

Abstract
Targeting tumor cells is an important strategy to improve the selectivity of cancer therapies. With the advanced studies in cancer biology, we know that cancer cells are usually under increased oxidative stress. The high level of reactive oxygen species in cancer cells has been exploited for developing novel therapeutic strategies to preferentially kill cancer cells. Our group, amongst others, have used boronic acids/esters as triggers for developing ROS-activated anticancer prodrugs that target cancer cells. The selectivity was achieved by combining a specific reaction between boronates and H2O2 with the efficient masking of drug toxicity in the prodrug via boronates. Prodrugs activated via ferrocene-mediated oxidation have also been developed to improve the selectivity of anticancer drugs. We describe how the strategies of ROS-activation can be used for further development of new ROS-targeting prodrugs, eventually leading to novel approaches and/or combined technology for more efficient and selective treatment of cancers.
Tumor genetics & targeting
A decade ago information and data regarding the initial human genome sequencing efforts became available [1,2]. This was followed a few years later by the full euchromatic sequence of the human genome [3]. It became clear that for diseases such as cancers, which evolve due to genetic changes, deciphering of the human tumor genome would transform how we identify, classify and treat malignancies [4]. At the same time, technological advances enable potential individual genome sequencing, an integral part of disease identification and treatment; this resulted in the genesis of personalized medicine [5]. Corollary to that is the development of genome-based chemistry to target such diseases. As the tumor genome is being deciphered, it becomes more and more attractive to identify driver mutations or lesions that are specific to cancer cells [6,7]. The expectation is that such information will provide a tumor-specific target. Such target identification allows the creation of small-molecule activators (for tumor suppressor genes and proteins) or inhibitors (for oncogenes).
Tumor genetics & therapeutics
Prime examples for successful genetic-based clinical cancer medicine are: imatinib, which targets the Bcr-Abl oncoprotein in chronic myelogenous leukemia; herceptin in Her-2-positive breast cancer; and tarceva targeting mutated EGFR in lung cancer. While these were paragons for such an approach, overall these efforts did not lead to a promising clinical future, as some tumors were unresponsive, while others responded and then became resistant as mutations occured in the object tumor proteins [8]. It is becoming clear that targeting single elements may not be the answer for most tumors. In addition, exploitation of differences between tumor and normal-cell biology has become the fundamental step in targeted therapeutics, but such an approach is not at its pinnacle, due to several reasons:
First, the genomic information is elementary, rudimentary and generally not complete. Data that becomes available is complex and inundating; and requires robust analyses in a timely fashion [7];
There are epigenetic modifications that change the genetic information;
It is becoming clear that non-coding RNAs override genetic sequence;
Targets are not available in many tumors, as driver mutations are not identified among many bystander lesions;
There are enormous amounts of intra-patient, inter-patient and intra-tumor heterogeneity, which does not allow for targeting a single lesion;
The tumor biology and pathophysiology are dependent on the micro- and macro-environment, an area that is still underdeveloped;
Targeting single elements generally does not produce desired clinical results.
These limitations underscore the need of cytotoxic agents, which, while toxic to some normal tissue, do result in stable diseases, partial and complete remissions, and even cures. In parallel, it also becomes clear that there should be efforts in changing cytotoxics to targeted cytotoxics.
Incidence of reactive oxygen species in tumor biology
Tumor biology has revealed that cancer cells are known to exhibit increased intrinsic oxidative stress. Compared with the normal counterparts, most cancer cells have inherently increased amounts of reactive oxygen species (ROS), such as superoxide, H2O2 and the hydroxyl radicals [9–12]. These oxygen-containing reactive chemicals react with nucleic acids, proteins and lipids. The high levels of ROS in cancer cells contributes to cancer-cell proliferation, DNA alterations, apoptosis, metastasis, angiogenesis and alternation in the cellular sensitivity to anticancer agents [13,14]. ROS can be found in the environment, but in cells the major source is through the mitochondrial respiratory chain [15]. There are additional sources and examples for ROS in cells and especially in cancer cells [16]. c-Myc, a commonly occurring oncogene, when activated triggers DNA damage and increases ROS [17]. Telomere dysfunction, which is frequently observed in cancer cells is associated with impaired mitochondrial biogenesis and function and increased ROS production [18]. Consistent with this report, it is worth noting that increased ROS is related to aging [19]. Increased ROS during aging may be associated with an age-related reduction in superoxide dismutase, an enzyme that neutralizes ROS [20].
ROS play a role in normal hematopoiesis and leukemogenesis with increased expression in myeloid leukemia blasts [21]. Similar to myeloid leukemia cells, comparison of chronic lymphocytic leukemia cells with normal lymphocytes revealed increased ROS in these quiescent malignant cells [22]. Several scientific groups have demonstrated that malignant transformation of normal cells mediated through Ras induced intracellular ROS production [23,24]. Similarly, modulation of intracellular ROS production was directly responsible for tumor development [25] and was differentially affected in normal versus tumor tissue [26]. Cells with increased ROS levels are prone to resistance to endogenous and radiation- or drug-induced cell death [27,28]. Such physiological survival phenomena lead to accumulation of cancer cells with higher ROS levels. Furthermore, ROS-mediated nuclear damage is associated with increased disease risk, progression and survival in cancer patients [29].
In cancer cells, ROS signaling plays a major role in survival, transcription, protein translation, and tumor formation and development. In general, redox signaling results in binding of several transcription factors to their cognate promoter sites. Such signaling leads to activation of genes that are associated with pathogenesis of specific tumors [30]. Superoxide and hydrogen peroxide are the primary determinant of such signaling. Studies have elucidated that these two species behave differently regarding signaling. While superoxide anions act as oncogenic ROS, hydrogen peroxide results in apoptosis of cancer cells [31]. Among several transcription factors, hypoxia induced factor 1 (HIF1) is not only identified as a primary target, but strategies to inhibit this factor have been successful [32,33]. In addition to genetic changes, epigenetic modification, especially genome-wide hypomethylation and hypermethylation of several of the promoter gene CpG islands, have been observed [34]. Such processes were directly associated with oxidative damage. Oxidative damage-induced formation and relocalization of a silencing complex to oncogenes may explain cancer-specific aberrant DNA methylation and transcriptional silencing [35]. These new observations further underscore the role of oxidative damage in diseases, such as cancer. Collectively, these investigations establish that tumor and normal cells have differential biological properties when it comes to hypoxia, oxidative pathways and ROS. These inherent differences between malignant cells and healthy cells could be exploited to provide treatment options.
Tumor biology & therapeutics
Cancer therapies are nearly as toxic to healthy cells as to cancer cells and a major focus in the development of new therapeutics is to exploit differences in cancer cells so that therapies can be highly targeted. In fact, hypoxia has been tested directly as a target and inhibitors, such as echinomycin, have been specifically developed [32,36]. Unfortunately, this compound was not useful as it has a dual effect on HIF1 activity under normoxic and hypoxic conditions [37], which was consistent with poor clinical results [38,39]. Another strategy was used by creating small-molecule chemotherapeutics that are activated only in this low-oxygen condition making them target cytotoxic agents [40]. A primary mechanism for such an approach is to create prodrugs that are activated by metabolic reduction in hypoxic conditions to change to cytotoxic agents specifically in tumor environments. One such example is PR-104, which is a DNA-crosslinking agent that is used as a prodrug, and has been shown to be active in murine and human tumor models [41,42]. Clinical investigations are ongoing with this molecule.
Similar to hypoxia, increased ROS could be exploited for therapeutic targeting of tumor tissue. As previously explained, since ROS induction, as well as decline below a threshold, impacts cancer cell killing, both strategies (e.g., pro-oxidant and antioxidant approaches) have been utilized [23,43,44]. The high level of ROS in cancer cells has been exploited for developing novel therapeutic strategies to preferentially kill cancer cells [11,16,45]. These have been reviewed by Hileman et al., Trachootham et al., Pelicano et al., Lopez-Lazaro, and Fang et al. [11,46–49]. Diverse chemotherapeutic agents have been developed to kill tumor cells by amplifying oxidant stress, such as agents that directly generate ROS or ones that inhibit antioxidant enzymes [50–52]. This is based on their vulnerability to further ROS insults. However, there was little clinical response to such agents, likely due to the fact that cancer cells were already adapted to higher levels of ROS. For example, an alternatively spliced isoform of pyruvate kinase M2 was identified in many cancer cells that maintains cellular redox homeostasis during metabolic stress [53].
An opposite approach is to use antioxidants to increase ROS-scavenging capacity [54–56]. Such agents are capable of abrogating ROS-signaling and suppressing tumor growth. However, several antioxidants used in clinical trials have been associated with increased cancer incidence. This was related to the inhibition of ROS-mediated apoptosis and the prevention of oxidative damage in tumors [57]. In addition, antioxidants were found to decrease the ROS-mediated anti-tumor activity of anticancer agents; for example, paclitaxel, bortezomib and radiation therapy [58,59]. Although the potential importance of the increased ROS stress in cancer cells as a therapeutic target has been appreciated a decade ago, no approach to date has been effective in moving beyond the status quo, which is little or no therapeutic selectivity. Tumor-cell redox balance and its modulation are ongoing efforts [60].
Tumor biology & rationale for prodrugs
Another attractive tactic to utilize increased ROS in cancer cells is to create agents that act as prodrugs for site-specific activation in the tumor environment due to the presence of ROS. Such an approach makes a cytotoxic agent become a targeted chemotherapeutic agent. Prodrug approaches have been used for the development of hypoxia-targeting anticancer drugs [40]. Scientists from the University of Auckland (Auckland, New Zealand) and others have been actively working in this field and several promising hypoxia-targeting anticancer prodrugs have been developed [40–42,61]. Several redox-modulating agents have also been developed as selective anticancer drugs [22,23,45,46,62–64], while there are very few reports about ROS-activated prodrugs. Cohen’s group reported the first H2O2-activated matrix metalloproteinase inhibitor (MMPi) by protecting the hydroxyl group of the zinc-binding group with a boronic ester [65]. Recently, the authors’ group have found that the prodrugs of nitrogen mustard coupled with an ROS trigger unit (e.g., an arylboronate or an arylboronic acid) can be triggered by H2O2 to release active anticancer drugs (effectors) [66]. Subsequently, Mokhir’s group showed that aminoferrocene-based prodrugs containing a phenylboronic acid pinacol ester can react with H2O2 to generate quinone methides as well as iron ions catalyzing the generation of hydroxyl radicals [67]. Prodrugs containing an oxidizable leaving group or a ferrocene moiety as the trigger units have also been reported [68–72]. These ROS-activated prodrugs demonstrated selective cytotoxicity towards cancer cells. In the remainder of this review, we discuss the present status and future prospects of ROS-activated anticancer prodrugs. We summarize how to use boron chemistry to develop novel ways for creating prodrugs that can be triggered by the high level of H2O2 found in cancer cells to release pharmacologically active species. Such agents have the potential to kill malignant cells while leaving healthy cells relatively untouched because they undergo tumor-specific activation. They also provide an excellent opportunity to evaluate the feasibility of the ROS-activated prodrug approach.
Boron-based ROS-activated prodrugs
Design of the trigger (ROS acceptor) unit for developing ROS-activated prodrugs
ROS-activated prodrugs should comprise two separate functional domains: an ROS-accepting moiety (‘trigger’) and an ‘effector’. The trigger unit should be joined with the effector by a ‘linker system’ so that the reaction of the trigger causes a large increase in the cytotoxic potency of the effector. The trigger units are expected to be ROS acceptors that can suppress the effector toxicity, while efficiently releasing the active species by reaction with ROS. Furthermore, they should be non-toxic to humans. The aryl boronic acids and their esters (1) can selectively react with H2O2 forming a boronate intermediate (2) that rapidly hydrolyzes to release the leaving groups resulting in the phenol (3) and borate ester or boric acid (Figure 1) [73]. Boronic acids and esters do not appear to have intrinsic toxicity issues, and the boric acid end product is considered non-toxic to humans [74]. Furthermore, the selective reactivity of boronic acids and esters towards H2O2 provides a chemospecific, biologically compatible reaction method for detecting endogenous H2O2 production. This approach allows for the development of highly selective fluorescent probes for imaging H2O2 in cells [75–78]. These properties coupled with their relative stability make aryl boronic acids and their esters good candidates as trigger units for the development of the efficient ROS-activated prodrugs. Recently, Cohen’s group has used boronic ester as the H2O2-sensitive trigger for developing hydrogen peroxide-activated MMPis [65]. These proinhibitors allow for efficient activation with H2O2 and demonstrated a dual mode of action in the prevention of reperfusion injury, by neutralizing ROS and generating an active MMPi.
Figure 1. Activation of boronates by hydrogen peroxide and release of quinone methide.
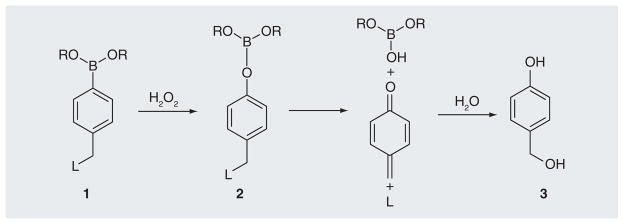
L: Leaving groups.
ROS-activated nitrogen mustard prodrugs
Initially, we used nitrogen mustard as an effector to develop an efficient ROS-responsive trigger unit and linker system. As the cytotoxicity of nitrogen mustards depend very much on the lone-pair electron at the mustard nitrogen, the prodrugs should contain an electron-withdrawing group linked to nitrogen mustards to decrease their electron density. A quaternary ammonium cation (linker A) is sufficient to mask the toxicity of the nitrogen mustard. Therefore, the nitrogen mustard (HN2) was coupled with an arylboronate generating ammonia salts 4a & b (Figure 2). An NMR study showed that 4a & b reacted with H2O2 to generate free HN2. Further studies with synthetic DNA indicated that 4a & b induced DNA interstrand cross-linking (ICL) and/or DNA alkylations upon H2O2 activation. However, in the absence of H2O2, no ICLs were observed [66]. These results proved that the toxicity of nitrogen mustard was efficiently masked in the prodrugs 4a & b, but can be released upon oxidative activation. The masked toxicity of the nitrogen mustard in 4a & b was caused by the positive charge developed on the nitrogen, which strongly decreased the electron density required for alkylation (Figure 2) [66]. The positive charge developed on the nitrogen also made the amino group a better leaving group. The tertiary amine HN2 is released upon the oxidation of the carbon–boron bond initiated by a nucleophilic attack by H2O2 (4a or 4a → 5a or 5b). Spontaneously, deboronation occurred leading to the formation of HN2 (5a & b → HN2). The presence of the lone-pair on HN2 facilitates the intramolecular displacement of the chloride with the amine nitrogen leading to the formation of a highly electrophilic aziridinium ring, which directly produced the DNA alkylation and ICLs.
Figure 2. The activation of prodrugs 4a and 4b by hydrogen peroxide and induced DNA damage.
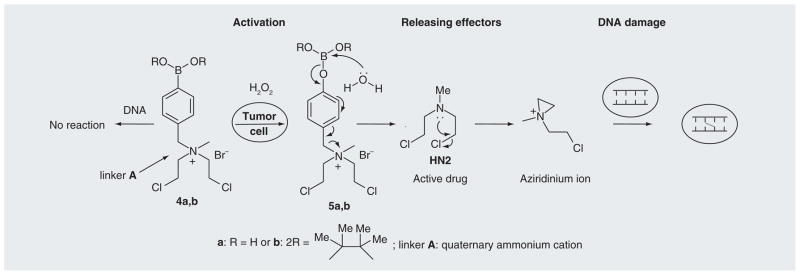
HN2: Nitrogen mustard.
Data taken from [66].
Compounds 4a & b showed approximately 90% inhibition toward SR cells (leukemia cells), 85% inhibition toward NCI-H460 (non-small-cell lung cancer cells), 66% inhibition toward CAKI-1 and 57% toward SN12C (renal cancer cells) (Figure 3a) [66]. However, normal lymphocytes were less affected (Figure 1B). Leukemia, lung cancer and renal cancer cells are believed to contain high levels of ROS [79–82]. It is highly likely that prodrugs 4a & b undergo oxidative activation in cancer cells with high levels of ROS.
Figure 3. Effect of compounds 4a and 4b on cancer cells and normal lymphocytes.

(A) Four human cancer cell lines (SR, NCI-H460, CAKI-1 and SN12C) were incubated with 10 μM of compounds 4a and 4b for 48 h. (B) Normal lymphocytes obtained from healthy donors (n = 3) are incubated without drug or 10 μM of 4a and 4b for 48 h. Cell death was measured by AV/PI staining which measures viable and non-viable cells (early and late apoptosis as well as necrosis).
AV/PI: Annexin V and propidium iodide.
Reproduced with permission from [66] © 2011 American Chemical Society.
Alternatively, a carboxyamide (B) was chosen as a linker unit [UWM Research Foundation, Inc., US Patent Application (2012)]. The electron-withdrawing property of the carbonyl group greatly reduced the toxicity of B. The release of the amine effector HN2 occurs upon the activation of B by H2O2 via an intermediate B-1 (Figure 4). The third strategy for the linker design is to use aniline boronate N-mustards (C) [UWM Research Foundation, Inc., US Patent Application (2012)]. The electron-withdrawing effect of the boronate group decreases the electron density of the benzene ring and makes the lone-pair of the mustard nitrogen delocalize to boron (C-1). The oxidation of the carbon-boron bond by H2O2, followed by transformation to a hydroxyl group, triggers increased electron release to the nitrogen of the mustard moiety (C-2 & C-3), greatly increasing its reactivity. The activity and selectivity of B and C were measured by crosslinking and alkylation of DNA [UWM Research Foundation, Inc., US Patent Application (2012)].
Figure 4.

The activation of prodrugs B and C by hydrogen peroxide.
ROS-activated quinone methide prodrugs
An alternative way to increase the potency of the ROS-activated prodrug is to identify a trigger unit that can couple with multiple potent effectors to maximize the cytotoxicity of pro-drugs upon activation. We have developed three prodrug building blocks that can couple with multiple effectors. Among these, compound 6 can be activated by H2O2 to release 2,5-bis(trimethylammonium)-benzyl-1,4-diol (7), which can generate biquinone methide under physiological conditions and lead to the efficient ICL formation and DNA alkylation (Figure 5) [83] [UWM Research Foundation, Inc., US Patent Application (2012)]. The oxidative activation of 6 by H2O2 produced an electron-rich aromatic ring, which facilitated the quinone methide (QM) formation and the release of the leaving group trimethylamine. Further investigation demonstrated that the electron-donating groups greatly increase QM formation [Cao S et al. Substituent effects on oxidation-induced quinone methides formation from their arylboronic ester precursors (2012). Submitted]. For example, the presence of the methoxy group in 9b led to 13.8% of the QM trapping product 10b when ethyl vinyl ether was used as a trapping agent, while no trapping product 10a was observed for the parent 9a (Figure 6). Therefore, a methoxy group can be introduced in 6 to increase the cross-linking yield. Compound 6 provides a novel building block for the development of H2O2-targeting anticancer prodrugs. Such a core structure is currently coupled with dual DNA or protein damaging agents (L) to produce a new generation of potent ROS-activated anticancer prodrugs. Such compounds will offer the major advantage that the cytotoxicity can be generated from the end product of the trigger unit – biquinone bimethide (effector 1) as well as the dual leaving groups (L: effector 2) (Figure 5). They are expected to be more potent than 4a & b for killing cancer cells. Numerous quinone-based anticancer drugs have been developed, such as mitomycin C and porfiromycin [84]. The leaving group contains bisalkylating or cross-linking agents that can damage DNA and/or protein. Therefore, an effective strategy has been developed to design and synthesize novel potent anticancer prodrugs that can be activated under tumor-specific conditions (high level of ROS) to release multiple active species by using compound 6 as a building block. Such a model will also be equally applicable to the development of prodrugs for the treatment of other diseases that are associated with H2O2.
Figure 5. Activation of quinone methide prodrugs.

ICL: Interstrand cross-link; L: NMe3, nitrogen mustard or other DNA-damaging functional groups (Effector 2).
Data taken from [83].
Figure 6.
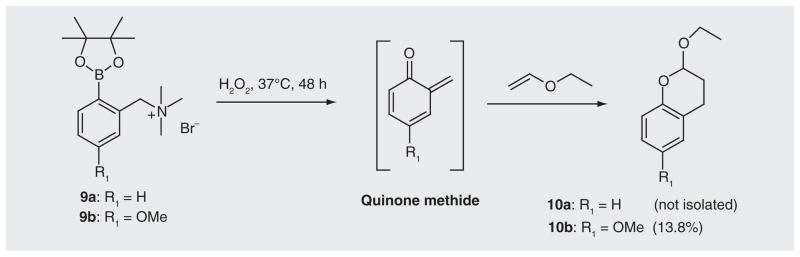
Quinone methide-trapping reactions of 9a and 9b in the presence of ethyl vinyl ether.
The arylboronate trigger unit has also been coupled with an aminoferrocene-generating, ferrocene-based prodrug 11 that can react with H2O2 to release two effectors, specifically, quinone methide and iron/ferrocenium ions (Figure 7) [67]. QMs alkylate glutathione, which inhibit the antioxidative system of the cells, while the iron ions induce catalytic generation of hydroxyl radicals. These prodrugs showed selective toxicity towards human promyelocytic leukemia and human glioblastoma-astrocytoma, but were non-toxic towards representative nonmalignant cells [67].
Figure 7. The activation of aminoferrocence-based prodrugs by H2O2 to release dual effectors.

ROS: Reactive oxygen species.
Data taken from [67].
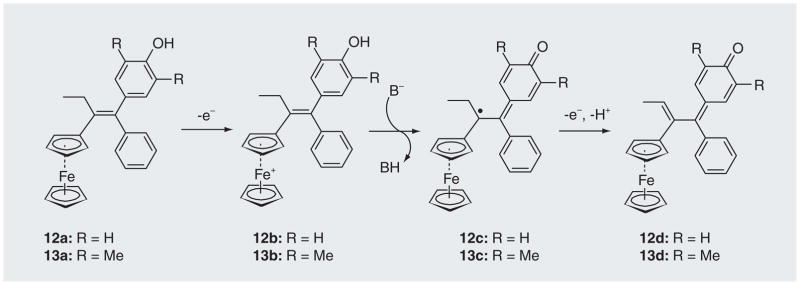
Other approaches used for triggering biologically active molecules via oxidative processes include the addition of a ferrocenyl moiety to polyaromatic phenols (an anti-estrogen drug skeleton) [69–72]. The groups of Amatore and Jaouen have developed several ferrocenyl phenols (e.g., 12a & 13a) that can undergo ferrocene-mediated oxidation to form cytotoxic species quinone methides [69–72]. The ferrocene triggered an intracellular oxidation of 12a & 13a to generate a potent cytotoxic quinone methide 12d or 13d (Figure 8). This process involves a base-promoted intramolecular electron transfer between the phenol and the ferrcenium cation (12b→12c & 13b→13c) [72]. Compounds 12a & 13a showed strong antiproliferative effect on hormone-independent breast cancer cells. These results indicated that the addition of a ferrocene to an anti-estrogen drug skeleton can induce cytotoxicity towards breast-cancer cells that are resistant to the common anti-estrogen drug [69,71].
Figure 8. Oxidation of ferrociphenols 12a or 13a to the quinone methides 12d or 13d.
Merino et al. presented another ROS-targeting strategy by designing DNA-modifying agents (e.g., 14) that contain an oxidizable leaving group (e.g., hydroquinone) and a nitrogen mustard moiety (Figure 9) [68]. Different from traditional nitrogen mustard, these agents contain a hydroquinone instead of a chlorine leaving group. The hydroquinone is a poor leaving group, which limits the reaction of 14 with biomolecules via a traditional mechanism of nitrogen mustard. However, such agents can be oxidized by hydrogen peroxide to form a nitrogen mustard fragment 15 and a strong electrophile 16. Both alkylate purine bases in DNA. These oxidatively activated DNA-modifying agents induced selective cytotoxicity towards renal cell carcinoma [68].
Figure 9. Oxidative activation of 14 forming DNA-modifying agents.
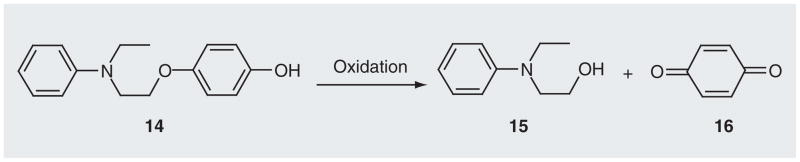
Data from [68].
Future perspective
Following the success of several ROS-activated prodrugs, there is renewed enthusiasm for further development of ROS-targeting prodrug approaches and the next decade promises significant advances in clinical impact. Future projects include defining the correlation between the inducible DNA damages and cellular cytotoxicity, the correlation between cellular toxicity and ROS level, ultimately developing produgs containing more potent effectors or coupling the efficient ROS-responsive trigger unit 6 with multiple potent effectors. With the availability of ROS-activated prodrugs, the combined technology has the potential to be developed for more efficient treatment of cancers.
Executive summary.
Tumor genetics & targeting
Deciphering the tumor genome facilitated the identification of tumor-specific targets, which allows the development of personalized gene-targeted cancer therapy.
Tumor genetics & therapeutics
While some genetic-based medicines, such as imatinib, herceptin and tarceva, were highly successful, this strategy is not ready for prime time since cancer genome research is at an early stage and driver mutations have not been identified.
Tumor biology & targeting
Compared with normal cells, tumor cells have higher levels of reactive oxygen species (ROS), which are caused by the active-energy metabolism associated with uncontrolled cell proliferation, malfunction of the mitochondrial respiration, telomere dysfunction and oncogenic stimulation. The increased oxidative stress in cancer cells could be exploited for developing cancer-targeting therapy.
Tumor biology & therapeutics
Cancer therapies are developed to target tumor-specific environments, such as tumor-hypoxia or the increased oxidative stress. Compounds are designed to increase ROS in cancer cells to the lethal level (pro-oxidant approach) or to abrogate ROS-signaling and suppress tumor growth (antioxidant approach). However, little or no therapeutic selectivity was achieved.
Tumor biology & rationale for prodrugs
Prodrug approaches are promising for tumor-specific destruction, such as hypoxia-targeting prodrugs. However, very few ROS-activated anticancer prodrugs are available, due to the obstacle of developing efficient and selective triggers that can be coupled with potent effectors via a linker, so that the reaction of the trigger with ROS causes a large increase in the cytotoxic potency of the effector.
Design of the trigger (ROS acceptor) unit for developing ROS-activated prodrugs
Arylboronates selectively react with hydrogen peroxide. They are used for developing fluorescent probes for imaging cellular H2O2 and for developing H2O2-activated matrix metalloproteinase inhibitors.
ROS-activated nitrogen mustard prodrugs
The first ROS-activated anticancer prodrugs have been developed by coupling nitrogen mustard with an arylboronate via an ammonia salt linker. These prodrugs can be triggered by H2O2 to release active drugs that can kill cancer cells, with little to no toxicity to normal cells. Other linkers, such as carboxyamides and aniline analogues are also effective to join arylboronates with nitrogen mustard in a way that the toxicity of the effector is masked in the prodrugs, while the active drugs are released upon reaction with H2O2.
ROS-activated quinone methide prodrugs
We have developed non-toxic prodrugs that can react with hydrogen peroxide to release biquinone methides, directly cross-linking and/or alkylating DNA. These agents can also crosslink/alkylate proteins as an important non-DNA mechanism of toxicity. The transformation of an electron-withdrawing boronate group to an electron-donating hydroxyl group greatly facilitates the formation of quinone methide. The potency of the quinone methide prodrugs can be further increased by introducing an electron-donating group on the core structure and/or coupling the core structure with dual DNA-damaging or protein-damaging functional groups.
Non boron-based strategies for ROS-activated prodrugs
Several ferrociphenol anticancer drugs have been developed. These compounds can be activated via ferrocene-mediated intramolecular oxidation to release active drugs, such as quinone methide derivatives. They showed a strong antiproliferative effect on hormone-independent breast cancer cells.
Key Terms
- Oxidative stress
Oxidative stress results from an imbalance between the production and detoxification of reactive oxygen species. The persistent oxidative stress can lead to cell damage through the oxidation of DNA, proteins and lipids. On the other hand, the intrinsic oxidative stress in cancer cells can be used for developing cancer-targeted therapies
- Reactive oxygen species
A variety of chemically reactive molecules and free radicals derived from molecular oxygen, such as H2O2, superoxide anion (O2−), hydroxyl radical (HO•), and hypochlorite ion (OCl−). The increased amount of reactive oxygen species in cancer cells lead to the increased intrinsic oxidative stress
- ROS-activated anticancer prodrugs
Compounds which are inactive in themselves but can be converted to active anticancer drugs upon activation by reactive oxygen species (e.g., H2O2). ROS-activated anticancer prodrugs can undergo tumor-specific activation, therefore, increasing the selectivity towards cancer cells
- Tumor-specific activation
The non-toxic prodrugs are only activated in the cancer cells through oxidation or reduction to release toxic species, while being kept intact in the normal cell environment
Footnotes
For reprint orders, please contact reprints@future-science.com
Financial & competing interests disclosure
The authors are grateful for financial support of this research from the NCI (1R15CA152914-01), UWM start-up funds and UWM research growth initiative grant (XP) (101X234). This research was supported in part by the NCI (CA136411), Lymphoma SPORE and (CA81534) CLL Consortium PO1 (VG). Additionally, the authors gratefully acknowledge funding by UWM research foundation for patent applications relating to technologies and compounds described in this article. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW. The sequence of human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Collins FS, Lander ES, Rogers J, et al. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 4.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 5▪.Mardis ER. A decade’s perspective on DNA sequencing technology. Nature. 2011;470:198–203. doi: 10.1038/nature09796. Paper published at the 10-year anniversary of the human genome being sequenced. It describes in detail how far have we come and how far we need to go. [DOI] [PubMed] [Google Scholar]
- 6.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 7.Chin L, Hahn WC, Getz G, Meyerson M. Making sense of cancer genomic data. Genes Dev. 2011;25:534–555. doi: 10.1101/gad.2017311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kenefick K. Small-molecule kinase inhibitors: from lab bench to clinic. Cell Notes. 2006;16:26–29. [Google Scholar]
- 9.Kawanishi S, Hiraku Y, Pinlaor S, Ma N. Oxidative and nitrative damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol Chem. 2006;387(4):365–372. doi: 10.1515/BC.2006.049. [DOI] [PubMed] [Google Scholar]
- 10▪.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. Evidence of the increased level of H2O2 in human cancer cells. [PubMed] [Google Scholar]
- 11▪.Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother Pharmacol. 2004;53:209–219. doi: 10.1007/s00280-003-0726-5. Publication of amplifying reactive oxygen species (ROS) to increase therapeutic selectivity. [DOI] [PubMed] [Google Scholar]
- 12.Toyokuni S, Okamoto K, Yodoi J, Hiai H. Persistant oxidative stress in cancer. FEBS Lett. 1995;358:1–3. doi: 10.1016/0014-5793(94)01368-b. [DOI] [PubMed] [Google Scholar]
- 13.Arnold RS, Shi J, Murad E, et al. Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc Natl Acad Sci USA. 2001;98:5550–5555. doi: 10.1073/pnas.101505898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishikawa K, Takenaga K, Akimoto M, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–664. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 15.King MS, Sharpley MS, Hirst J. Reduction of hydrophilic ubiquinones by the flavin in mitochondrial NADH: ubiquinone oxidoreductase (complex I) and production of reactive oxygen species. Biochemistry. 2009;48:2053–2062. doi: 10.1021/bi802282h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsang WP, Chau SP, Kong SK, Kwok TT. Reactive oxygen species mediate doxorubicin induced p53-independent apoptosis. Life Sci. 2003;73:2047–2058. doi: 10.1016/s0024-3205(03)00566-6. [DOI] [PubMed] [Google Scholar]
- 17.Vafa O, Wade M, Kern S, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–1044. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 18.Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–65. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Afanas’ev I. Reactive oxygen species signaling in cancer: comparison with aging. Aging Dis. 2011;2:219–230. [PMC free article] [PubMed] [Google Scholar]
- 20.Elchuri S, Oberley TD, Qi WB, et al. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 21.Hole PS, Darley RL, Tonks A. Do reactive oxygen species play a role in myeloid leukemias? Blood. 2011;117:5816–5826. doi: 10.1182/blood-2011-01-326025. [DOI] [PubMed] [Google Scholar]
- 22.Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407:390–395. doi: 10.1038/35030140. [DOI] [PubMed] [Google Scholar]
- 23▪.Trachootham D, Zhou Y, Zhang H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. Demonstrates that plant product acting as antioxidant is selective to tumor cells. [DOI] [PubMed] [Google Scholar]
- 24.Young TW, Mei FC, Yang G, Thompson-Lanza JA, Liu J, Cheng X. Activation of antioxidant pathways in ras-mediated oncogenic transformation of human surface ovarian epithelial cells revealed by functional proteomics and mass spectrometry. Cancer Res. 2004;64:4577–4584. doi: 10.1158/0008-5472.CAN-04-0222. [DOI] [PubMed] [Google Scholar]
- 25.Laurent A, Nicco C, Chéreau C, et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005;65:948–956. [PubMed] [Google Scholar]
- 26.Nicco C, Laurent A, Chereau C, Weill B, Batteux F. Differential modulation of normal and tumor cell proliferation by reactive oxygen species. Biomed Pharmacother. 2005;59:169–74. doi: 10.1016/j.biopha.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 27.Choi J, Liu RM, Forman HJ. Adaptation to oxidative stress: quinone-mediated protection of signaling in rat lung epithelial L2 cells. Biochem Pharmacol. 1997;53:987–993. doi: 10.1016/s0006-2952(96)00867-2. [DOI] [PubMed] [Google Scholar]
- 28.Kim GJ, Chandrasekaran K, Morgan WF. Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: a review. Mutagenesis. 2006;21:361–367. doi: 10.1093/mutage/gel048. [DOI] [PubMed] [Google Scholar]
- 29.Sheridan J, Wang LM, Tosetto M, et al. Nuclear oxidative damage correlates with poor survival in colorectal cancer. Br J Cancer. 2009;100:381–388. doi: 10.1038/sj.bjc.6604821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mates JM, Sanchez-Jimenez FM. Role of reactive oxygen species in apoptosis: implications for cancer therapy. Int J Biochem Cell Biol. 2000;32:157–170. doi: 10.1016/s1357-2725(99)00088-6. [DOI] [PubMed] [Google Scholar]
- 31.Pervaiz S, Clement MV. Superoxide anion: oncogenic reactive oxygen species? Int J Biochem Cell Biol. 2007;39:1297–1304. doi: 10.1016/j.biocel.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 32.Kong D, Park EJ, Stephen AG, et al. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005;65:9047–9055. doi: 10.1158/0008-5472.CAN-05-1235. [DOI] [PubMed] [Google Scholar]
- 33.Wang R, Zhou S, Li S. Cancer therapeutic agents targeting hypoxia-inducible factor-1. Curr Med Chem. 2011;18:3168–3189. doi: 10.2174/092986711796391606. [DOI] [PubMed] [Google Scholar]
- 34.Tsai HC, Baylin SB. Cancer epigenetics: linking basic biology to clinical medicine. Cell Res. 2011;21:502–517. doi: 10.1038/cr.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Hagan HM, Wang W, Sen S, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20:606–619. doi: 10.1016/j.ccr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell. 2011;8:399–411. doi: 10.1016/j.stem.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vlaminck B, Toffoli S, Ghislain B, Demazy C, Raes M, Michiels C. Dual effect of echinomycin on hypoxia-inducible factor-1 activity under normoxic and hypoxic conditions. FEBS J. 2007;274:5533–5542. doi: 10.1111/j.1742-4658.2007.06072.x. [DOI] [PubMed] [Google Scholar]
- 38.Muss HB, Blessing JA, Hanjani P, Malfetano JH, Kemp GM, Webster K. Echinomycin (NSC 526417) in recurrent and metastatic nonsquamous cell carcinoma of the cervix. A Phase II trial of the gynecologic oncology group. Am J Clin Oncol. 1992;15:363–364. doi: 10.1097/00000421-199208000-00019. [DOI] [PubMed] [Google Scholar]
- 39.Muss HB, Blessing JA, DuBeshter B. Echinomycin in recurrent and metastatic endometrial carcinoma. A Phase II trial of the Gynecologic Oncology Group. Am J Clin Oncol. 1993;16:492–493. doi: 10.1097/00000421-199312000-00006. [DOI] [PubMed] [Google Scholar]
- 40.Brown JM, Wilson WR. Exploiting tumor hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 41.Benito J, Shi Y, Szymanska B, et al. Pronounced hypoxia in models of murine and human leukemia: high efficacy of hypoxia-activated prodrug PR-104. PloS ONE. 2011;6:e23108. doi: 10.1371/journal.pone.0023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patterson AV, Ferry DM, Edmunds SJ, et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin Cancer Res. 2007;13:3922–3932. doi: 10.1158/1078-0432.CCR-07-0478. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther. 2008;7:1875–1884. doi: 10.4161/cbt.7.12.7067. [DOI] [PubMed] [Google Scholar]
- 44.Alexandre J, Nicco C, Chéreau C. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J Natl Cancer Inst. 2006;98:236–244. doi: 10.1093/jnci/djj049. [DOI] [PubMed] [Google Scholar]
- 45.Pelicano H, Feng L, Zhou Y, et al. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human cells by a reactive oxygen species-mediated mechanism. J Biol Chem. 2003;278:37832–37839. doi: 10.1074/jbc.M301546200. [DOI] [PubMed] [Google Scholar]
- 46.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic appoach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 47▪.Pelicano H, Carney D, Huang P. ROS stress in cancer cells and the therapeutic implications. Drug Resist Update. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. Summarizes the therapeutic implications of ROS stress in cancer cells, and strategies that take advantage of the increased ROS in cancer cells to enhance therapeutic activity and selectivity. [DOI] [PubMed] [Google Scholar]
- 48.Lopez-Lazaro M. Dual role of hydrogen peroxide in cancer: possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007;252:1–8. doi: 10.1016/j.canlet.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 49.Fang J, Seki T, Maeda H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv Drug Deliv Rev. 2009;61:290–302. doi: 10.1016/j.addr.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 50.Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10:175–176. doi: 10.1016/j.ccr.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 51.Jing Y, Dai J, Chalmers-Redman RM, Tatton WG, Waxman S. Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood. 1999;94:2102–2111. [PubMed] [Google Scholar]
- 52.Miyajima A, Nakashima J, Yoshioka K, Tachibana M, Tazaki H, Murai M. Role of reactive oxygen species in cis-diclorodiammineplatinum-induced cytotoxicity on bladder cancer cells. Br J Cancer. 1997;76:206–210. doi: 10.1038/bjc.1997.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anastasious D, Poulogiannis G, Asara JM, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee SH, Oe T, Blair IA. Vitamin C-induced decomposition of lipid hydroperoxides to endogenous genotoxins. Science. 2001;292:2083–2086. doi: 10.1126/science.1059501. [DOI] [PubMed] [Google Scholar]
- 55.Lee KW, Lee HJ, Kang KS, Lee CY. Preventive effects of vitamin C on carcinogenesis. Lancet. 2002;359:172. doi: 10.1016/S0140-6736(02)07358-0. [DOI] [PubMed] [Google Scholar]
- 56.Duvoix A, Blasius R, Delhalle S, et al. Chemopreventive and therapeutic effects of curcumin. Cancer Lett. 2005;223:181–190. doi: 10.1016/j.canlet.2004.09.041. [DOI] [PubMed] [Google Scholar]
- 57.Halliwell B. The antioxidant paradox. Lancet. 2000;355:1179–1180. doi: 10.1016/S0140-6736(00)02075-4. [DOI] [PubMed] [Google Scholar]
- 58.Alexandre J, Batteux F, Nicco C, et al. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. Int J Cancer. 2006;119:41–48. doi: 10.1002/ijc.21685. [DOI] [PubMed] [Google Scholar]
- 59.Llobet D, Eritja N, Encinas M, et al. Antioxidants block proteasome inhibitor function in endometrial carcinoma cells. Anticancer Drugs. 2008;19:115–124. doi: 10.1097/CAD.0b013e3282f24031. [DOI] [PubMed] [Google Scholar]
- 60.Doroshow JH. Redox modulation of chemotherapy-induced tumor cell killing and normal tissue toxicity. J Natl Cancer Inst. 2006;98:223–225. doi: 10.1093/jnci/djj065. [DOI] [PubMed] [Google Scholar]
- 61.Denny WA. Prodrugs strategies in cancer therapy. Eur J Med Chem. 2001;36:577–595. doi: 10.1016/s0223-5234(01)01253-3. [DOI] [PubMed] [Google Scholar]
- 62.Raj L, Ide T, Gurkar AU, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63.Doering M, Ba LA, Lilienthal N, et al. Synthesis and selective anticancer activity of organochalcogen based redox catalysts. J Med Chem. 2010;53:6954–6963. doi: 10.1021/jm100576z. [DOI] [PubMed] [Google Scholar]
- 64.Bair JS, Palchaudhuri R, Hergenrother PJ. Chemistry and biology of deoxynyboquinone, a potent inducer of cancer cell death. J Am Chem Soc. 2010;132:5469–5478. doi: 10.1021/ja100610m. [DOI] [PubMed] [Google Scholar]
- 65▪.Major Jourden JL, Cohen SM. Hydrogen peroxide activated matrix metalloproteinase inhibitors: a prodrug approach. Angew Chem Int Ed. 2010;49:6795–6797. doi: 10.1002/anie.201003819. Publication of the first H2O2-activated prodrug by using arylboronates as the trigger units. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66▪▪.Kuang Y, Balakrishnan K, Gandhi V, Peng X. Hydrogen peroxide inducible DNA cross-linking agents: targeted anticancer prodrugs. J Am Chem Soc. 2011;133:19278–19231. doi: 10.1021/ja2073824. First evidence that ROS-activated anticancer prodrugs showed selective toxicity towards cancer cells. Both chemical synthesis and biological investigation are described. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hagen H, Marzenell P, Jentzsch E, et al. Aminoferrocene-based prodrugs activated by reactive oxygen species. J Med Chem. 2012;55(2):924–934. doi: 10.1021/jm2014937. [DOI] [PubMed] [Google Scholar]
- 68.Li G, Bell T, Merino EJ. Oxidatively activated DNA-modifying agents for selective cytotoxicity. Chem Med Chem. 2011;6:869–875. doi: 10.1002/cmdc.201100014. [DOI] [PubMed] [Google Scholar]
- 69▪.Hillard E, Vessieres A, Thouin L, Jaouen G, Amatore C. Ferrocene-mediated proton-coupled electron transfer in a series of ferrocifen-typed breast cancer drug candidates. Angew Chem Int Ed. 2006;45:285–290. doi: 10.1002/anie.200502925. Publication of anticancer prodrugs activated by ferrocene-mediated oxidation. [DOI] [PubMed] [Google Scholar]
- 70.Vessieres A, Top S, Pigeon P, et al. Modification of the estrogenic properties of diphenols by the incorporation of ferrocene. Generation of antiproliferative effects in vitro. J Med Chem. 2005;48:3937–3940. doi: 10.1021/jm050251o. [DOI] [PubMed] [Google Scholar]
- 71.Hamels D, Dansette PM, Hillard EA, et al. Ferrocenyl quinone methides as strong antipfoliferative agents: formation by metabolic and chemical oxidation of ferrocenyl phenols. Angew Chem Int Ed. 2009;48:9124–9126. doi: 10.1002/anie.200903768. [DOI] [PubMed] [Google Scholar]
- 72.Messina P, Labbe E, Buriez O, et al. Deciphering the activation sequence of ferrociphenol anticancer drug candidates. Chem Eur J. 2012;18(21):6581–6587. doi: 10.1002/chem.201103378. [DOI] [PubMed] [Google Scholar]
- 73.Kuivila HG, Armour AG. Electrophilic displacement reactions. IX Effects of substituents on rates of reactions between hydrogen peroxide and benzeneboronic acid. J Am Chem Soc. 1957;79:5659–5662. [Google Scholar]
- 74.Yang WQ, Gao X, Wang BH. In: Boronic Acids. Hall DG, editor. Wiley-VCH; Weinheim, Germany: 2005. pp. 481–512. [Google Scholar]
- 75.Dickinson BC, Chang CJ. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J Am Chem Soc. 2008;130:9638–9639. doi: 10.1021/ja802355u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76▪.Miller EW, Albers AE, Pralle A, Isacoff EY, Chang CJ. Boronate-based fluorescent probes for imaging cellular hydrogen peroxide. J Am Chem Soc. 2005;127:16652–16659. doi: 10.1021/ja054474f. Describes the aryboronates used as trigger units in fluorescent probes for selectively imaging cellular H2O2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miller EW, Tulyathan O, Isacoff EY, Chang CJ. Moleaular imaging of hydrogen peroxide produced for cell signalling. Nat Chem Biol. 2007;3:263–267. doi: 10.1038/nchembio871. [DOI] [PubMed] [Google Scholar]
- 78.Srikun D, Miller EW, Domaille DW, Chang CJ. An ICT-based approach to ratiometric fluorescence imaging of hydrogen peroxide produced in living cells. J Am Chem Soc. 2008;130:4596–4597. doi: 10.1021/ja711480f. [DOI] [PubMed] [Google Scholar]
- 79.Kaynar H, Meral M, Turhanb H, Kelesc M, Celikb G, Akcayb F. Glutathione peroxidase, glutathione-S-transferase, catalase, xanthine oxidase, Cu–Zn superoxide dismutase activities, total glutathione, nitric oxide, and malondialdehyde levels in erythrocytes of patients with small cell and non-small cell lung cancer. Cancer Lett. 2005;227:133–139. doi: 10.1016/j.canlet.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 80.Battisti V, Maders LDK, Bagatini MD, et al. Measurement of oxidative stress and antioxidant status in acute lymphoblastic leukemia patients. Clin Biochem. 2008;41:511–518. doi: 10.1016/j.clinbiochem.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 81.Ganesamoni R, Bhattacharyya S, Kumar S, et al. Status of oxidative stress in patients with renal cell carcinoma. J Urol. 2012;187:1172–1176. doi: 10.1016/j.juro.2011.11.105. [DOI] [PubMed] [Google Scholar]
- 82.Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003;101:4098–4104. doi: 10.1182/blood-2002-08-2512. [DOI] [PubMed] [Google Scholar]
- 83▪▪.Cao S, Wang Y, Peng X. ROS-inducible DNA cross-linking agent as a new anticancer prodrug building block. Chem Eur J. 2012;18:3850–3854. doi: 10.1002/chem.201200075. First publication of ROS-activated prodrugs that can release DNA cross-linking agent quinone methides by reacting with H2O2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Seddon B, Kelland LR, Workman P. Bioreductive prodrugs for cancer therapy. Methods Mol Med. 2004;90:515–542. doi: 10.1385/1-59259-429-8:515. [DOI] [PubMed] [Google Scholar]