circled. B. General structure of a dimeric type V triazole HL and corresponding building blocks. Trimeric derivatives were also synthesized; see text.
Results and Discussion
To access the triazole HLs of types I–IV (Figure 1A) via CuACC reactions, we first synthesized L-HL building blocks bearing azide or alkyne functionalities. These building blocks were then used as handles for reaction with complementary alkyne or azide–functionalized units, respectively. The CuAAC strategy was also used to synthesize triazole type V compounds containing two or three HL moieties (Figure 1B), as discussed below.
Synthesis of building block type 1: azidoacetyl HL (3)
Azidoacetyl HL 3 was readily available from L-HL, using a procedure involving bromoacetylation and subsequent displacement of the bromide with N3 (Scheme 1). In our initial two-step procedure to generate 3, bromide 2 was synthesized under Schotten-Baumann conditions and isolated by simple extraction. Subsequent reaction of 2 with sodium azide in DMSO, aqueous work-up, and extraction provided the azide 3 in only moderate overall yield (41% over two steps). We noticed, however, that both steps proceeded with clean conversion of the starting material as indicated by TLC. We therefore hypothesized that the observed moderate yield was merely due to the high solubility of 2 and 3 in water, which was required for the numerous extractions and troublesome removal of DMSO in the second step. These observations prompted us to attempt a one-pot reaction, where L-HL was directly converted to the azide building block under Schotten–Baumann conditions in the presence of both bromoacetylbromide and sodium azide (Scheme 1). To our satisfaction, this one-pot procedure provided the azidoacetyl HL 3 in excellent yield (89%) after simple extractive aqueous work-up.
Scheme 1.
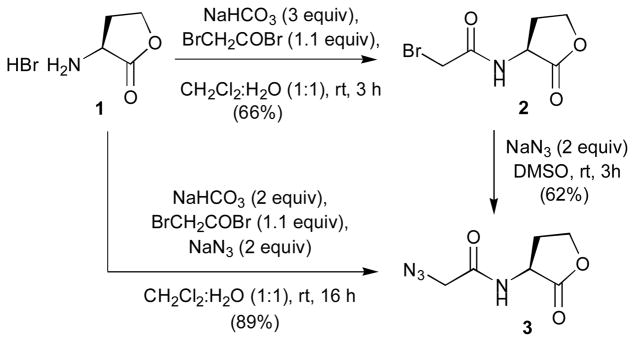
Synthesis of azidoacetyl L-HL 3.
Synthesis of building block type 2: 4-pentynoyl L-HL (4)
A range of standard amide coupling reagents was surveyed for the reaction of 4-pentynoic acid with L-HL. These studies revealed PyBOP as the optimal reagent for the synthesis of alkyne derivative 4 (>95% yield; Scheme 2).
Scheme 2.
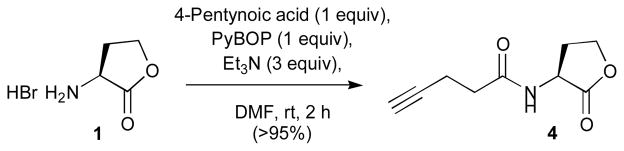
Synthesis of 4-pentynoyl L-HL 4.
Synthesis of building block types 3 and 4: alkyne- and azidobenzoyl L-HLs
Acylation of L-HL by 4- and 3-iodobenzoyl chloride, generated in situ by treating the corresponding acids with oxalyl chloride and catalytic amounts of DMF, provided intermediates 7 and 8 (Scheme 3). These intermediates were then subjected to Sonogashira cross-coupling with TMS-acetylene to give coupling products 9 and 10 in excellent yields (91% and 94%, respectively).58 Treatment of 9 and 10 with TBAF afforded the desired alkyne-functionalized benzoyl L-HL building blocks 11 and 12.
Scheme 3.

Synthesis of alkyne- and azide-functionalized benzoyl L-HLs 11–14.
Iodides 7 and 8 were also subjected to the azidation conditions reported by Andersen et al. (Scheme 3).59 Sodium azide, in the presence of copper iodide and N,N′-dimethylethylene diamine ligand, provided 4- and 3-azidobenzoylated L-HL building blocks 13 and 14 in satisfactory yields (72% and 78%, respectively).
Synthesis of triazole HL libraries (types I–IV)
The L-HL building blocks synthesized above were next subjected to CuACC reactions with a range of complementary, commercially available alkyne and azide–functionalized units to generate the triazole HL libraries. After screening a range of reaction conditions, we settled on a straightforward and general CuACC protocol using CuI and DIPEA in acetonitrile (a representative reaction is shown in Scheme 4). In all cases, complete conversion of starting material was observed after overnight reaction (~16 h, at room temperature), typically with some precipitation of the desired triazole product. A first product yield was easily isolated by simple filtration of the reaction mixture. A second quantity of product was then obtained by evaporation of the filtrate, redissolution of the resulting residue in hot acetic acid or methanol, subsequent filtration, and precipitation with diethylether. We applied this CuACC reaction and isolation protocol (described fully in the Supplementary Information) to construct the four triazole HL libraries (types I–IV) shown in Figure 2.
Scheme 4.
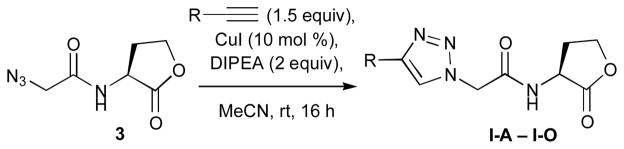
Representative synthesis of type I triazole HLs (I-A–I-O) from building block 3.
Figure 2.

Structures of the type I IV triazole HL libraries.
In general, the triazole HL library members were isolated in good to excellent yields and purities after precipitation with diethylether. However, compounds resulting from the reaction of azidoacetyl L-HL 3 with aliphatic acetylenes were of slightly lower purity (~85%) than all of the other triazoles synthesized (typically >95%). Reaction yields also varied somewhat within and between individual libraries. For example, the reaction of azido building blocks 3, 13, and 14 with aromatic acetylenes resulted in higher yields than reactions with aliphatic acetylenes (see Supplementary Information for full details).
Synthesis of multivalent triazole AHL library (type V)
Having developed efficient methodology for the synthesis of monomeric triazole HL libraries, we reasoned that the above L-HL building blocks could also serve as precursors for the construction of novel, multivalent compounds incorporating several L-HL moieties. Multivalent scaffolds remain largely unexamined in prior studies of AHL analogs,24, 45, 60 and could provide an interesting means to increase the effective concentration of ligand in a receptor local environment, thus potentially enhancing ligand activity (agonistic or antagonistic). Receptor geometry also must be considered, and the relatively imbedded native ligand-binding site of LuxR-type proteins (assuming non-native analogs target this same site) could preclude such a mechanism.14, 61 Nevertheless, in order to make multivalent compounds for testing, we investigated the reaction of azido-functionalized L-HL building blocks with di- and triynes, as well as their reaction with alkyne-functionalized L-HL building blocks.
Starting with azidoacetyl L-HL building block 3, we examined the CuAAC reaction with diynes, triynes, and L-HL building block 4. Isolation of the desired triazole products from these reactions, however, proved quite difficult. Even though reaction stoichiometries were tailored to each specific reaction, purities of the precipitated products typically proved much lower than the purities of the above type I–IV AHL analogs. One exception was V-G, stemming from reaction of 3 with 1,3-diethynylobenzene, which was isolated in satisfactory purity (85%) after precipitation with diethylether (see Supplementary Information).
Next, we examined similar reactions with azidobenzoyl building blocks 13 and 14. To our satisfaction, the reaction with these building blocks proved much improved relative to azidoacetyl building block 3. As was the case for the reactions with 3, reactant stoichiometries had to be optimized for each specific reaction. In most cases, however, the desired products could be isolated in good purities and yields using the diethylether precipitation protocol described above.
LasR antagonism by triazole HLs
We first evaluated the ability of the five triazole HL libraries to modulate the activity of the major LuxR-type QS receptor in P. aeruginosa, LasR. P. aeruginosa uses LasR to control virulence factor production (e.g., elastase production), motility, and biofilm formation, and is an extremely attractive target for small molecule modulation.61 LasR activity is regulated by its cognate AHL signal, N-(3-oxo-dodecanoyl)-L-homoserine lactone (OdDHL). We screened the five triazole HL libraries for LasR antagonism using an Escherichia coli strain harboring both a LasR expression vector and a LasR β-galactosidase reporter.62 Compounds were evaluated for receptor antagonism at 100 μM (except where limited by solubility, see Table 4) against OdDHL at 10 nM. The primary screening data for each library are listed in Tables 1 and 3–5. Triazole HLs exhibiting >50% LasR antagonism were subjected to dose response analyses to determine their IC50 values (listed in Table 2).
Table 4.
Primary agonism and antagonism assay data of the type IV triazole HL library in LasR.a
| P. aeruginosa – LasR | ||||
|---|---|---|---|---|
| compound | Activation [%] | Inhibition [%] | ||
| 100 μM | 10 μMa | 100 μM | 10 μM | |
| IV-A | 1 | – | 46 | 12 |
| IV-B | 1 | – | 29 | – |
| IV-C | 0 | – | 60 | 13 |
| IV-D | 0 | – | 36 | – |
| IV-E | – | 0 | – | 44 |
| IV-F | – | 0 | – | 18 |
| IV-G | – | 0 | – | 50 |
| IV-H | – | 0 | – | 21 |
| IV-I | 0 | – | 54 | 10 |
| IV-J | 0 | – | 46 | 14 |
| IV-K | – | 1 | – | 21 |
| IV-L | – | 0 | – | 20 |
| IV-M | – | 0 | – | 50 |
| IV-N | – | 0 | – | 21 |
| IV-O | – | 0 | – | 18 |
| IV-P | – | 0 | – | 17 |
| IV-Q | 0 | – | 38 | – |
| IV-R | 0 | – | 21 | – |
| IV-S | – | 0 | – | 1 |
| IV-T | – | 0 | – | 1 |
| IV-U | – | 3 | – | 5 |
| IV-V | 2 | – | 14 | – |
| IV-W | – | 1 | – | 17 |
| IV-X | – | 0 | – | 18 |
| IV-Y | – | 1 | – | 9 |
| IV-Z | – | 0 | – | 21 |
| IV-AA | – | 0 | – | 13 |
| IV-AB | – | 0 | – | 20 |
| IV-AC | – | 6 | – | 11 |
| IV-AD | – | 0 | – | 22 |
| IV-AE | 0 | – | 62 | 47 |
| IV-AF | – | 0 | 6 | – |
Table 1.
Primary agonism and antagonism assay data of the type I and type II triazole HL libraries in LasR.a
| P. aeruginosa – LasR | ||
|---|---|---|
| compound | Activation [%]b | Inhibition [%]c |
| I-A | 0 | 55 |
| I-B | 0 | 75 |
| I-C | 17 | 55 |
| I-D | 39 | 23 |
| I-E | 0 | 52 |
| I-F | 0 | 70 |
| I-G | 1 | 72 |
| I-H | 47 | 14 |
| I-I | 5 | 56 |
| I-J | 0 | 21 |
| I-K | 0 | 71 |
| I-L | 0 | 49 |
| I-M | 0 | 38 |
| I-N | 49 | 7 |
| I-O | 0 | 61 |
| II-A | 3 | 75 |
| II-B | 0 | 77 |
| II-C | 3 | 72 |
| II-D | 42 | 21 |
| II-E | 1 | 65 |
| II-F | 13 | 58 |
See Experimental Section for details of reporter strains and methods. All assays performed in triplicate. Error ≤10%.
AHLs evaluated at 100 μM and normalized to OdDHL at 100 μM (100%).
AHLs evaluated at 100 μM against OdDHL at 10 nM.
Table 3.
Primary agonism and antagonism assay data of the type III triazole HL library in LasR.a
| P. aeruginosa – LasR | ||
|---|---|---|
| compound | Activation [%] | Inhibition [%] |
| III-A | 1 | 23 |
| III-B | 1 | 7 |
| III-C | 0 | 48 |
| III-D | 0 | 40 |
| III-E | 1 | 14 |
| III-F | 0 | 16 |
| III-G | 1 | 19 |
| III-H | 0 | 11 |
| III-I | 0 | 16 |
| III-J | 1 | 17 |
See Table 1 for footnotes.
Table 5.
Primary agonism and antagonism assay data for the type V library in LasR.a
| P. aeruginosa LasR | ||
|---|---|---|
| compound | Activation [%] | Inhibition [%] |
| V-A | 0 | 14 |
| V-B | 1 | 22 |
| V-C | 1 | 14 |
| V-D | 1 | 10 |
| V-E | 0 | 16 |
| V-F | 0 | 16 |
| V-G | 8 | 6 |
| V-H | 16 | 1 |
| V-I | 2 | 5 |
See Table 1 for footnotes.
Table 2.
IC50 values for the most potent LasR antagonists in this study.a
| compound | IC50 value (μM)b |
|---|---|
| I-B | 17.6 |
| I-C | 3.27 |
| I-F | 16.3 |
| I-G | 12.4 |
| I-I | 15.9 |
| I-K | 13.7 |
| II-A | 16.5 |
| II-C | 10.9 |
| II-F | 4.03 |
| IV-AE | 2.64 |
See Supplementary Information for plots of antagonism dose response curves.
IC50 values determined by testing AHLs over a range of concentrations (10 nM – 100 μM) against OdDHL at 10 nM.
The type I and II triazole HL libraries contained the majority of the active LasR modulators identified in this study. These two triazole libraries were typified by short acyl chain segments (one or two carbon atoms) separating the L-HL and functionalized triazole units (Figure 2). In general, the LasR antagonism trends by the type I and II triazole HL libraries were analogous to those observed for non-native acyl HLs with similar acyl groups. Notably, these type I and II triazole HL analogs demonstrated enhanced potency relative to the recently reported AHL analogs with triazole and tetrazole units in place of the native amide bond.56
In the type I library, the acyl chain segment was a single methylene unit (Figure 2). The triazole ring, derived from the azidoacetyl building block 3, was oriented differently than it was in the type II ligands. The 2-N atom of the triazole ring then mimics the 3-oxo functionality found in OdDHL, the native AHL for LasR. We observed that the type I triazole HLs with alkyl chains extending from the triazole ring (i.e., I-A–I-D) displayed trends parallel to previously reported 3-oxo acyl-HLs;23, 30 that is, triazole HLs with short alkyl chains were LasR antagonists, while triazole HLs with longer chains were LasR agonists (see below). As the alkyl chain increased in length from propyl (I-A, 55%) to butyl (I-B, 75%, IC50 value = 17.6 μM), inhibitory activity increased from moderate to strong (Table 1). Upon further elongation of the chain from pentyl (I-C, 55%, IC50 value = 3.2 μM)) to hexyl (I-D, 23%), LasR antagonism diminished and concomitant LasR agonistic activity increased (17 and 39%, respectively; see below). Interestingly, this transition from antagonist to weak agonist occurred as the entire length of the acyl chain (counting the shortest distance across the triazole ring) transitioned from 10 to 11 atoms, a chain length approaching that of the native LasR ligand, OdDHL (with 12 carbon atoms). This result suggests that acyl chain lengths that mimic native AHL ligands can lend agonistic activity to triazole HLs.
Of the cycloalkyl triazole derivatives in the type I triazole HL library (I-E–I-H), those with larger ring structures were stronger LasR antagonists than those with smaller rings (Table 1). For example, the cyclopropyl derivative (I-E) was a moderate LasR inhibitor (52%), but the cyclopentyl (I-F, 70%, IC50 value = 16.3 μM) and cyclohexyl (I-G, 72%, IC50 value = 12.4 μM) derivatives were both more potent LasR inhibitors. We note that the inclusion of an additional methylene unit between the triazole and cyclohexyl group (as in I-H) led to weaker LasR inhibition (14%) and stronger LasR activation instead; we return to this interesting activity switch below.
Type I triazole HLs with aryl acyl groups were found to antagonize LasR (Table 1). These results correlate well with activity trends for phenylacetanoyl HLs (PHLs; e.g., C14, Figure 4), which we have shown to strongly inhibit many LuxR-type receptors.23, 30 The unsubstituted phenyl-triazole HL (I-I, 56%, IC50 value = 15.9 μM) was a moderate antagonist of LasR. Compared to I-I, the 3,5-difluoro derivative (I-K, 71%, IC50 value = 13.7 μM) was a slightly better LasR antagonist; however, triazole HLs with bulkier substituents, such as the methoxynaphthyl derivative (I-M, 38%) and the biphenylether derivative (I-J, 21% inhibition), were less active. In turn, the thiophene derivative (I-O, 61%) was a moderate LasR antagonist, and similar inhibitory activity was observed previously with the structurally-related thiophene AHL D3 (Figure 4).23, 30
Figure 4.
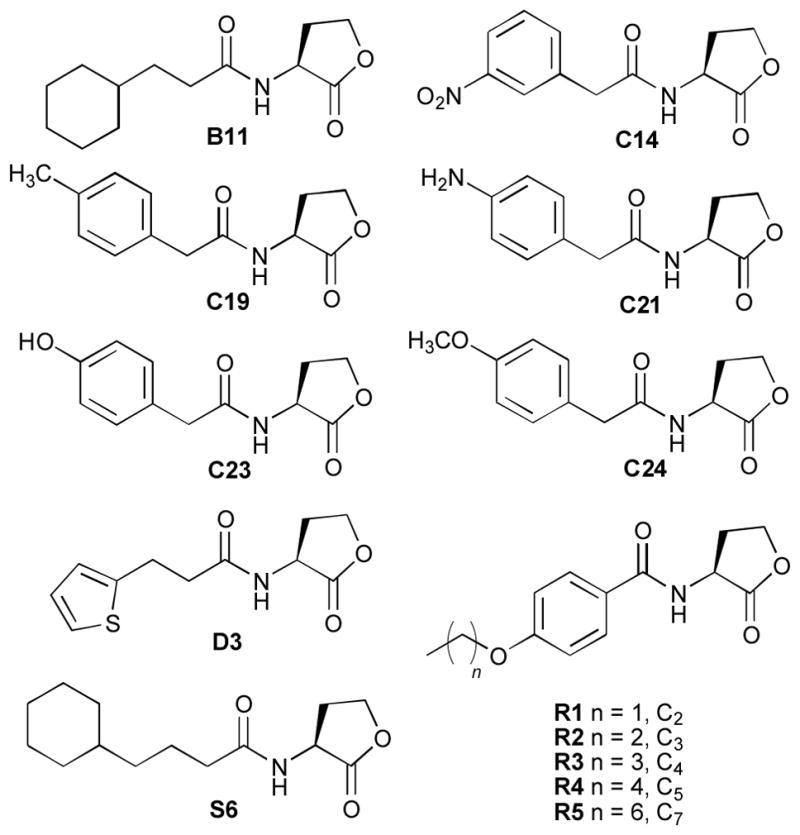
Selected non-native AHLs previously evaluated for LasR agonism and antagonism23, 30 with structures similar to the compounds reported in this study.
In general, the LasR antagonism activity trends for the type II triazole HLs were consistent with those observed for type I ligands with similar structural scaffolds (Tables 1 and 2). The para-aniline derivative (II-B, 77%) inhibited LasR somewhat more than meta-aniline (II-F, 58%) in the primary screen; however, dose response analysis revealed that II-F maintained its inhibitory activity at lower concentrations (IC50 value = 4.03 μM) relative to II-B (IC50 value > 20 μM). Modest inhibition was also observed for phenol derivative II-E (65%), yet an IC50 value could not be obtained due to a large drop off in activity at lower concentrations. In contrast, the anisole derivative (II-A, 75%, IC50 value = 16.5 μM) and the toluene derivative (II-C, 72%, IC50 value = 10.9 μM) continued to inhibit LasR at low concentrations. Our previous studies had shown that PHLs with similar acyl groups as these triazole HLs (C19, C21, C23, and C24; Figure 4) were only poor LasR antagonists (< 20%).23, 30 This result indicates that inclusion of triazole functionality into the phenylacetanoyl-HL scaffold can augment their inhibitory activity against LasR.
Type III triazole HL library
Within the type III triazole HL library, the phenylthioether derivatives III-C and III-D were the most potent LasR antagonists (Table 3), with the para-triazole HL III-C slightly more active than the meta-triazole HL III-D. Other members of this library were only weak LasR antagonists, but the para-triazole HL isomers were still typically the most potent (e.g., the indole (III-A, 23% vs. III-B, 7%) and phenol derivatives (III-G, 19% vs. III-H, 11%)).
Type IV triazole HL library
As a result of their alternate synthesis, the type IV triazole HLs, synthesized from azidobenzoyl HL building blocks 13 and 14 instead of alkyne-functionalized benzoyl HL building blocks 11 and 12, had different triazole linkages relative to the type III triazole HLs, with the variable substituent installed at the 5-position as opposed to the 1-position of the triazole (Figure 2). Interestingly, this structural modification yielded compounds with enhanced antagonistic activities against LasR relative to their type III analogs (Table 4). Similar to the type III triazole HLs, the para-substituted type IV triazole HLs were generally stronger LasR antagonists than their meta-substituted analogs. We note that certain type IV triazole HLs had limited DMSO solubility, so these compounds were evaluated at 10 μM instead of 100 μM in the reporter assays (as indicated in Table 4).
The first sub-set of the type IV library (IV-A–IV-H) was designed to evaluate the effects of alkyl substituents of variable chain lengths (see Figure 2). For the para-substituted series, LasR antagonism clearly increased with increasing alkyl chain length (from three to six carbons). The meta-substituted analogs, while less active overall, exhibited a similar inhibitory trend against LasR. These results are in accord with activity trends for our previously reported, structurally related acyl-HLs (R1–R6, Figure 4), where LasR antagonism likewise increased with alkyl substituent length.34
The second sub-set of type IV triazole HLs (IV-I–IV-P) was investigated to explore the influence of cycloalkyl substituents on compound activity (Figure 2). These derivatives were modest to moderate antagonists of LasR (Table 4). Within the sub-set of compounds, the para-substituted cyclopropyl derivative (IV-I, 54%) was a relatively strong LasR inhibitor at 100 μM. However, when evaluated at the same concentration as the rest of the cycloalkyl series (10 μM), the larger cyclopentyl (IV-K, 21%) and cyclohexyl derivatives (IV-M, 50%) were more effective LasR inhibitors than the cyclopropyl derivative (IV-I, 10%). A similar inhibition trend was observed for cycloalkyl-substituted analogs in the type I triazole HL library (as discussed above). This trend was repeated with the meta-substituted cycloalkyl library members; however, similar to the other triazoles in this library, the antagonistic activities of the meta-substituted derivatives were slightly diminished relative to the para-substituted derivatives.
The third sub-set of type IV triazole HLs contained relatively bulky aryl substituents (IV-Q–IV-Z, Figure 2), and were found to be only modest LasR antagonists (Table 4), suggesting that there may be a limitation to the size of substituents on the type IV triazole HL scaffolds for effective LasR inhibition. The difluorophenyl derivatives (IV-S, 1%, and IV-T, 1%) were far less active here relative to the structurally related (and potent) type I triazole HL (I-K, 71%, IC50 value = 13.7 μM; see above). The unsubstituted phenyl derivatives (IV-Q, 38% at 100 μM, and IV-R, 21% at 100 μM) were modest LasR inhibitors, but the further modifications of thephenyl ring tested led to diminished activity. Interestingly, in this sub-set of derivatives, the meta-substituted ligands were actually more active than the para-substituted ligands, opposing the trend observed with the rest of the type IV triazole HL library.
The fourth and final sub-set of type IV triazole HLs contained heterocyclic moieties or related heteroatom substituents (IV-AA–IV-AF, Figure 2). These derivatives were less active than their corresponding type I analogs (Table 4), with the one striking exception – the aryl thioether IV-AE. This type IV triazole HL was a stronger LasR antagonist (62%, IC50 value = 2.64 μM) than its isomeric analog in the type III library (III-C, 8%). Notably, IV-AE was the strongest LasR antagonist identified in this study, and is comparable in activity to some of the most potent acyl-HL-derived LasR antagonists we have recently reported.23, 30 Intriguingly, the structurally related type I ligand (I-N) was a strong LasR agonist instead (see below). Similar to other derivatives in this library, LasR antagonism was severely diminished for the meta-substituted thioether analog of IV-AE, IV-AF (6%).
Lastly, the type V triazole HL library contained derivatives with multiple HL rings linked together by a triazole ring, a phenyl ring, or a tertiary amine (Figure 3). In general, these multivalent scaffolds were only modestly active as LasR antagonists (Table 5), and the dimeric compounds (V-A–V-F) were more potent than the two trimeric compounds (V-I and V-H). The most active compound (V-B, 22%) was derived from meta-alkyne benzoyl HL and a para-azidobenzoyl HL. Other variants, linked through the opposite alkyne/azide coupling partners (V-C, 14%) or meta-substituted (V-D, 10%) or para-substituted (V-A, 14%) aromatic rings were less active. Larger triazole HL dimers linked via a phenyl ring between two triazole-benzoyl HLs (V-E, 16%, and V-F, 16%) displayed similar modest activities against LasR.
Figure 3.

Structures of the type V triazole L-HL library.
LasR agonism by triazole HLs
We next examined the ability of the five triazole HL libraries to agonize LasR using the E. coli reporter strain introduced above. Each compound was tested for LasR activation at 100 μM (except where limited by solubility, see Tables 1 and 3–5 and Experimental Section). Relative to OdDHL at 100 μM (set to 100%), only seven triazole HLs were capable of > 10% LasR agonism (I-C, 17%; I-D, 39%; I-H, 47%; I-N, 49%; II-D, 42%; II-F, 13%; and V-H, 16%; see Figures S-1–S-3 in the Supplementary Information for full primary screening data). Due to their limited activities, none of these synthetic AHLs were active and soluble over a range of concentrations that enabled the determination of accurate EC50 values. Still, several of these compounds and their related SAR trends are worthy of further discussion.
Four of the seven LasR agonists were uncovered in the type I triazole HL library. While short alkyl-substituted triazoles (propyl or butyl chains) were LasR antagonists (as discussed above), two with longer alkyl chains had diminishing antagonistic activity and enhanced agonistic activity in LasR (i.e., pentyl triazole (I-C, 17% agonism) and hexyl triazole (I-D, 39% agonism)). Again, this activity transition occurred as the entire length of the triazole acyl chain approached that of the OdDHL acyl chain (12 atoms). Notably, a third type I agonist (I-H, 47%) only deviated from triazole I-G, one of the top 10 LasR antagonists (IC50 value = 12.4 μM) identified herein (see above), by a methylene unit between the cyclohexyl ring and the triazole unit (Figure 2). Interestingly, acyl HLs structurally similar to I-H and previously studied in our laboratory (e.g., S6 and B11, Figure 4) do not display LasR agonistic activities, although they are agonists of other LuxR-type receptors.30, 34 We postulate that the triazole ring in I-H, a 3-oxo mimic that is absent in both B11 and S6, could play a role in its agonistic activity in LasR. The fourth type I LasR agonist identified was the aryl thioether derivative (I-N, 49%). This triazole HL represented the most potent LasR agonist uncovered in this study, and is amongst the most potent non-native LasR agonists that we have identified to date.20, 23, 30 Two type II triazole HLs were modest to moderate LasR agonists, meta-anisoyl HL (II-F, 13%) and the meta-benzaldehyde-substituted HL (II-D, 42%). Lastly, the para-substituted trimeric HL derivative (V-H) was a modest LasR agonist (16%) – additional studies would be required to determine if multivalency plays any role in this activation effect. The meta-substituted trimeric HL analog (V-I, 2%) was inactive, however, corroborating trends observed in the triazole libraries discussed above.
AbaR antagonism and agonism by triazole HLs
We next examined the ability of the five triazole HL libraries to modulate the AbaR receptor in A. baumannii. A. baumannii is a human pathogen that regulates biofilm formation and surface motility through one AHL signal, (R)-N(3′-hydroxydodecanoyl)-L-homoserine lactone (OH-dDHL) and one putative LuxR-type receptor, AbaR.52, 63, 64 Both of these phenotypes are linked to virulence in A. baumannii. We recently reported a set of non-native AHLs that inhibit motility and biofilm formation in this pathogen;52 however, to date, few small molecules are known to modulate QS in A. baumannii, and we seek additional SAR information to develop optimized AbaR inhibitors. While the LasR and AbaR receptors recognize structurally similar natural ligands (OdDHL and OH-dDHL, respectively), we found that the activity trends for identical non-native AHLs in these two receptors vary greatly.30, 52 Therefore, we hypothesized that screening the triazole AHL libraries for activity in both LasR and AbaR could reveal additional, unique structure–activity information for these important QS receptors.
We evaluated the five triazole HL libraries for AbaR antagonism and agonism in an A. baumannii (ΔabaI) mutant strain harboring an AbaR β-galactosidase reporter (see Experimental Section for a detailed description of this reporter system and assay protocols).64 Overall, we identified fewer modulators of AbaR in the triazole HL libraries relative to LasR (23 triazole HLs displayed > 20% inhibition, and one triazole HL displayed > 10% agonism; listed in Tables 6 and 7). Several of the active compounds merit additional discussion.
Table 6.
Primary agonism and antagonism assay data of the most potent triazole HLs in AbaR.a
| A. baumannii – AbaR | ||
|---|---|---|
| compound | Activation [%]d | Inhibition [%]e |
| I-A | 0 | 47 |
| I-B | 0 | 42 |
| I-C | 0 | 29 |
| I-D | 0 | 41 |
| I-E | 4 | 42 |
| I-F | 0 | 61 |
| I-G | 0 | 68 |
| I-H | 0 | 50 |
| I-I | 0 | 59 |
| I-K | 0 | 25 |
| I-N | 0 | 32 |
| I-O | 5 | 55 |
| II-A | 9 | 51 |
| II-B | 0 | 21 |
| II-C | 2 | 58 |
| II-D | 0 | 33 |
| II-F | 0 | 22 |
See Experimental Section for details of reporter strains and methods. All assays performed in triplicate. Error = ±10%.
AHLs evaluated at 100μM and normalized to OH-dDHL at 100 μM (100%).
AHLs evaluated at 100 μM against OH-dDHL at 700 nM.
Table 7.
Primary agonism and antagonism assay data of the triazole type IV library in AbaR.a
| A. baumannii – AbaR | ||||
|---|---|---|---|---|
| compound | Activation [%] | Inhibition [%] | ||
| 100 μM | 10 μM | 100 μM | 10 μM | |
| IV-C | 1 | 36 | ||
| IV-D | 0 | 25 | ||
| IV-E | 0 | 30 | ||
| IV-F | 0 | 26 | ||
| IV-I | 1 | 22 | ||
| IV-Z | 0 | 42 | ||
| IV-AE | 26 | 2 | ||
Most of the AbaR modulators were type I triazole HLs (12 of the 24 compounds), and within this structure class, the disparity between their SAR for LasR and AbaR antagonism was relatively clear. Type I triazole HLs I-A–I-D closely mimic the structure of OdDHL (the natural ligand of LasR), and these triazoles exhibited SAR in LasR that paralleled that for acyl-HLs (as discussed above). However, the length of the acyl chain did not influence their ability to inhibit AbaR (I-A, 47%; I-B, 42%; I-C, 29%; I-D, 41%), and none of these ligands activated this receptor. These results corroborate finding from our earlier study of AbaR modulators, which demonstrated that AbaR is very selective with regards to activation by acyl-HLs: only OH-dDHL strongly activates AbaR and AHLs with other acyl chain lengths and oxidation states at the 3-position were significantly less active.52 In turn, one sub-set of type I triazole HLs displayed similar SAR in AbaR as in LasR: compounds with large cycloalkyl substituents (I-F, 61%; I-G, 68%) were more potent inhibitors than those with smaller ring substituents (I-E, 42%). We highlighted above that the addition of a methylene spacer between the cyclohexyl group and the triazole group of I-G (to give I-H) had converted a LasR inhibitor (I-G; 72% antagonism) into a LasR activator (I-H; 47% agonism). In AbaR, however, I-H exhibited antagonism only (50%).
Five type II triazole HLs displayed moderate AbaR antagonism in the reporter gene assay (Table 6). The toluyl derivative (II-C), harboring a weakly electron–donating substituent, was a moderate AbaR inhibitor (58%). As the substituents on the phenyl group became more electron withdrawing, antagonistic activity diminished (e.g., anisole (II-A, 51%) > benzaldehyde (II-D, 33%) > aniline (II-B, 21%; II-F, 22%) > phenol (II-E, 9%) derivative). Similar to LasR, the type III triazole HLs were only modest modulators of AbaR (see Figure S-6 in Supplementary Information for full data). The type IV triazole HL library was more active in AbaR, however, and contained six antagonists and the only AbaR agonist revealed by the primary screen. The majority of the AbaR antagonists of this type were alkylated triazoles (IV-A–IV-H; 25–36%). Interestingly, the lone AbaR agonist identified (aryl thioether IV-AE, 26%) is also the most potent LasR antagonist identified in this study (62%, IC50 value = 2.64 μM). This divergence in activity is striking, and suggests that IV-AE could be modulating AbaR and LasR activity by different mechanisms. We note that the most potent LasR agonist identified in this study was also an aryl thioether derivative (I-N, 49%), albeit with a type I scaffold. This convergence of activity for aryl thioester-substituted triazole HLs suggests that further analysis of this substructure is warranted in new AHL analog design. Finally, while triazole HL IV-AE was only a moderately weak AbaR agonist, its discovery is significant, as few non-native agonists of AbaR have been reported to date (only seven including IV-AE).52
Summary and Conclusions
In conclusion, we have developed simple and efficient methodology for the rapid synthesis of AHL mimics using CuAAC reactions. Efficient protocols were developed for the synthesis of L-HL building blocks that contain alkyne or azido handles. These building blocks smoothly underwent cycloaddition reactions with readily–available alkyne and azide counterparts. A straightforward protocol was developed for the rapid isolation of triazole products via simple precipitation in generally good to excellent yields and purities. This methodology enabled the construction of a series of four triazole libraries (type I-IV) where the AHLs were linked to various hydrophobic groups via a 1,2,3-triazole moiety. In addition, the building blocks were also used for the construction of a series of multivalent triazole AHL analogs (type V) incorporating two or three L-HL head groups. The resulting 72 compounds were systematically evaluated in cell-based, reporter gene assays for their ability to agonize or antagonize two QS receptors – LasR and AbaR – in the clinically relevant pathogens P. aeruginosa and A. baumannii, respectively. Several potent and novel agonists and antagonists were identified.
A considerable number of SARs were apparent from the results of the biological screens (Figure 5). Of the five triazole types, the type I and II triazole HLs were the most active in general, comprising all but one of the most potent antagonists identified in this study (the outlier being the type IV triazole HL, IV-AE). The type I and II scaffolds most closely resembled the AHLs that naturally activate LasR and AbaR (i.e., OdDHL and OH-dDHL, respectively). The strongest LasR antagonists contained short alkyl chains (I-A–I-D); these triazole derivatives transitioned from inhibitory to activating in LasR as the chain length approached that of OdDHL. This trend was not unique to this scaffold, as alkylated type IV ligands (IV-A–IV-H) displayed parallel activities. Cycloalkyl and heterocyclic type I and II triazole HLs were active inhibitors of both receptors: derivatives with large saturated rings (I-G) inhibited both receptors strongly. Interestingly, the potent inhibitor I-G was converted to a strong LasR agonist upon the inclusion of a single methylene unit between the triazole and cyclohexyl group (I-H). This divergence in activity was not observed in the AbaR screen. Unlike previous studies of structurally related phenylacetanoyl-HLs (i.e., PHLs) in LasR, triazole-derived phenylacetanoyl-HLs (II-A–II-F) were modest LasR antagonists, suggesting that the inclusion of triazole functionalities into certain scaffolds heightens inhibitory activity against LasR.
Figure 5.

General SAR trends for LasR and AbaR modulators identified in this study.
Of the type III and IV triazole HL libraries, the type IV compounds, with variable substituents installed at the 5-position as opposed to the 1-position of the triazole (e.g., III-C and IV-AE), were more active (Figure 5). Within these libraries, alkyl-substituted derivatives (IV-A–IV-H) displayed activities parallel to the type I triazole HLs (I-A–I-D). Benzoyl derivatives were typically inhibitory; para-substituted derivatives were generally more active than their meta-substituted analogs in LasR (e.g., IV-A vs. IV-B). Inhibitory activity, particularly that towards AbaR, was limited by the steric bulk of acyl group. Intriguingly, the lone AbaR agonist (IV-AE) identified in this study was also the most potent LasR antagonist in this study. This contrasting activity trend for IV-AE between to the two receptors is notable, as LasR and AbaR share very similar native ligands (OdDHL and OH-dDHL, respectively), and have high sequence homology in their ligand-binding sites (note, this site is only putative in the case of AbaR).61, 65 Additional biochemical studies are warranted to probe this disparity further. Lastly, the type V triazoles containing multiple HL groups displayed modest LasR agonistic and antagonistic activities at best, suggesting that while additional study of such multivalent structures could be fruitful, ligand redesign (e.g., to include larger carbon spacers to limit bulk in the imbedded LuxR-type receptor ligand-binding site) may be necessary.
Taken together, the results of this study indicate that triazole HLs present a unique scaffold for the modulation of LuxR-type receptor activity. The ease with which these derivatives can be synthesized in solution and on scale underscore their potential as new tools to study QS. Ongoing studies are directed at examining other robust coupling reactions beyond CuACC for the solution-phase synthesis of new AHL analogs, and will be reported in due course.
Experimental
General chemical methods and instrumentation
Unless otherwise stated, all reactions were run under an argon atmosphere. Glassware was dried over a Bunsen flame under vacuum. Solvents were typically freshly distilled or dried over molecular sieves. All reactions were monitored by thin-layer chromatography (TLC) and/or reversed-phase high-performance liquid chromatography (RP-HPLC). All solvents were of RP-HPLC quality, and commercially available reagents were used without further purification. New compounds have been characterized by 1H NMR, RP-HPLC, MS (ESI) and IR (see Supplementary Information for full characterization). For selected compounds, TLC (Rf), HRMS (ESI), optical rotations, and 13C NMR data have also been included. As necessary, 13C signals were determined by 1H-13C gHSQC and 1H-13C gHMBC methods.
Analytical TLC was conducted on Merck aluminium sheets covered with silica gel (C60). The plates were either visualized under UV-light or stained by dipping in a developing agent followed by heating. Flash column chromatography was performed using a CombiFlash® (Teledyne ISCO) with Matrex 60 Å, 35–70 μ silica gel. Analytical RP-HPLC was performed on a Waters Alliance 2695 RP-HPLC system using a Symmetry 60 Å C18 column (d 3.5 μm, 4.6 × 75 mm; column temp: 25 °C; flow: 1 mL/min) with detection at 215 nm and 254 nm. Eluents A (0.1% TFA in H2O) and B (0.1% TFA in acetonitrile) were used in a linear gradient (100% A to 100% B) in a total run time of 13 min.
1H NMR and 13C NMR spectra were recorded on either a Bruker Aspect-3000 spectrometer (operating at 200 MHz for proton and 50 MHz for carbon), a Varian Mercury-300 spectrometer (operating at 300 MHz for proton and 75 MHz for carbon), or a Varian Unity Inova-500 spectrometer (operating at 500 MHz for 1H NMR). 1H-13C gHSQC and 1H-13C gHMBC were also recorded on a Varian Unity Inova-500 spectrometer. The chemical shifts (δ) are reported in parts per million (ppm) and the coupling constants (J) in Hz.
Analytical LC-MS (ESI) analysis was performed on a Waters AQUITY RP-UPLC system equipped with a diode array detector using an AQUITY UPLC BEH C18 column (d 1.7 μm, 2.1 × 50 mm; column temp: 65 °C; flow: 0.6 mL/min). Eluents A (0.1% HCO2H in H2O) and B (0.1% HCO2H in acetonitrile) were used in a linear gradient (5% B to 100% B) in a total run time of 2.6 min. The LC system was coupled to a SQD mass spectrometer. Analytical LC-HRMS (ESI) analysis was performed on an Agilent 1100 RP-LC system equipped with a diode array detector using a Phenomenex Luna C18 column (d 3 μm, 2.1 × 50 mm; column temp: 40 °C; flow: 0.4 mL/min). Eluents A (0.1% HCO2H in H2O) and B (0.1% HCO2H in acetonitrile) were used in a linear gradient (20% B to 100% B) in a total run time of 15 min. The LC system was coupled to a Micromass LCT orthogonal time-of-flight mass spectrometer equipped with a Lock Mass probe operating in positive electrospray mode.
IR analysis was performed on a Bruker Alpha FT-IR spectrometer. Measurement of optical rotation was carried out using a Perkin-Elmer polarimeter 341. The temperature for all recordings was approximately 20 °C.
Synthesis of L-HL hydrobromide salt (1)49
L-Methionine (30.4 g, 204 mmol) and bromoacetic acid (30.8 g, 222 mmol) were dissolved in i-PrOH:H2O:AcOH (5:5:2, 300 mL) in a round-bottomed flask fitted with a magnetic stirring bar. The reaction was refluxed overnight with stirring, after which the volatiles were removed in vacuo. The resulting brown oil was partly dissolved in a 4:1 mixture of i-PrOH:HBr (33% in acetic acid) (200 mL), and a white precipitate formed. This precipitate was collected by filtration and washed with isopropanol. The mother liquor was concentrated in vacuo and resubjected to precipitation using a 4:1 mixture of i-PrOH:HBr (33% in acetic acid) (200 mL). This procedure was repeated twice. The different crops of precipitate were combined to give the title compound as a white solid (31.9 g, 86% yield). Mp: 240–244 °C; 1H NMR (300 MHz, CH3OD) δ 4.53 (dt, J = 9.1, 1.1 Hz, 1H), 4.42-4.33 (m, 2H), 2.74 (dddd, J = 12.5, 8.9, 5.9, 1.2 Hz, 1H), 2.33 (dddd, J = 12.5, 11.7, 11.0, 9.1 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 173.2, 66.3, 47.7, 26.9; IR (neat) cm−1: 2986, 2880, 1775, 1496, 1210, 1155, 1009; [α]D20: −25.00° (c: 0.020, DMSO).
Synthesis of N-Bromoacetyl-L-HL (2)
L-HL hydrobromide salt (1) (4.0 g, 22.0 mmol) and sodium bicarbonate (5.5 g, 66 mmol) were dissolved in CH2Cl2:H2O (1:1, 50 mL) in a round-bottomed flask fitted with a magnetic stirring bar. Bromoacetyl bromide (2.1 mL, 4.69 g, 23.2 mmol) was then added dropwise under stirring. The reaction mixture was left under stirring for 3 h at rt, whereupon the dichloromethane was removed in vacuo. The remaining aqueous phase was extracted with ethyl acetate (3 × 40 mL). The combined organic layers were dried over sodium sulfate, and the solvent was removed in vacuo to give the title compound as white crystals (3.2 g, 66% yield). 1H NMR (300 MHz, DMSO-d6) δ 8.83 (d, J = 7.7 Hz, 1H), 4.60 (dt, J = 10.6, 8.5 Hz, 1H), 4.36 (t, J = 8.8 Hz, 1H), 4.21 (ddd, J = 9.8, 9.0, 6.4 Hz, 1H), 3.92 (s, 2H), 2.47-2.38 (m, 1H), 2.22-2.08 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 174.7, 166.1, 65.3, 48.3, 28.8, 27.9.
Synthesis of N-Azidoacetyl-L-HL (3) from N-bromoacetyl-L-homoserine lactone
N-Bromoacetyl-L-HL (2) (1.0 g, 4.50 mmol) and sodium azide (0.59 g, 9.0 mmol) were dissolved in DMSO (25 mL) in a round-bottomed flask fitted with a magnetic stirring bar. The reaction mixture was left under stirring at rt for 16 h, after which it was transferred to a separation funnel with brine (90 mL) and ethyl acetate (90 mL). The organic layer was separated, and the aqueous phase was extracted with ethyl acetate (3 × 120 mL). The combined organic layers were washed with water (180 mL) and brine (180 mL), and dried over sodium sulfate. The volatiles were then removed in vacuo to give the title compound as white crystals (0.5 g, 62% yield). Mp: 82–85 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.66 (d, J = 7.8 Hz, 1H) 4.60 (td, J = 10.6, 8.5 Hz, 1H), 4.36 (dt, J = 8.9, 1.4 Hz, 1H), 4.21 (ddd, J = 9.8, 9.0, 6.4 Hz, 1H), 3.92 (s, 2H) 2.46-2.37 (m, 1H) 2.25-2.11 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 175.0, 167.6, 65.4, 50.6 48.1, 28.1; IR (neat) cm-1: 3281, 2111, 1778, 1660, 1530, 1384, 1224, 1173, 1026 ; Rf = 0.30 (ethyl acetate, vanillin); [α]D20: −24,4° (c: 0.043, DMSO).
Synthesis of N-1-oxo-4-pentyn-L-HL (4)
4-Pentynoic acid (1.0 g, 10.2 mmol), triethylamine (4.25 mL, 3.09 g, 30.6 mmol) and PyBOP (5.30 g, 10.2 mmol) were dissolved in dry DMF (50 mL) in a round-bottomed flask fitted with a magnetic stirring bar. The reaction mixture was left under stirring for 15 min at rt, after which L-HL hydrobromide salt (1) (1.85 g, 10.2 mmol) in dry DMF (20 mL) was added. The reaction was then left under stirring for 2 h. The volatiles were removed in vacuo, and water was added (100 mL). The aqueous phase was extracted with ethyl acetate (3 × 300 mL). The combined organic layers were dried over magnesium sulfate and concentrated in vacuo. The residue was then purified by flash column chromatography on silica gel (ethyl acetate, vanillin, Rf = 0.51) to give the title compound as a white powder (1.77 g, >95% yield). Mp: 117–119 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.45 (d, J = 7.8 Hz, 1H), 4.56 (ddd, J = 10.9, 8.9, 8.3 Hz, 1H), 4.34 (dt, J = 8.8, 1.8 Hz, 1H), 4.20 (ddd, J = 10.5, 8.7, 6.4 Hz, 1H), 2.78 (dd, J = 2.6, 2.0 Hz, 1H), 2.45-2.26 (m, 5H), 2.19-2.04 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 175.2, 170.3, 83.4, 71.3, 65.2, 47.8, 33.7, 28.3, 13.9; IR (neat) cm−1: 3339, 3253, 1782, 1648, 1539, 1377, 1224, 1177, 1020; Rf = 0.51 (ethyl acetate, vanillin); [α]D20: −28.7° (c: 0.044, DMSO).
Representative procedure (I): Coupling of L-HL hydrobromide salt to substituted benzoic acids – N-(4-Iodobenzoyl)-L-HL (7)
4-Iodobenzoic acid (12.40 g, 50.0 mmol) was dissolved in dichloromethane (200 mL) in a round-bottomed flask fitted with a magnetic stirring bar. Oxalyl chloride (10.0 mL, 14.64 g, 115.4 mmol) and dry DMF (192 μL, 0.18 g, 2.5 mmol) were added. The reaction mixture was stirred for 5 h at rt, after which the volatiles were removed in vacuo. The residue was dissolved in dry DMF (130 mL) and triethylamine (16.0 mL, 11.68 g, 115.4 mmol). After 15 min, L-HL hydrobromide salt (1) (7.00 g, 38.5 mmol) dissolved in dry DMF (70 mL) was added. The reaction mixture was stirred for 2 h at rt. The volatiles were removed in vacuo, and the residue was purified by flash column chromatography on silica gel (ethyl acetate followed by dichloromethane) to give a crystalline compound containing the title compound as the major component (as observed by NMR). The material was dissolved in methanol and undissolved residues were filtered off. The filtrate was concentrated in vacuo to give the title compound as an off-white solid (9.67 g, 76% yield). Mp: 260-264 °C; RP-HPLC purity: > 95 % (Rt = 6.16 min); 1H NMR (300 MHz, DMSO-d6) δ 9.05 (d, J = 8.1 Hz, 1H),7.90-7.87 (m, 1H), 7.64-7.61 (m, 1H), 4.76 (td, J = 10.7, 8.7 Hz, 1H), 4.41 (dt, J = 8.8, 1.9 Hz, 1H), 4.27 (ddd, J = 10.3, 8.8, 6.6 Hz, 1H), 2.50-2.40 (m, 1H), 2.38-2.24 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 175.1, 165.3, 137.3, 132.7, 129.1, 99.4, 65.3, 49.2, 48.4, 27.9; IR (neat) cm−1: 3255, 3068, 2992, 2916, 1773, 1643, 1584, 1537, 1478, 1375, 1316, 1216, 1159, 1003, 914, 840, 756, 712, 671, 535, 427; [α]D20: −16.65° (c: 0.020, DMSO); MS (ESI) calcd for C11H11INO3 [M + H]+: 332.0, found: 332.1.
Representative procedure (II): Sonogashira cross-coupling of TMS-acetylenes to aromatic iodides – N-(4-Trimethylsilylethynylobenzoyl)-L-HL (9)
N-(4-Iodobenzoyl)-L-HL (7) (166 mg, 0.5 mmol), tetrakistriphenylphosphine palladium (11.6 mg, 0.01 mmol) and copper(I) iodide (3.8 mg, 0.02 mmol) were charged in a flame-dried Schlenck-flask fitted with a magnetic stirring bar. After three successive vacuum/argon cycles, a mixture of NEM:DMF (1:1, 5 mL) was added, followed by trimethylsilylacetylene (85 μL, 59 mg, 0.6 mmol). A white precipitate formed immediately. The reaction mixture was stirred overnight at rt. The volatiles were then removed by in vacuo. The black residue was dissolved in dichloromethane (20 mL) and treated with water (20 mL) and aqueous HCl (1 M, 4 mL) for 2 min. The organic layer was separated, and the aqueous phase was extracted with dichloromethane (2 × 20 mL). The combined organic layers were dried over magnesium sulfate, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (gradient 100 % heptane to 100 % ethyl acetate) to give the title compound as a beige solid (137 mg, 91% yield). Mp: 165-167 °C; RP-HPLC purity: > 95 % (Rt = 8.19 min); 1H NMR (300 MHz, DMSO-d6) δ 9.09 (d, J = 7.9 Hz, 1H), 7.86 (d, J = 8.2 Hz, 2H), 7.58 (d, J = 8.1 Hz, 2H), 4.78 (td, J = 10.3, 8.7 Hz, 1H), 4.41 (t, J = 8.6 Hz, 1H), 4.27 (ddd, J = 9.8, 8.7, 6.6 Hz, 1H), 2.46 (m, 1H), 2.34 (m, 1H), 0.24 (d, J = 1.2 Hz, 9H); IR (neat) cm−1: 3288, 2958, 2159, 1774, 1643, 1541, 1497, 1381, 1322, 1248, 1222, 1179, 1013, 839, 758; [α]D20: −17.90° (c: 0.020, DMSO); MS (ESI) calcd for C16H20NO3Si [M + H]+: 302.1, found: 302.2.
Representative procedure (III): TMS-deprotection – N-(4-Ethynylobenzoyl)-L-HL (11)
N-(4-Trimethylsilylethynylobenzoyl)-L-HL (9) (1.12 g, 3.72 mmol), TBAF (2.70 mL, 2.43 g, 9.31 mmol) and THF (19 mL) were added to a round-bottomed flask fitted with a magnetic stirring bar. The reaction mixture was stirred overnight at rt. The reaction mixture was poured into a separation funnel containing water (130 mL) and dichloromethane (150 mL). The organic layer was separated, and the aqueous phase was extracted with dichloromethane (2 × 150 mL). The combined organic layers were dried over magnesium sulfate, filtered, and concentrated in vacuo. The residue was then filtered through a short silica gel column, and the filtrate was concentrated in vacuo to give the title compound as a white solid (591 mg, 58% yield). Mp: 194-196 °C; RP-HPLC purity: > 95 % (Rt = 5.46 min); 1H NMR (300 MHz, DMSO-d6) δ 9.08 (d, J = 8.0 Hz, 1H), 7.86-7.83 (m, 2H), 7.61-7.58 (m, 2H), 4.81-4.72 (m, 1H), 4.44-4.37 (m, 2H), 4.26 (ddd, J = 10.3, 8.7, 6.6 Hz, 1H), 2.50-2.40 (m, 1H), 2.37-2.25 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 175.9, 165.9, 134.1, 132.5, 128.2, 125.6, 83.8, 83.5, 66.1, 49.2, 28.6; IR (neat) cm−1: 3413, 3277, 2913, 1771, 1662, 1534, 1498, 1380, 1316, 1180, 1016, 952, 854, 764, 658; [α]D20: −2.27° (c: 0.015, DMSO); MS (ESI) calcd for C13H12NO3 [M + H]+: 230.1, found: 230.2.
Representative procedure (IV): Azidation of aromatic iodides59 – N-(4-Azidobenzoyl)-L-HL (13)
N-(4-Iodobenzoyl)-L-HL (7) (3.00 g, 9.06 mmol), sodium azide (1.18 g, 18.12 mmol), copper(I) iodide (350 g, 1.81 mmol), and sodium ascorbate (179 mg, 0.91 mmol) were added to a round-bottomed flask fitted with a magnetic stirring bar followed by degassed DMSO:H2O (5:1, 60 mL) and N,N′-dimethylethylenediamine (293 μL, 240 mg, 2.718 mmol). The reaction mixture was stirred overnight at rt, after which it was poured into a separation funnel containing brine (170 mL) and ethyl acetate (170 mL). The organic layer was separated, and the aqueous phase was extracted with ethyl acetate (3 × 170 mL). The combined organic layers were dried over magnesium sulfate, filtered, and concentrated in vacuo. The residue was then filtered through a short silica gel column, and the filtrate was concentrated in vacuo to give the title compound as a light yellow solid (1.61 g, 72% yield). Mp: 165 °C; RP-HPLC purity: > 95 % (Rt = 5.49 min); 1H NMR (300 MHz, DMSO-d6) δ 9.00 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.4 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 4.77 (td, J = 17.6, 8.9 Hz, 1H), 4.41 (dt, J = 8.7, 1.5 Hz, 1H), 4.31-4.23 (m, 1H), 2.54-2.40 (m, 1H), 2.36-2.26 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 175.3, 165.0, 142.7, 129.9, 129.1, 119.0, 65.3, 49.2, 48.4, 27.9; IR (neat) cm−1: 3272, 8408, 2089, 1764, 1638, 1600, 1536, 1596, 1382, 1279, 1220, 1178, 1011, 848, 761, 691; [α]D20: −22.30° (c: 0.020, DMSO); MS (ESI) calcd for C11H11N4O3 [M + H]+: 247.1, found: 247.2.
Representative procedure (V): Triazole formation using aliphatic acetylenes and azidoacetyl HL building block 3 – Triazole I-A
1-Pentyne (241 μL, 166 mg, 2.44 mmol) and azide 3 (300 mg, 1.63 mmol) were dissolved in acetonitrile (6 mL) in a round-bottomed flask fitted with a magnetic stirring bar. Copper(I) iodide (31 mg, 0.16 mmol) and N,N-diisopropylethylamine (558 μL, 421 mg, 3.26 mmol) were then added, and the reaction was left under stirring at rt. The reaction was monitored by TLC for full conversion of the azide (ethyl acetate, vanillin, Rf = 0.3). Upon full conversion of starting material (16 h), the volatiles were removed in vacuo. The residue was dissolved in boiling acetic acid, and then filtered by hot gravity filtration. The acetic acid was removed in vacuo. The residue was dissolved in hot methanol. Diethyl ether was added, upon which a precipitate formed. The precipitate was then isolated and washed with diethyl ether, to give the title compound as a green powder (288 mg, 70% yield). Mp: 123–126 °C; RP-HPLC purity: 83 % (Rt = 4.27 min); 1H NMR (300 MHz, DMSO-d6) δ 8.84 (d, J = 7.7 Hz, 1H), 7.81 (s, 1H), 5.11 (s, 2H), 4.64 (td, J = 10.8, 8.8 Hz, 1H), 4.35 (dt, J = 8.8, 1.4 Hz, 1H), 4.22 (ddd, J = 10.1, 8.9, 6.4 Hz, 1H), 2.59 (t, J = 7.5 Hz, 2H), 2.46-2.39 (m, 1H), 2.23-2.09 (m, 1H), 1.67-1.52 (m, 2H,), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (DMSO-d6, from 1H-13C gHSQC and 1H-13C gHMBC, 500 MHz) δ 174.8, 165.6, 146.5, 123.1, 65.2, 51.0, 47.8, 28.1, 26.5, 21.9, 13.6; IR (neat) cm−1: 3316, 2925, 2852, 1780, 1674, 1556, 1448, 1387, 1169, 1000, 945; [α]D20: −27.0° (c: 0.079, DMSO); MS (ESI) calcd for C11H17O3N4 [M + H]+: 253.1, found: 253.2.
Representative procedure (VI): Triazole formation using aromatic acetylenes and azidoacetyl HL building block 3 – Triazole I-I
Phenylacetylene (268 μL, 250 mg, 2.44 mmol) and azide 3 (300 mg, 1.63 mmol) were dissolved in acetonitrile (16 mL) in a round-bottomed flask fitted with a magnetic stirring bar. Copper(I) iodide (31 mg, 0.16 mmol) and N,N-diisopropylethylamine (558 μL, 421 mg, 3.76 mmol) were then added, and the reaction was left under stirring at rt. The reaction was monitored by TLC for full conversion of the azide (ethyl acetate, vanillin, Rf = 0.3). Upon full conversion of starting material (16 h), the volatiles were removed in vacuo. The residue was dissolved in boiling acetic acid, and then filtered by hot gravity filtration. Diethyl ether was added, upon which a precipitate formed. The precipitate was then isolated and washed with diethyl ether, to give the title compound as a white powder (341 mg, 73% yield). Mp: 196–199 °C; RP-HPLC purity: > 95 % (Rt = 5.21 min); 1H NMR (300 MHz, DMSO-d6) δ 8.92 (d, J = 7.9 Hz, 1H), 8.55 (s, 1H), 7.87 (dd, J = 8.2, 1.2 Hz, 2H), 7.46 (app. t, J = 7.5 Hz, 2H), 7.34 (dt, J = 7.0, 1.3 Hz, 1H), 5.24 (s, 2H), 4.68 (td, J = 11.0, 8.9 Hz, 1H), 4.36 (dt, J = 8.7, 1.5 Hz, 1H), 4.23 (ddd, J = 10.5, 8.7, 6.4 Hz, 1H), 2.48-2.43 (m, 1H), 2.25-2.11 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 174.8, 165.6, 146.1, 130.6, 128.8, 127.8, 125.0, 122.9, 65.3, 51.4, 48.1, 28.2; IR (neat) cm−1: 3302, 1778, 1555, 1489, 1360, 1227, 1171, 1001; [α]D20: −18.3 (c: 0.035, DMSO); MS (ESI) calcd for C14H15O3N4 [M + H]+: 287.1, found: 287.2.
Representative procedure (VII): Triazole formation from alkyne-containing HL building blocks 11 and 12 – Triazole III-A
5-Azidoindole (62 mg, 0.39 mmol), alkyne 11 (60 mg, 0.26 mmol), copper(I) iodide (7.5 mg, 0.04 mmol), N,N-diisopropylethylamine (134 μL, 101 mg, 0.79 mmol) were dissolved in acetonitrile (2.5 mL) in a round-bottomed flask fitted with a magnetic stirring bar. The reaction was left under stirring at room rt. The reaction was monitored on RP-HPLC for full conversion of the azide. Upon full conversion of starting material (16 h), the reaction mixture was concentrated in vacuo. The residue was dissolved in boiling acetic acid, and filtered by hot gravity filtration. The acetic acid was removed in vacuo, and the residue was dissolved in hot methanol. Diethyl ether was added and a precipitate was formed. The precipitate was isolated and dried in vacuo, to give the title compound as a black solid (89 mg, 88% yield). Mp: 269–271 °C; RP-HPLC purity: > 95 % (Rt = 6.14 min); 1H NMR (300 MHz, DMSO-d6) δ 11.47 (s, 1H), 9.35 (s, 1H), 9.07 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 8.2 Hz, 1H), 7.99 (d, J = 8.4 Hz, 2H), 7.66-7.59 (m, 2H), 7.54-7.52 (m, 1H), 6.61-6.59 (m, 1H), 4.81 (td, J = 10.3, 8.9 Hz, 1H), 4.43 (dt, J = 8.6, 1.6 Hz, 1H), 4.33-4.25 (m, 1H), 2.58-2.46 (m, 1H), 2.44-2.29 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 175.0, 165.2, 145.8, 135.2, 133.3, 132.3, 129.0, 127.7, 127.3, 124.8, 120.6, 113.9, 112.0, 111.7, 101.6, 65.0, 48.1, 27.7; IR (neat) cm−1: 3413, 3340, 3112, 1772, 1638, 1516, 1484, 1228, 1183, 809, 765; MS (ESI) calcd for C21H18N5O3 [M + H]+: 388.1, found: 388.3.
Representative procedure (VIII): Triazole formation from HL building blocks 13 and 14 – Triazole IV-A
1-Pentyne (90 μL, 62 mg, 0.91 mmol), azide 13 (150 mg, 0.61 mmol), copper(I) iodide (17 mg, 0.09 mmol), N,N-diisopropylethylamine (313 μL, 236 mg, 1.83 mmol) and acetonitrile (8 mL) were added to a round-bottomed flask fitted with a magnetic stirring bar, and the reaction was left under stirring at rt. The reaction was monitored on RP-HPLC for full conversion of the azide. Full conversion of starting material was observed after 16 h, after which a first crop of product had precipitated from the reaction mixture. This crop was collected by filtration. The filtrate was concentrated in vacuo. The residue was dissolved in boiling acetic acid, and filtered by hot gravity filtration. The acetic acid was removed in vacuo, and the residue was dissolved in hot methanol. Diethyl ether was added, and the formed precipitate was isolated. The different crops of precipitate were combined and dried in vacuo to give the title compound as a beige solid (179 mg, 94% yield). Mp: 226–228 °C; RP-HPLC purity: >95 % (Rt = 5.75 min); 1H NMR (300 MHz, DMSO-d6) δ 9.15 (d, J = 8.0 Hz, 1H), 8.70 (s, 1H), 8.05 (s, 4H), 4.82 (m, 1H), 4.43 (td, J = 8.5, 4.3 Hz, 1H), 4.29 (m, 1H), 2.69 (t, J = 7.5 Hz, 2H), 2.49 (m, 1H), 2.35 (m, 1H), 1.69 (m, 2H), 0.96 (t, J = 7.3 Hz, 3H); IR (neat) cm−1: 3271, 2958, 1768, 1635, 1542, 1513, 1174, 1012, 851, 767; MS (ESI) calcd for C16H19N4O3 [M + H]+: 315.1, found: 315.3.
Biological reagents and strain information
All biological reagents were purchased from Fisher Scientific and used according to enclosed instructions. Luria-Bertani (LB) medium was prepared as instructed with pH = 7.0. Buffers and solutions (Z Buffer, 0.1% (m/v), aqueous SDS, and phosphate buffer) for Miller absorbance assays were prepared as described.66 The LasR reporter strain was E. coli DH5α harbouring the LasR expression vector pJN105L and a plasmid-born lasI–lacZ fusion (pSC11).62 The AbaR reporter strain used in this study was A. baumannii abaI::lacZ.64 Bacterial cultures were grown in a standard laboratory incubator with shaking (200 rpm) unless noted otherwise. Absorbance measurements were obtained using a Biotek Synergy 2 microplate reader using Gen5 data analysis software. All bacteriological assays were performed in triplicate.
Compound handling
Stock solutions of synthetic compounds (10 mM or 1 mM) were prepared in DMSO and stored at 4 °C in Eppendorf tubes. The amount of DMSO used in small molecule screens did not exceed 2% (v/v). Solvent resistant polypropylene or polystyrene 96-well multititer plates were used when appropriate for small molecule screening. The concentrations of synthetic triazole ligand used in the primary antagonism and agonism assays and the relative ratios of synthetic ligand to native ligand (LasR, 100 μM:10 nM; AbaR, 100μM:700 nM) in the antagonism assays were chosen to provide the greatest dynamic range between inhibitors and activators for each bacterial reporter strain. The concentration of native ligand used in the antagonism assays was approximately equal to its EC50 value in the bacterial reporter strain. No compound displayed significant growth inhibitory activity over the time course of these assays (see Supplementary Information for OD600 data).
LasR reporter gene assay (β-galactosidase)
An overnight culture of E. coli was diluted 1:10 with fresh LB containing 100 μg mL−1 ampicillin and 15 μg mL−1 gentamicin. The subculture was incubated with shaking at 37 °C until the optical density of 200 μL reached 0.27 (approximately 90 min). Arabinose (4 mg mL−1) was then added to the culture to induce production of LasR. A 198-μL portion of the diluted culture was then added to each well of a multititer containing triazoles prepared in the same way outlined above. Plates were incubated statically at 37 °C until the optical density of the wells reach 0.45 (approximately 2 h). The cultures were then assayed for β-galactosidase activity. Briefly, the OD600 of each well of the 96-well multititer plate was recorded. Next, 50-μL aliquots from each well were transferred to a solvent resistant 96-well multititer plate contain 200 μL Z buffer, 8 μL CHCl3, and 4 μL 0.1% (w/v) aqueous SDS. This suspension was mixed via repetitive pipetting (~30 ×), after which the CHCl3 was allowed to settle. A 100-μL aliquot from each well was transferred to a fresh 96-well multititer plate, and 20 μL of substrate (ONPG, 4 μg mL−1 in phosphate buffer) was added at time zero. After 20 min incubation at 25 °C, the reaction was terminated by the addition of 50 μL of 1 M Na2CO3. Absorbance at 420 and 550 nm were measured for each well using a plate reader, and Miller units were calculated according to standard methods.66 Primary LasR antagonism assays were performed in an identical manner except that the triazole was screened against 10 nM OdDHL. Assay data for all compounds in LasR is shown in Figures S-1–S-3 in the Supplementary Information.
AbaR reporter gene assay (β-galactosidase)
For primary agonism assays, 2 μL of concentrated control or triazole stock solution (to give a final concentration of 100 or 10 μM) was added to wells in a 96-well multititer plate. An overnight culture of A. baumannii abaI::lacZ was diluted 1:100 with fresh LB medium. A 198-μL portion of the diluted culture was added to each well of the multititer plate containing compounds. Plates were incubated statically at 37 °C for 18–24 h. The cultures were then assayed for β-galactosidase activity following the method outlined above for LasR with two exceptions: a 150 μL aliquot from each well was transferred to a fresh 96-well multititer plate and the 20 min incubation with ONPG was at 55 °C. Primary AbaR antagonism assays were performed in an identical manner except that triazoles were screened at 100 μM against 700 nM (R)-OH-dDHL. Assay data for all compounds in AbaR is shown in Figures S-6–S-8 in the Supplementary Information.
Dose response reporter gene assays
The dose response reporter gene assays were performed according to the protocol outlined above except that the concentrations of triazole ligands were varied between 1 × 10−3 and 1 × 105 nM. IC50 and EC50 values were calculated using GraphPad Prism software (v. 4.0) using a sigmoidal curve fit; plots are shown in the Supplementary Information.
Supplementary Material
Acknowledgments
H.E.B acknowledges the NIH (AI063326), Burroughs Wellcome Fund, and Johnson & Johnson for generous financial support for this work. T.E.N is grateful to the Carlsberg Foundation and Danish Council for Strategic Research (Center for Antimicrobial Research) for financial support. D.M.S. was funded in part by an NSF Graduate Fellowship (DGE-0718123).
Footnotes
Electronic Supplementary Information Available: Full characterization data for all new compounds, full primary screening data, and dose response curves. See DOI:
Notes and References
- 1.Ng WL, Bassler BL. Annu Rev Genet. 2009;43:197–222. doi: 10.1146/annurev-genet-102108-134304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waters C, Bassler B. Annu Rev Cell Dev Biol. 2005;21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- 3.Gamby S, Roy V, Guo M, Smith JAI, Wang J, Stewart JE, Wang X, Bentley WE, Sintim HO. ACS Chem Biol. 2012;7:1023–1030. doi: 10.1021/cb200524y. [DOI] [PubMed] [Google Scholar]
- 4.Camilli A, Bassler BL. Science. 2006;311:1113–1116. doi: 10.1126/science.1121357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stevens AM, Schuster M, Rumbaugh KP. J Bacteriol. 2012;194:2131–2141. doi: 10.1128/JB.00143-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eberhard A, Burlingame AL, Eberhard C, Kenyon GL, Nealson KH, Oppenheimer NJ. Biochemistry. 1981;20:2444–2449. doi: 10.1021/bi00512a013. [DOI] [PubMed] [Google Scholar]
- 7.Smith RS, Iglewski BH. Curr Opin Microbiol. 2003;6:56–60. doi: 10.1016/s1369-5274(03)00008-0. [DOI] [PubMed] [Google Scholar]
- 8.Zhu J, Oger PM, Schrammeijer B, Hooykaas PJ, Farrand SK, Winans SC. J Bacteriol. 2000;182:3885–3895. doi: 10.1128/jb.182.14.3885-3895.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rumbaugh KP, Diggle SP, Watters CM, Ross-Gillespie A, Griffin AS, West SA. Curr Biol. 2009;19:341–345. doi: 10.1016/j.cub.2009.01.050. [DOI] [PubMed] [Google Scholar]
- 10.Bjarnsholt T, Givskov M. Philos Trans R Soc, B. 2007;362:1213–1222. doi: 10.1098/rstb.2007.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Njoroge J, Sperandio V. EMBO Mol Med. 2009;1:201–210. doi: 10.1002/emmm.200900032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sintim HO, Smith JA, Wang J, Nakayama S, Yan L. Future Med Chem. 2010;2:1005–1035. doi: 10.4155/fmc.10.185. [DOI] [PubMed] [Google Scholar]
- 13.Stanley SA, Hung DT. Biochemistry. 2009;48:8776–8786. doi: 10.1021/bi9009083. [DOI] [PubMed] [Google Scholar]
- 14.Galloway WRJD, Hodgkinson JT, Bowden SD, Welch M, Spring DR. Chem Rev. 2011;111:28–67. doi: 10.1021/cr100109t. [DOI] [PubMed] [Google Scholar]
- 15.Clatworthy AE, Pierson E, Hung DT. Nat Chem Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 16.Dong Y-h, Wang L-y, Zhang L-H. Philos Trans R Soc, B. 2007;362:1201–1211. doi: 10.1098/rstb.2007.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hentzer M, Wu H, Andersen JB, Riedel K, Rasmussen TB, Bagge N, Kumar N, Schembri MA, Song Z, Kristoffersen P, Manefield M, Costerton JW, Molin S, Eberl L, Steinberg P, Kjelleberg S, Høiby N, Givskov M. EMBO J. 2003;22:3803–3815. doi: 10.1093/emboj/cdg366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuqua C, Greenberg E. Nat Rev Mol Cell Bio. 2002;3:685–695. doi: 10.1038/nrm907. [DOI] [PubMed] [Google Scholar]
- 19.Fuqua C, Parsek MR, Greenberg EP. Annu Rev Genet. 2001;35:439–468. doi: 10.1146/annurev.genet.35.102401.090913. [DOI] [PubMed] [Google Scholar]
- 20.Geske GD, O’Neill JC, Blackwell HE. Chem Soc Rev. 2008;37:1432–1447. doi: 10.1039/b703021p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amara N, Mashiach R, Amar D, Krief P, Spieser SAH, Bottomley MJ, Aharoni A, Meijler MM. J Am Chem Soc. 2009;131:10610–10619. doi: 10.1021/ja903292v. [DOI] [PubMed] [Google Scholar]
- 22.Hodgkinson JT, Galloway WRJD, Wright M, Mati IK, Nicholson RL, Welch M, Spring DR. Org Biomol Chem. 2012;10:6032–6044. doi: 10.1039/c2ob25198a. [DOI] [PubMed] [Google Scholar]
- 23.Sabag-Daigle A, Soares JA, Smith JN, Elmasry ME, Ahmer BMM. Appl Environ Microbiol. 2012;78:5424–5431. doi: 10.1128/AEM.00046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevens AM, Queneau Y, Soulère L, von Bodman S, Doutheau A. Chem Rev. 2011;111:4–27. doi: 10.1021/cr100064s. [DOI] [PubMed] [Google Scholar]
- 25.Smith K, Bu Y, Suga H. Chem Biol. 2003;10:563–571. doi: 10.1016/s1074-5521(03)00107-8. [DOI] [PubMed] [Google Scholar]
- 26.Givskov M, de Nys R, Manefield M, Gram L, Maximilien R, Eberl L, Molin S, Steinberg PD, Kjelleberg S. J Bacteriol. 1996;178:6618–6622. doi: 10.1128/jb.178.22.6618-6622.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hjelmgaard T, Persson T, Rasmussen TB, Givskov M, Nielsen J. Bioorg Med Chem. 2003;11:3261–3271. doi: 10.1016/s0968-0896(03)00295-5. [DOI] [PubMed] [Google Scholar]
- 28.Antunes LCM, Ferreira RBR. Crit Rev Microbiol. 2009;35:69–80. doi: 10.1080/10408410902733946. [DOI] [PubMed] [Google Scholar]
- 29.Geske GD, Mattmann ME, Blackwell HE. Bioorg Med Chem Lett. 2008;18:5978–5981. doi: 10.1016/j.bmcl.2008.07.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geske GD, O’Neill JC, Miller DM, Mattmann ME, Blackwell HE. J Am Chem Soc. 2007;129:13613–13625. doi: 10.1021/ja074135h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geske GD, O’Neill JC, Blackwell HE. ACS Chem Biol. 2007;2:315–319. doi: 10.1021/cb700036x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattmann ME, Geske GD, Worzalla GA, Chandler JR, Sappington KJ, Greenberg EP, Blackwell HE. Bioorg Med Chem Lett. 2008;18:3072–3075. doi: 10.1016/j.bmcl.2007.11.095. [DOI] [PubMed] [Google Scholar]
- 33.Praneenararat T, Geske GD, Blackwell HE. Org Lett. 2009;11:4600–4603. doi: 10.1021/ol901871y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mattmann ME, Shipway PM, Heth NJ, Blackwell HE. ChemBioChem. 2011;12:942–949. doi: 10.1002/cbic.201000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mcinnis CE, Blackwell HE. Bioorg Med Chem. 2011;19:4812–4819. doi: 10.1016/j.bmc.2011.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mcinnis CE, Blackwell HE. Bioorg Med Chem. 2011;19:4820–4828. doi: 10.1016/j.bmc.2011.06.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palmer AG, Streng E, Jewell KA, Blackwell HE. ChemBioChem. 2011;12:138–147. doi: 10.1002/cbic.201000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Welch M, Dutton JM, Glansdorp FG, Thomas GL, Smith DS, Coulthurst SJ, Barnard AML, Salmond GPC, Spring DR. Bioorg Med Chem Lett. 2005;15:4235–4238. doi: 10.1016/j.bmcl.2005.06.066. [DOI] [PubMed] [Google Scholar]
- 39.Glansdorp FG, Thomas GL, Lee JJK, Dutton JM, Salmond GPC, Welch M, Spring DR. Org Biomol Chem. 2004;2:3329–3336. doi: 10.1039/B412802H. [DOI] [PubMed] [Google Scholar]
- 40.Reverchon S, Chantegrel B, Deshayes C, Doutheau A, Cotte-Pattat N. Bioorg Med Chem Lett. 2002;12:1153–1157. doi: 10.1016/s0960-894x(02)00124-5. [DOI] [PubMed] [Google Scholar]
- 41.Castang S, Chantegrel B, Deshayes C, Dolmazon R, Gouet P, Haser R, Reverchon S, Nasser W, Hugouvieux-Cotte-Pattat N, Doutheau A. Bioorg Med Chem Lett. 2004;14:5145–5149. doi: 10.1016/j.bmcl.2004.07.088. [DOI] [PubMed] [Google Scholar]
- 42.Frezza M, Castang S, Estephane J, Soulere L, Deshayes C, Chantegrel B, Nasser W, Queneau Y, Reverchon S, Doutheau A. Bioorg Med Chem. 2006;14:4781–4791. doi: 10.1016/j.bmc.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 43.Frezza M, Soulere L, Reverchon S, Guiliani N, Jerez C, Queneau Y, Doutheau A. Bioorg Med Chem. 2008;16:3550–3556. doi: 10.1016/j.bmc.2008.02.023. [DOI] [PubMed] [Google Scholar]
- 44.Boukraa M, Sabbah M, Soulere L, El Efrit ML, Queneau Y, Doutheau A. Bioorg Med Chem Lett. 2011;21:6876–6879. doi: 10.1016/j.bmcl.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 45.Lowery CA, Salzameda NT, Sawada D, Kaufmann GF, Janda KD. J Med Chem. 2010;53:7467–7489. doi: 10.1021/jm901742e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan J, Ren D. Expert Opin Ther Pat. 2009;19:1581–1601. doi: 10.1517/13543770903222293. [DOI] [PubMed] [Google Scholar]
- 47.Olsen JA, Severinsen R, Rasmussen TB, Hentzer M, Givskov M, Nielsen J. Bioorg Med Chem Lett. 2002;12:325–328. doi: 10.1016/s0960-894x(01)00756-9. [DOI] [PubMed] [Google Scholar]
- 48.Koch B, Liljefors T, Persson T, Nielsen J, Kjelleberg S, Givskov M. Microbiology. 2005;151:3589–3602. doi: 10.1099/mic.0.27954-0. [DOI] [PubMed] [Google Scholar]
- 49.Persson T, Hansen TH, Rasmussen TB, Skindersø ME, Givskov M, Nielsen J. Org Biomol Chem. 2005;3:253–262. doi: 10.1039/b415761c. [DOI] [PubMed] [Google Scholar]
- 50.Mattmann ME, Blackwell HE. J Org Chem. 2010;75:6737–6746. doi: 10.1021/jo101237e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palmer AG, Streng E, Blackwell HE. ACS Chem Biol. 2011;6:1348–1356. doi: 10.1021/cb200298g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stacy DM, Welsh MA, Rather PN, Blackwell HE. ACS Chem Biol. 2012;7:1719–1728. doi: 10.1021/cb300351x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meldal M, Tornøe C. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 54.Rostovtsev V, Green LG, Fokin VV, Sharpless KB. Angew Chem, Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 55.Tornøe C, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 56.We note that two recent reports have demonstrated that certain triazole- and tetrazole-containing AHL analogs, with the amide bond of native AHLs replaced by heterocyclic rings, have some LuxR-type modulatory activity. See: Brackman G, Risseeuw M, Celen S, Cos P, Maes L, Nelis HJ, Van Calenbergh S, Coenye T. Bioorg Med Chem. 2012;20:4737–4743. doi: 10.1016/j.bmc.2012.06.009.Sabbah M, Fontaine F, Grand L, Boukraa M, Efrit ML, Doutheau A, Soulère L, Queneau Y. Bioorg Med Chem. 2012;20:4727–4736. doi: 10.1016/j.bmc.2012.06.007.
- 57.Non-AHL-derived azoles have been reported as QS modulators: alkyl-tetrazoles inhibited LuxR-type activity in P. aeruginosa, and alkyl-triazoles and alkyl-tetrazoles inhibited QS activity in the opportunistic fungus Candida albicans. See: Müh U, Schuster M, Heim R, Singh A, Olson ER, Greenberg EP. Antimicrob Agents Chemother. 2006;50:3674–3679. doi: 10.1128/AAC.00665-06.Shchepin R, Navarathna DHMLP, Dumitru R, Lippold S, Nickerson KW, Dussault PH. Bioorg Med Chem. 2008;16:1842–1848. doi: 10.1016/j.bmc.2007.11.011.
- 58.Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975;16:4467–4470. [Google Scholar]
- 59.Andersen J, Madsen U, Björkling F, Liang X. Synlett. 2005;14:2209–2213. [Google Scholar]
- 60.Zhu J, Beaber JW, More MI, Fuqua C, Eberhard A, Winans SC. J Bacteriol. 1998;180:5398–5405. doi: 10.1128/jb.180.20.5398-5405.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schuster M, Greenberg EP. In: Chemical Communication among Bacteria. Winans SC, Bassler BL, editors. ASM Press; Washington, DC: 2008. pp. 133–144. [Google Scholar]
- 62.Lee JH, Lequette Y, Greenberg EP. Mol Microbiol. 2006;59:602–609. doi: 10.1111/j.1365-2958.2005.04960.x. [DOI] [PubMed] [Google Scholar]
- 63.Clemmer KM, Bonomo RA, Rather PN. Microbiology. 2011;157:2534–2544. doi: 10.1099/mic.0.049791-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Niu C, Clemmer KM, Bonomo RA, Rather PN. J Bacteriol. 2008;190:3386–3392. doi: 10.1128/JB.01929-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liang HH, Kong WN, Shen T, Duan JL, Duan KM. Chin Sci Bull. 2012;57:2413–2418. [Google Scholar]
- 66.Miller JH. Experiments in Molecular Genetics. 1972:352–355. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.