

Abstract
Cruzain is a member of the papain/cathepsin-L family of cysteine proteases, and the major cysteine protease of the protozoan Trypanosoma cruzi, the causative agent of Chagas’ disease. We report an auto-induction methodology that provides soluble-cruzain at high yields (> 30 mg per liter in minimal media). These increased yields provide sufficient quantities of active enzyme for use in NMR-based ligand mapping. Using CD and NMR spectroscopy, we also examined the solution-state structural dynamics of the enzyme in complex with a covalently bound vinyl sulfone inhibitor (K777). We report the backbone amide and side chain carbon chemical shift assignments of cruzain in complex with K777. These resonance assignments were used to identify and map residues located in the substrate binding pocket, including the catalytic Cys25 and His162. Selective 15N-Cys, 15N-His, and 13C-Met labeling was performed to quickly assess cruzain-ligand interactions for a set of eight low molecular weight compounds exhibiting micromolar binding or inhibition. Chemical shift perturbation mapping verifies that six of the eight compounds bind to cruzain at the active site. Three different binding modes were delineated for the compounds, namely covalent, non-covalent, and non-interacting. These results provide examples of how NMR spectroscopy can be used to screen compounds for fast evaluation of enzyme-inhibitor interactions in order to facilitate lead compound identification and subsequent structural studies.
Chagas’ disease is the leading cause of heart disease in Latin America, with an estimated 8-11 million people infected and another 100 million at risk.1 An estimated 50,000 deaths per year are attributable to cardiomyopathy and gastrointestinal damage caused by chronic Chagas’ disease.2,3 This neglected tropical disease is caused by the protozoan Trypanosoma cruzi, and is transmitted by the introduction of fecal matter of reduviid bugs into the blood stream of mammalian hosts after being bitten by the insect. Although normally confined to Latin America, cases of Chagas’ disease are appearing in Europe and the southern United States.1,4 The only currently approved treatments for Chagas’ disease, nifurtimox and benznidazole, have numerous deleterious side effects, and are only available in North America in limited quantities from the US Centers for Disease Control. Both drugs are generally prescribed during the early acute stages of the disease, and are less effective for treatment of the chronic condition.5
A promising target for potential therapeutics against Chagas’ disease is the major cysteine protease expressed by T. cruzi, cruzain. Cruzain is a member of the papain superfamily of cysteine proteases, and is most sequentially and structurally homologous to cathepsin L. In the case of Chagas’ disease, cruzain is present in all life stages of the trypanosome and is implicated in cellular entry and digestion of immunoglobins.1 The active site of cruzain contains two catalytic residues (Cys25 and His162), with possibly a third (Asn182) residue contributing to proteolysis (sequential residue numbering,6 instead of residue numbering matching papain,7 is used for cruzain throughout this report). A fourth residue (Gln19) serves to stabilize the oxyanion.8 Similar to many other cysteine protease family members, cruzain is initially expressed as an inactive zymogen, which undergoes a pH, temperature, and concentration dependent self-activation, transforming into the mature, catalytically active protease.8,9
One lead compound that targets the mature form of cruzain and is currently in pre-clinical trials is the vinyl sulfone, K777 (Table 1, previously designated K11777).10 K777 inhibits cruzain by irreversibly binding to the thiol group of the catalytic Cys25. K777 is reported to cure T. cruzi-infected mice, and has shown efficacy against multiple strains of nifurtimox- and benznidazole-resistant T. cruzi, validating cruzain as a therapeutic target for Chagas’ disease.11 However, the limited potency of K777 has generated interest in identifying new chemical scaffolds that are more effective and are non-covalent competitive inhibitors of the enzyme.
Table 1.
Cruzain inhibitors used for NMR titration experiments.
| Compound Structure |
aZINC/SMDC Number |
Inhibitor Class |
Inhibitory/Binding Constant against Cruzain (μM) |
Ref. |
|---|---|---|---|---|
|
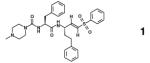
K777 (55865535) |
covalent, irreversible |
Kd = 1.9 | 6 |
|
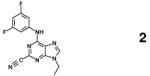
36379291 | covalent, reversible |
IC50 = 0.01 | 24*** (cmpd 32) |

|
746694 | non- covalent; intermediate of cmpd 2 |
n/a |
24
(cmpd 32 intermediate) |
|
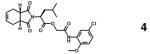
8691187 | non- covalent |
Ki = 2.0 IC50 = 1.0 |
13 (cmpd 4) |

|
3363859 | non- covalent |
Ki = 6.0 IC50 = 7.0 |
13 (cmpd 5) |
|
|
943080 | non- covalent |
Ki = 2.0 | 13 (cmpd 27) |

|
2236859 | non- covalent |
Ki = 2.0 | 13 (cmpd 29) |

|
43120874 | non- interacting/ aggregator |
n/a |
Ellman personal comm.; 12 |
ZINC database;46 SMDC = UCSF Small Molecule Discovery Center.
Previous structure-activity relationship (SAR) studies focusing on cruzain-inhibitor complexes have historically relied on x-ray crystallography. However, due to concentration-dependent self-proteolysis, no apo structures of active cruzain are currently available. Although high-throughput screening efforts have identified potential lead compounds, not all cruzain-inhibitor complexes yielded well-behaved crystals.12,13 In addition, the previous expression protocol14,15 involved production of cruzain via inclusion bodies and the subsequent unfolding and refolding of the protease, severely limiting overall yields. With these factors in mind, we: (i) developed an expression and purification protocol that provided sufficient quantities of cruzain for NMR studies; (ii) used NMR-based methods to quickly verify and map binding modes of newly discovered inhibitors, especially those for which there is no available crystallographic data; and (iii) initiated solution-state biophysical studies to gain further insights into apo and inhibited cruzain structure-function relationships. Importantly, cruzain serves as a model for the cysteine cathepsin proteases, a large family of important enzymes.
This report represents the first NMR study to examine the solution behavior of an active cathepsin L-like cysteine protease. Using auto-induction methods,16 we were able to increase the overall yields of activated, mature cruzain by greater than 20 fold over the original inclusion body preparation.15 This new optimized protocol also enabled the expression of isotopically labeled samples for NMR studies, allowing us to obtain backbone and side chain resonance assignments of a covalently inhibited form of cruzain. These assignments, along with selective 15N-Cys, 15N-His, and 13C-Met cruzain labeling, were critical in mapping and characterizing inhibitor binding modes of eight small-molecule inhibitors, including K777. The overall results described herein provide a methodology for rapidly enhancing our understanding of structure-function relationships within the cysteine cathepsin family.
Materials and Methods
Design of the His6-tagged Zymogen
The gene sequence encoding the C-terminal truncated procruzain ( Δc; GenBank accession code M84342.1)14,15 was used as the template for the construct described in this study. The forward primer was designed to encode an NdeI restriction site, followed by a His6 sequence, a BamHI restriction site, and an N-terminal pro-cruzain sequence starting at Cys(-104). The reverse primer encodes the cruzain-Δc terminal residue Gly215, followed by stop codons and a HindIII restriction site. Primer sequences (Integrated DNA Technologies, Inc.) are as follows: forward, 5′-GACGCCATATGCATCACCATCACCATCACGGATCCTGCCTCGTCCCCGCGGCGACG-3′; reverse, 5′-CGTGCAAGCTTCTATTAACCGACCACCGCGGAGCT-3′. The resulting oligonucleotide encoding the His6-tagged procruzain sequence was ligated into a pET21a vector (Novagen/EMD), then transformed into ArcticExpress (DE3)RIL Competent Cells (Agilent). The genes were verified through DNA sequencing. The resulting protein sequence is displayed in Figure 1a.
Figure 1.

Domain diagram of cruzain, activation and inhibition of its zymogen form, and 15N-1H HSQC spectrum (a) The domain diagram and one-letter sequence of the zymogen (procruzain). The active catalytic domain (cruzain) used in this study is highlighted as boldface. Residues of an engineered N-terminal His6-tag (underlined) and the truncated pro-region are numbered −112 to −1. Residue numbers of cruzain are numbered 1 to 215. (b) Incubation at 37 °C and addition of DTT to PMSF- and MMTS-inhibited zymogen (~ 37 kDa) initiates auto-proteolysis of the N-terminal pro-region (~ 14 kDa) from the catalytically active domain (~ 23 kDa), as monitored by SDS-PAGE. (c) Addition of the vinyl sulfone K777 to procruzain impedes, but does not completely inhibit, self-activation of the zymogen. (d) The 15N-1H HSQC spectrum of cruzain inhibited with K777 indicates a well-folded and stable enzyme structure.
Protein Expression
Starter cultures used to express His6-tagged procruzain consisted of 5 mL Luria broth containing carbenicillin (100 Δg/mL) and gentamycin (20 Δg/mL). After > 15 hrs shaking at 37 °C, the overnight LB starter culture was added directly to 2 × 1 L auto-induction minimal media contained in baffled 4 L shaker flasks. Modified versions of auto-induction media recipes N-5052 and PA-505216 were used for uniform 15N- and selective 15N or 13C-labeling, respectively. In the case of uniform 15N-labeling, 2.7 g 15NH4Cl (Cambridge Isotope Laboratories or Sigma-Aldrich) per liter of media was added. In the case of selective labeling, three separate protease samples were produced by adding one of the following to one liter of auto-induction minimal media: (i) 100 mg 15N-Cys (Cambridge Isotope Laboratories), (ii) 100 mg 15N3-His (Ajinomoto), or (iii) 250 mg 13C-methyl Met (Cambridge Isotope Laboratories). For uniform 13C/15N/2H-labeling, a modified version of auto-induction media recipe C-750501 was used.16 Here, 1 L uniformly 13C/15N/2H-labeled rich media (E. coli-OD2 CDN, Silantes GmbH) was diluted with 1 L D2O (Cambridge Isotope Laboratories) and split into 2 × 1 L aliquots. All auto-induction minimal media solutions contained carbenicillin (100 Δg/mL) and gentamycin (20 Δg/mL) as antibiotics. Further specific details regarding the modifications to the standard auto-induction recipes are reported in the Supporting Information. After shaking at 37 °C for approximately 5 hrs (until OD600 > 0.5), the temperature was reduced to 20 °C, and the culture was allowed to shake for an additional 72 hrs. The cultures were then centrifuged, and the cell pellets were stored at −20 °C until ready for lysis. Pre-conditioning of LB starter cultures into small volumes (50 mL) of auto-induction minimal media overnight was tried and demonstrated to be ineffective, and is therefore not recommended.
Protein Purification and Zymogen Activation
Frozen cell pellets were thawed, resuspended in 50 mL lysis buffer per L expression media, and lysed via sonication: 20 sec pulse followed by a 120 sec recovery time for 2 min total pulsing time. The lysis buffer consisted of 50 mM Tris (pH 10), 300 mM NaCl, 10 mM imidazole, 1 mM CaCl2, 1 mM MgSO4, 1 ΔM of DNase I (Roche), 1 mM phenylmethanesulfonyl fluoride (PMSF), and 2 mM methyl methanethiosulfonate (MMTS). PMSF and MMTS are used as covalent cysteine protease inhibitors, and are reversible by addition of DTT. The resulting lysate was centrifuged, and the supernatant was passed through a 0.22 Δm filter prior to FPLC purification. The initial purification was performed at 4 °C using two 5 mL HisTrapFF Crude nickel columns (GE Healthcare LifeSciences) attached in series. The following buffers were used: wash/binding, 50 mM Tris (pH 10), 300 mM NaCl, 10 mM imidazole; elution, 50 mM Tris (pH 10), 300 mM NaCl, 500 mM imidazole. Elution occurs with 30% elution buffer. MMTS (5 mM final concentration) was added to fractions containing His6-tagged procruzain upon elution from the column.
Major fractions were combined and dialyzed at 4 °C in activation buffer (50 mM acetate pH 5.0, 100 mM NaCl, 0.1 mM EDTA) containing no reducing agents. After overnight dialysis, the resulting cloudy solution was transferred to 50 mL conical centrifuge tubes. Auto-proteolysis of the pro-region (~ 14 kDa) from the zymogen (~ 37 kDa) was initiated by adding DTT (1 mM final concentration) to the solution and incubating in a 37 °C water bath. Transformation of the inactive zymogen into the catalytically active domain (cruzain, ~ 23 kDa) was monitored by removing 50 ΔL aliquots of the solution at selected time points. These samples were analyzed either through SDS-PAGE under denaturing conditions (Figure 1b), or via a fluorescence-based activity assay using the peptide substrate Z-Phe-Arg-AMC (Biotium, Inc.).14 Total reaction time to process procruzain into active cruzain was generally 20 to 40 minutes, as indicated by the solution becoming clear. Longer activation times of up to 2 hrs were required to completely proteolyze the smaller pro-domain fragments. To prevent further degradation of the mature cruzain, the reaction was quenched by placing the sample tubes on ice and adding MMTS to 1 mM final concentration.
The solution was next buffer exchanged to 10X PBS (pH 5.0), and a second round of FPLC purification was performed at 4 °C using a preparative-scale Superdex75 (26/60) size exclusion column (GE Healthcare LifeSciences). The FPLC buffers used in this purification round did not contain reducing agents. Unless otherwise specified below, MMTS (1 mM final concentration) was added to each of the major fractions immediately upon elution from the column. The fractions were then collected, flash frozen, and stored at −80 °C until ready for use.
Estimated final enzyme concentrations were determined using a NanoDrop 2000c UV spectrophotometer (Thermo Scientific) with the following extinction coefficients: His6-GS-procruzain, ε280 = 68910 M−1 cm−1 (oxidized Cys), 68410 M−1 cm−1 (reduced Cys); cruzain, ε280 = 60430 M−1 cm−1 (oxidized Cys), 59930 M−1 cm−1 (reduced Cys). Final estimated yields of the inactive zymogen procruzain and the purified active cruzain are reported in the results section below.
NMR Spectroscopy
All samples were buffer exchanged from 10X PBS (pH 5.0) to NMR buffer prior to data acquisition. The standard NMR buffer consisted of 20 mM potassium phosphate (pH 5.0), no salt, 1 mM DTT, and 10% (v/v) 2H2O. The apo or inhibited states of the active site Cys25 and protease concentrations of the NMR samples varied depending on the experiment performed and are indicated below. All data was acquired at 27 °C on Bruker Avance 500 MHz and 800 MHz instruments equipped with triple-resonance cryoprobes. External 13C, 15N, and 1H chemical shift referencing was performed as previously reported.17 All spectral data were processed with NMRpipe18 and analyzed with Sparky.19
NMR Resonance Assignments
To prevent further self-proteolysis of the uniformly 13C/15N/2H-labeled apo cruzain, major fractions eluting from the size exclusion purification step were immediately inhibited with greater than stoichiometric ratios of the vinyl sulfone K777. Standard K777 stocks consisted of 10 mM unlabeled inhibitor dissolved in d6-DMSO (Cambridge Isotope Laboratories). NMR samples consisted of ~ 0.5 mM uniformly 13C/15N/2H-labeled cruzain-K777 complex in 0.45 – 0.50 mL NMR buffer. The suite of three-dimensional triple-resonance experiments used to acquire backbone amide and side chain carbon resonance assignments consisted of HNCACB, HN(CO)CACB, HNCO, and CC(CO)NH.20 A 3D-15N/1H-NOESY-HSQC acquired on a uniformly 15N/1H-labeled cruzain-K777 sample was used to assign the methionine ε-methyl groups. General spectral parameters and acquisition times are listed in the Supporting Information. Chemical shift assignments are listed in Supplemental Table S1.
NMR-based Inhibitor Binding Assays
NMR samples used for the 15N-1H HSQC and 13C-1H HSQC experiments consisted of 0.025 – 0.050 mM uniformly or selectively 15N-His, 15N-Cys, or 13C-Met labeled cruzain in 0.40 – 0.45 mL solution. Inhibitors were dissolved in d6-DMSO (Cambridge Isotope Laboratories) resulting in 10 mM stock solutions. Titrations were performed by adding ΔL aliquots of the 10 mM stock solution in a step-wise manner until 10 – 20 fold stoichiometric excess with respect to cruzain was achieved. A total of 5 titration points (apo, 2.5x, 5x, 10x, and 20x) for each inhibitor were acquired. NMR solutions were supplemented with 1 mM DTT (final concentration) prior to acquiring the first (apo) titration point. Final DMSO concentrations did not exceed 5% (v/v). Further spectral parameters and data processing protocols are reported in the Supporting Information. Total chemical shift perturbation values were calculated as previously described.21
Circular Dichroism
All circular dichroism (CD) data were acquired on a JASCO J-715 Spectropolarimeter at room temperature and using a 0.2 cm pathlength QS cuvette. A single scan ranging from 260 to 200 nm was acquired for each sample. All other spectral parameters and estimation of fractional helicity (fH) values are as previously described.21 Concentrated cruzain-K777 or procruzain stocks were 500 ΔM in 20 mM potassium phosphate buffer containing no salt, at pH 5.0, pH 7.0 and pH 10.0. For each titration point, 10 ΔL of concentrated enzyme stock was diluted with 490 ΔL buffer containing various concentrations of guanidinium hydrochloride (GdnHCl), for a final cruzain concentration of 10 ΔM. Final GdnHCl concentrations in the assay solutions ranged from 0 to 8 M (cruzain-K777), and 0 to 5 M (procruzain). Assay solutions were mixed by gently pipetting the mixtures and incubating them at room temperature for 30 minutes prior to data acquisition.
Structure representations
All structure representations were generated using the x-ray crystal structure of the cruzain-K777 complex (PDB accession 2OZ2)6 with PyMol Version 1.5.0 (Schrödinger, LLC).
Results
High level expression of soluble cruzain
Extensive modification of previously reported cruzain expression protocols14,15 provided sufficient quantities of purified, active cruzain for use in NMR-based studies. The original protease construct expressed as an insoluble fusion protein, requiring urea denaturation and multiple refolding and dialysis steps. Reported yields from the original protocol using rich media were 15 – 20 mg of the fusion protein per liter bacterial culture, resulting in approximately 1 – 1.5 mg active cruzain after the self-activation and purification steps.14,15 In the current protocol, a His6-tag sequence is placed at the N-terminus of the cruzain Δc construct15 (Figure 1a) for ease of purification over a nickel sepharose column. The construct expressed as a soluble-fraction zymogen using auto-induction methods described by Studier.16 Compared to the original inclusion body preparation using rich media,14 auto-induction in both minimal and rich media is a significant improvement. Yields of the zymogen form of cruzain were > 200 mg per liter of rich auto-induction media (A. Tochowitz and M. Merski, personal communication) and 150 mg per liter of minimal auto-induction media.
These higher protein yields allowed for faster auto-activation of the inactive zymogen into the catalytically active cruzain (Figure 1b): 20 – 40 minutes, compared to approximately 4 – 7 hours in the previous protocol.14 This result also verifies that self-activation of procruzain is concentration dependent. Notably, incubation of procruzain with K777 impeded, but did not completely abolish, in vitro self-activation of the zymogen (Figure 1c and Supporting Information). Fluorescence-based activity assays using the peptide substrate Z-Phe-Arg-AMC (data not shown) verified the cleaved product to be the active form of cruzain. Final yields of purified active cruzain were 80 – 100 mg per liter of auto-induction rich media and 30 – 50 mg per liter of auto-induction minimal media. The corresponding 15N/1H HSQC spectra of the K777-inhibited and apo (Figure 1d and Supplemental Figs. S1-S2) cruzain display well-dispersed resonance peaks, verifying the presence of a folded protein.
NMR assignments of the cruzain-K777 complex
Examination of the cruzain 15N/1H HSQC spectra indicated that the apo state was highly susceptible to continued self-proteolysis under the current NMR conditions. Spectra of apo cruzain acquired at time intervals over a period of less than 1 day at 27 °C exhibited resonance peaks between 7.0 and 8.5 ppm in the 1H dimension which were not present in the time “zero” spectrum (data not shown). The presence of these peaks, which grew in intensity over a period of time, is a hallmark of protein degradation and/or unfolding. Storing apo cruzain samples at 4 °C did not alleviate the problem, as the degradation peaks were also present in their respective HSQC spectra.
To obtain backbone and side chain resonance assignments, standard triple-resonance data sets20 were acquired using uniformly 13C/15N/2H-labeled cruzain inhibited with unlabeled K777. However, the 15N/1H HSQC spectra of the deuterated cruzain-K777 complex exhibited several missing peaks in comparison with that of the protonated analog (Supplemental Fig. S1), indicating incomplete 2H to 1H back-exchange of the amide protons during protein purification. Attempts to partially denature the cruzain-K777 complex at pH 5.0 with 2 M GdnHCl, followed by rapid dilution and refolding yielded no appreciable amide proton back exchange, as observed in the HSQC spectra (data not shown). Fortunately, the resulting triple-resonance spectra yielded backbone amide and side chain resonance assignments for 165 of the 206 (approximately 80%) non-proline residues in the cruzain-K777 complex (Supplemental Table 1). The majority of the absent backbone amide assignments correspond to residues in the central α-helix from Phe28 to Leu40, as well as portions of the adjacent β-sheets. Importantly, with the exception of Asn182 and Trp184, the backbone amide resonances of residues located within the active site and substrate binding pocket of the cruzain-K777 complex are assigned and provide sufficient coverage to map the binding modes of protease inhibitors.
The cruzain-K777 complex is exceptionally stable
Circular dichroism (CD) studies were performed to examine the structural stabilities of the cruzain-K777 complex and apo procruzain (Figure 2). Overall, the CD signatures acquired in the absence of denaturant at pH 5 exhibit significantly greater helical content than at pH 7 or 10 (Supplemental Fig. S3). The CD spectrum of procruzain at pH 5 was not acquired due to significant protein aggregation. In acidic (pH 5) and neutral conditions (pH 7) the CD signatures of inhibited cruzain remain relatively stable under increasing denaturant concentrations, with the θ222 band only losing half of its signal at 8 M GdnHCl. These results explain the lack of 2H to 1H back exchange observed for the uniformly labeled 13C/15N/2H cruzain-K777 NMR sample described above, as low denaturant concentrations are normally used to partially unfold deuterated proteins.22 Conversely, at pH 10, the cruzain-K777 complex becomes structurally labile at greater than 5 M GdnHCl concentrations. The apo-form of procruzain at pH 10 displayed less structural stability at low GdnHCl concentrations relative to the cruzain-K777 complex under similar conditions. In this case, the θ222 bands of the apo procruzain CD spectra were more negative than that of the cruzain-K777 complex at 0 M GdnHCl, indicating greater helical content. However, by 2 M GdnHCl, the θ222 band of the procruzain CD curve has lost more than half of its intensity.
Figure 2.

Circular dichroism denaturation study of cruzain-K777 and procruzain. Minimal perturbations of the CD spectra of the cruzain-K777 complex with guanidinium hydrochloride (0 to 8 M GdnHCl final concentration, colored as indicated), and measured at (a) pH 5, (b) pH 7, and (c) pH 10 reflects the structural stability of the inhibited protease against chemical denaturation. (d) The CD spectra of procruzain in the absence of inhibitor, and measured at pH 10 indicates weaker overall stability against GdnHCl denaturation (0 to 5 M final concentration, colored as indicated). Comparisons of the θ222 band and estimated fractional helicity data are displayed in Supplemental Figure S3.
The structural stability of the cruzain-K777 complex at pH 5 is also reflected in the heteronuclear NOE data, which measures backbone mobility in terms of NOE ratios (Supplemental Fig. S4). Including the N- and C-terminal residues, the majority of the backbone amides of the inhibited protease exhibit NOE ratios greater than 0.6, indicating a lack of overall backbone mobility. The regions which display lower NOE ratios, associated with higher levels of conformational mobility, are in loop regions, for example residues 85-107 located between two β-strands. Taken together, the CD and heteronuclear NOE studies indicate that the cruzain-K777 complex has an extraordinarily stable structure, even in the presence of high denaturant concentrations, and is more structured under acidic conditions.
Choosing 15N-Cys, 15N-His, and 13C-Met labels as cruzain inhibitor binding probes
Selective labeling of cruzain with 15N-Cys, 15N-His, and 13C-Met was performed to more quickly assess potential inhibitor-protease interactions via NMR spectroscopy. This labeling scheme was chosen since the two catalytic residues are Cys25 and His162. In addition, there are relatively few cysteine (eight), histidine (four) and methionine (four) residues within the cruzain sequence, which are all well-dispersed within the cruzain structure (Figure 3) and in their respective HSQC spectra (Supplemental Fig. S2). Six of the eight cysteine residues are sequestered in disulfide bonds (Cys22-Cys63, Cys56-Cys101, and Cys155-Cys203). Of the free cysteine residues, one (Cys36) is buried in the central helical core, while the catalytic Cys25 is surface exposed. In the case of the cruzain-K777 crystal structure,6 only the Cys22-Cys63 pair is located within 8 Å of the catalytic Cys25. Three of the four histidine residues (His43, His106, and His115) are located on the distal side of the molecule with respect to the catalytic His162. Although all the methionine residues are positioned greater than 8 Å from the active site Cys25, Met68 and Met145 are present and surface-exposed in the substrate binding pocket, while Met152 is located in a proximal loop. Importantly, Met68 forms a portion of the S2 pocket of cruzain. This binding pocket subsite, which interacts with the P2 position (two residues N-terminal to the scissile bond) of the peptide substrate, is known to be a determinant of cysteine cathepsin enzyme specificity.23 The loop containing Met145 and Met152 is separated from the catalytic His162 by a cluster of tryptophan residues (Trp144, Trp184 and Trp188). Therefore, any perturbations in the conformation of His162 upon inhibitor binding may also propagate through the tryptophan cluster towards the two methionine residues.
Figure 3.

Choosing cysteine, histidine, and methionine residues as cruzain inhibitor binding probes. The distribution of (a) cysteine (yellow balls), (b) histidine (cyan balls), and (c) methionine (orange and magenta balls) residues mapped on the crystal structure of the cruzain-K777 complex (PDB 2OZ2).6 The catalytic Gln19 (dark blue), Cys25 (red), and His162 (cyan) residues are indicated by boxes. The putative catalytic residue Asn182 is omitted for clarity. Met68 and Met145 (orange), both located on the surface of the substrate binding pocket, and the relatively unstructured N-terminal region (residues 1-24, brown cartoon), are also indicated. (d) The cruzain active site, represented as a 90° rotation about the horizontal axis with respect to parts (a-c). The positions of the catalytic Gln19 (blue), Cys25 (red), His162 (cyan), and Asn182 (green) amide groups are indicated with balls. Orange balls denote positions of the Met68 and Met145 ε-methyl groups; purple balls denote positions of the Met52 and Met152 ε-methyl groups. A cluster of tryptophan residues (Trp184, dark red; Trp188, brown; and Trp144, pink) connects Met145 and Met152 with His162. Glu208 (light green), which forms a portion of the S2 pocket, is also displayed. The van der Waal surfaces of the tryptophan cluster and Glu208 are indicated by meshes. Surface representation of the cruzain active site with (e) and without (f) the K777 structure. Residue colors are the same as part (d).
Classification of cruzain-inhibitor interactions using selectively labeled probes
The selectively labeled cruzain samples were titrated with several inhibitors with known binding modes as a proof of principle. All the listed compounds (Table 1) had been previously identified through either chemical modification of known peptide substrates or via computational docking experiments and verified as having some inhibitory effect on protease activity.6,12,13,24 The cysteine, histidine, and methionine residues were segregated into three categories according to their respective distance to the active site: (1) “active site”, (2) “proximal”, and (3) “remote”. The “active site” residues (Cys25, His162, Met68, and Met145) are surface exposed and are located in the substrate binding pocket within 10 Å of the catalytic Cys25 thiol group. The “proximal” residues (Cys22, Cys63, and Met152) border the active site region and are located between 10 – 15 Å of the Cys25 thiol. The remaining Cys/His/Met residues, which are generally located greater than 15 Å from the Cys25 thiol, are categorized as “remote”.
Covalent, irreversible inhibitor
HSQC titrations with the vinyl sulfone K777, a known covalent, irreversible inhibitor, yielded dramatic chemical shift perturbations of peaks corresponding to the catalytic residues (Cys25 and His162) and residues located within, or proximally to, the active site and substrate binding pocket (Cys22, Cys63, Met68, Met145, and Met152) (Figure 4a-c). Residues located remotely to this region displayed negligible shift perturbations. The apparent maximum shift perturbations were reached at low stoichiometric inhibitor ratios (< 2.5 fold molar excess with respect to cruzain) and were not significantly affected with increasing K777 concentrations (Figure 4d-f).
Figure 4.

Chemical shift perturbation data of cruzain reporter groups with K777. The HSQC titration spectra of selectively labeled (a) 15N-Cys, (b) 15N-His, and (d) 13C-Met cruzain with 0 (black) and 10 or 20 (red) molar equivalents of K777. Solid boxes indicate residues located in the substrate binding pocket. Dotted lines correspond to other perturbed residues. The chemical shift perturbations relative to the apo state at 2.5 (black) and 10 or 20 (red) molar equivalents K777 observed for the selectively labeled (d) 15N-Cys, (e) 15N-His, and (f) 13C-Met cruzain resonances. Residues located in (asterisks and boxed) or proximal to the substrate binding pocket of cruzain exhibit the largest shift perturbations.
Covalent, reversible inhibitor
Spectral data of the nitrile compound 2,24 a covalent, reversible inhibitor, displayed similar chemical shift perturbation profiles to those observed for the K777 titration (Supplemental Figs. S5-S7). However, precipitate formation within the NMR tube occurred during the titration of compound 2. Since none of the HSQC resonance peaks concurrently broadened as inhibitor concentration increased, compound 2, not cruzain, is likely the precipitating/aggregating species in solution. Major differences in the overall perturbation patterns between the two data sets, especially noticeable for the methionines, may be attributed to the different chemical moieties present in K777 and compound 2, as well as their relative positioning on the enzyme. For example, in the crystal structure of the cruzain-compound 2 complex (3I06.pdb)24 the inhibitor is in contact with Met68, but is positioned greater than 10 Å from Met145. Conversely, in the cruzain-K777 complex, both methionine methyl groups are in contact with the inhibitor. Moreover, the terminal sulfyl phenyl ring of K777 is in contact with Trp184, allowing for conformational perturbations of the tryptophan indole ring to affect the observed Met145 and Met152 chemical shifts.
Non-covalent inhibitors
In comparison to the covalently bound K777 and compound 2, titration with high molar equivalents of the known non-covalent cruzain inhibitors 4 – 713 display significantly reduced, sometimes negligible, 15N-Cys chemical shift perturbations (Supplemental Fig. S5). In the case of the 15N-His and 13C-Met spectra, only the catalytic His162, as well as the Met68 and Met145, displayed any strong chemical shift perturbations (Supplemental Figs. S6-S7). In general, the shift perturbations of the 15N-His and 13C-Met resonances do not reach a maximum value at greater than 20-fold molar excess inhibitor concentrations. Additionally, in the case of the titration with compound 4, the His162 amide resonance exhibited extensive peak broadening at low inhibitor concentrations while the intensities of the other three histidine peaks remained relatively unchanged. This broadening of a specific amino-acid resonance peak is indicative of an “intermediate” NMR time-scale conformational exchange, rather than protein aggregation. Similar peak broadening was observed for the catalytic Cys25 at low concentrations of compound 5. Multiple peaks, likely corresponding to degradation products were also observed in both the 15N-Cys and 15N-His HSQC spectra of the compound 5 titration. These results confirm that the non-covalent inhibitors have overall weaker binding affinity to cruzain than their covalent counterparts, as reflected in their micromolar Ki and IC50 values (Table 1).
Non-interacting compounds
The oxadiazole 8,12 represents a scaffold subsequently considered to be a non-interacting compound and/or aggregator (J. Ellman, personal communication). Titration of the cruzain samples with compound 8 resulted in aggregates forming in the NMR tubes. Overall chemical shift perturbations for the three reporter groups were minor in comparison to those observed for the other compounds (Supplemental Figs. S5-S7). Interestingly, peaks corresponding to the catalytic Cys25 and His162 appear to exchange broaden more dramatically than the other cysteine and histidine residues. As with compound 2, the remaining resonance peaks in the HSQC spectra did not significantly broaden over the course of the titration. In addition, the Met145 ε-methyl group is the only resonance among the other methionine residues that is perturbed upon addition of compound 8. Together, these results suggest that, although the compound is aggregating in solution, it may have a limited binding affinity to cruzain.
The only scaffold listed in Table 1 with a previously unknown binding mode is compound 3, a chlorinated intermediate of compound 2. In this case, compound 3 was added to the cruzain NMR samples to test the hypothesis that the nitrile group of compound 2 was necessary for covalent binding to Cys25. Similarly to compounds 2 and 8, titration of compound 3 resulted in precipitate formation in solution. In this case, however, negligible effects on resonance shift perturbations or peak broadening were observed in the 15N-Cys and 15N-His HSQC spectra (Supplemental Fig. S5-S6). As with compound 8, minimal perturbations are observed for the Met145 peak (Supplemental Fig. S7). Direct comparison of the overall and average reporter group chemical shift perturbations between compounds 2 and 3 (Figure 5) indicate that the nitrile derivative interacts more strongly to cruzain than the chlorinated derivative. If the Cys25 thiol were to covalently bind directly to the purine ring of compound 3, significant chemical shift perturbations on par with those observed for compound 2 would have been observed for the reporter groups. Collectively, the NMR titration data suggests a possible weak transient non-covalent interaction between cruzain and compound 3. The NMR data also verifies the previously reported cruzain-compound 2 binding mode:24 the Cys25 thiol sulfur is covalently bonded to the nitrile group, and not directly to the purine group of the inhibitor.
Figure 5.

Summary of the cruzain-inhibitor NMR titration data. Chemical shift perturbations of the selectively labeled 15N-Cys, 15N-His, and 13C-Met cruzain resonances upon addition of the inhibitors listed in Table 1. The bar charts represent the summation of the chemical shift perturbations, and are separated as Cys/His/Met residues located (a) in the substrate binding pocket, (b) proximally to the substrate binding pocket, and (c) outside the substrate binding pocket. Average shift perturbations corresponding to the same residue classifications (d-f) are also plotted. The inhibitors are categorized as covalent irreversible (black), covalent reversible (red), non-covalent (blue), and non-interacting/aggregators (yellow). Asterisks indicate exchange broadening of the resonance peaks upon addition of inhibitor. Shift perturbations of the Cys, His, and Met residues plotted individually are displayed in Supplemental Figs. S6-S8.
General trends
For the set of compounds listed in Table 1, all of which exhibit binding or inhibitory constants in the low micromolar range, the total summed and average 15N-Cys, 15N-His, and 13C-Met shift perturbations are largest for the covalently bound inhibitors (K777 and compound 2), and smallest for the non-interactors/aggregators (Figure 5). As expected for these competitive inhibitors, which were designed to target the active site, residues located within the substrate binding site (Cys25, His162, Met68 and Met145) display larger total and average chemical shift perturbations relative to the “proximal” (Cys22, Cys63, and Met152) and “remote” residues. The exception to these trends appears for compounds 4 and 5, in which extensive peak broadening of the catalytic residues would limit any contribution to the observed total chemical shift perturbation.
Maximum chemical shift perturbations of the “catalytic” cysteine, histidine and methionine residues were observed at less than two fold excess compound relative to cruzain in the cases of the covalently bound inhibitors. Conversely for the non-covalent inhibitors, greater than 20 fold excess compound was required before apparent maximum chemical shift perturbations were reached. A comparison of the 15N-Cys data (Supplemental Figure S5) reveals distinct patterns: the covalently bound inhibitors impart chemical shift perturbations on the catalytic Cys25 greater than 0.5 ppm, while the non-covalent compounds display values less than 0.15 ppm for all the cysteine reporter groups. In the case of the 15N-His data (Supplemental Figure S6), the chemical shift perturbation values of the catalytic His162 observed for the K777 and compound 2 titrations appear to have reached a maximum value at 2.5 fold excess compounds, with no significant differences at 20 fold excess inhibitor. Conversely, although significant perturbations are observed for His162 upon addition of 2.5 equivalents of the non-covalent inhibitors, such as compounds 4, 6, and 7, further changes in the chemical shift resonances are observed at 20 fold excess. Similar patterns are observed in the Met68 (Supplemental Figure S7).
Discussion
We have established an expression and purification protocol resulting in high yields of activated cruzain. The optimized auto-induction protocol has not only assisted in completing recent crystallography efforts,13,24 but also allowed for the cost-effective production of isotopically labeled cruzain for use in NMR studies. Due to its inherent instability, active cruzain has not yet been crystallized in its apo form. All previously published cruzain SAR studies had relied on obtaining high-resolution x-ray crystal structures, usually with an inhibited protease. With the exception of compound 6 (3KKU.pdb),13 none of the non-covalent inhibitors listed in Table 1 have been successfully co-crystallized with cruzain. The existing crystallographic structures of cruzain will continue to be utilized for in silico docking and modeling studies, and other high-throughput screening (HTS) methods such as fluorescence activity assays will help triage compound libraries in order to identify potential lead inhibitors. However, neither the docking or HTS studies will verify and characterize inhibitor binding sites or allow for more thorough biophysical studies of cruzain.
As we have illustrated here, with the advantages of selective isotopic labeling, binding modes of these inhibitors can be examined in a relatively fast manner using NMR-based methods. In particular, the use of selectively 15N-Cys, 15N-His, and 13C-Met labeled cruzain was helpful in quickly verifying the binding sites of eight small-molecule inhibitors. With the exception of the non-interactors/aggregators, titration of each compound using these cruzain samples perturbed the chemical shifts of residues at or near the active site. Importantly, these NMR titrations verified previously reported in silico docking results of selected non-covalently binding compounds,13 including those for which there are no existing crystallographic data sets. Using current NMR automation technology with samples contained in standard 5 mm NMR tubes, a binary binding experiment (i.e. apo versus greater than 20 fold excess compound) can be performed in 1 to 2 hours per sample. The rate of data acquisition can be further enhanced using NMR flow-cells and 96 well plate sampling attachments, which requires smaller sample volumes.25 This has the added advantage of allowing for higher protein and inhibitor concentrations relative to the standard 5 mm tubes, and more importantly, faster data acquisition times.
Results from the NMR titrations can be used to verify whether these candidate compounds bind to cruzain and if they are competitive inhibitors. In addition, NMR-based titrations may verify recently proposed allosteric binding sites located greater than 10 Å from the catalytic Cys25.26 Insights gained from the NMR titrations of potential inhibitors, especially those that exhibit weak binding affinities to cruzain, may also help determine whether efforts at optimizing the compounds for further SAR studies are warranted. Although the NMR titration assays are not able to detect subtle differences between various chemical scaffolds, such as relative orientation within the binding pocket, they may be useful in determining differences between chemical analogs. For example, clear differences in the HSQC spectra are observed between compounds 2 (a covalent binder) and 3 (a non-interactor/aggregator). Both share the same chemical scaffold, but differ in the functional group attached to the purine ring: a nitrile group in compound 2 and a chloro group in compound 3. In these cases, NMR spectroscopy and x-ray crystallography can be used in conjunction to identify and optimize potential therapeutic leads for cruzain.
There has been intense and long-standing interest in cysteine proteases, particularly due to their involvement in a wide range of human diseases.8,9,27,28 In particular, cysteine cathepsins, which make up 11 of the 15 canonical cathepsin family members, are known to play roles in extracellular matrix remodeling in humans, leading to the development of various pathologies including cancer, cardiovascular and inflammatory diseases.9,28 However, there are relatively few NMR-based studies that focus on their catalytic domains. One early effort focused on pH studies of active papain and relied on one-dimensional proton spectra.29 A second study presented two-dimensional NMR data of the zymogen procathepsin L, but did not report any useful resonance assignments.30 The majority of the previous NMR-based structure calculations have centered on protein inhibitors such as chagasin (cruzain)31 or p41icf (cathepsin L).32
More recently, NMR-based structural studies focusing on cysteine proteases such as Streptopain,33,34 Foot and Mouth Disease Virus Leader Protease,35 Ubiquitin C-terminal hydrolases,36,37 and SARS Coronavirus Main Protease (SARS CoV Mpro)38 have appeared in the literature. Importantly, in these enzyme constructs the catalytic cysteine had been substituted with either serine or alanine, or in the case of SARS CoV Mpro, truncated, rendering the proteases inactive. To date, there are few examples of active papain-like cysteine proteases that have been extensively studied via NMR, including the Josephin domain of ataxin-3,39-41 the NlpC/P60 domain of lipoprotein Spr,42 and Sortase A.43 All of these proteases contain the catalytic cysteine and histidine residues positioned in approximately the same relative positions as papain. However, all have low overall sequential (< 15 %) and structural homology (backbone RMSD > 3.5 Å) with respect to papain and other members of the cathepsin cysteine protease family. Thus, to our knowledge, the data presented herein represents the first NMR-based study focusing on the mature, active form of a papain/cathepsin-like cysteine protease in its wild-type state.
On a more basic level, production of NMR-ready cruzain will allow for more extensive study into structure-function relationships. For example, backbone dynamics of both the zymogen and mature cruzain can now be examined in order to more fully understand how conformational mobility influences proteolytic activity. In addition, selectively labeled samples may also be utilized to determine the pKa values of residues such as the catalytic Cys25 and His162 (Supporting Information and Supplemental Figs. S8-S11). Such information may be useful in further dissecting the mechanism of the enzyme.
Further insight into the mechanism of in vitro procruzain self-activation is provided by our observation that K777 impedes, but does not completely abolish, cleavage of the pro-region from the catalytic domain. Examination of the structurally homologous Cys25 to Ser25 variant of procathepsin L (1CJL, backbone RMSD for the catalytic domain with 2OZ2 = 0.582 Å)44 shows a short helix positioned orthogonally at the active site occluding the catalytic residues. However, the 44 residues immediately preceding the N-terminus of the catalytic domain lie in the substrate binding cleft and are relatively unstructured. As has been suggested for all members of the papain/cathepsin cysteine protease family,44 the pro-region in cruzain may likewise adopt a similar conformation and mode of auto-inhibition. The relatively unstructured state of the pro-region may allow for transient interactions with the catalytic domain, which in turn, allows for the active site to be exposed in a catalytically competent state. Once an “active” enzyme is available, it would be ready to proteolyze other procruzain molecules, initiating a cascade of auto-activation events.
The expression and purification strategies described herein may also be adapted to papain and the other eleven known structurally homologous cysteine cathepsins, opening the possibility of characterizing their solution states via NMR spectroscopy or other biophysical methods. For example, K777 was recently demonstrated to inhibit the activities of cathepsins B, L, and S isolated from pancreatic extracts.45 Results from these studies could further the understanding of mechanisms which govern enzymatic activity, as well as assist the discovery of new inhibitors for this large family of closely related cysteine proteases.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. Brian K. Shoichet, Prof. Jonathan Ellman, Dr. Rafaela S. Ferriera, Dr. Clifford Bryant, Dr. Anthony O’Donoghue, Dr. Anna M. Tochowitz, Dr. Matthew Merski, and Ms. Karen Andrade for technical support. We also thank Prof. John D. Gross, Prof. Adam R. Reslo and Prof. Michelle R. Arkin for helpful discussions, and Mr. Jonathan E. Gable for a thorough reading of this manuscript.
Funding Sources: This work was funded in part by grants from the National Institutes of Health (P01-A135707 and R01-AI090592, to JHM; P50-GM-082250, to CSC) the National Cancer Institute (P30-CA-82103-13, to CSC), and the Sandler Foundation.
Abbreviations
- AMC
7-amino-4-methylcoumarin
- DTT
dithiothreitol
- GdnHCl
guanidinium hydrochloride
- HSQC
heteronuclear single quantum coherence spectroscopy
- MMTS
methyl methanethiosulfonate
- NOE
nuclear Overhauser effect
- PMSF
phenylmethane sulfonyl fluoride
- RMSD
root mean squared deviation
- SAR
structure-activity relationship
- Z or N-CBZ
N-carbobenzyloxy
Footnotes
ACCESSION NUMBERS The available chemical shift resonance assignments of the cruzain-K777 complex have been submitted to the Biological Magnetic Resonance Data Bank (http://www.bmrb.wisc.edu/) with the accession code 18236.
SUPPORTING INFORMATION AVAILABLE Additional methodology and experimental results; a table listing the available chemical shift assignments for the U-13C/15N/2H cruzain-K777 complex; a table listing the apparent fitted 15N-Cys, 15N-His, and 13C-Met pKa values; HSQC overlay of protonated and deuterated cruzain-K777; HSQC annotated with available resonance assignments; HSQC overlays of uniformly and selectively labeled cruzain ± K777; θ222 band and estimated fractional helicity bar charts from the CD denaturation study; steady state 1H-{15N} NOE ratios for cruzain-K777; chemical shift perturbation bar charts summarizing the inhibitor cruzain titrations; 15N-His, 15N-Cys and 13C-Met pH titration curves of MMTS- and K777-inhibited cruzain. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.Sajid M, Robertson SA, Brinen LS, McKerrow JH. Cruzain : the path from target validation to the clinic. Adv Exp Med Biol. 2011;712:100–115. doi: 10.1007/978-1-4419-8414-2_7. [DOI] [PubMed] [Google Scholar]
- 2.Le Loup G, Pialoux G, Lescure FX. Update in treatment of Chagas disease. Curr Opin Infect Dis. 2011;24:428–434. doi: 10.1097/QCO.0b013e32834a667f. [DOI] [PubMed] [Google Scholar]
- 3.Munoz-Saravia SG, Haberland A, Wallukat G, Schimke I. Chronic Chagas’ heart disease: a disease on its way to becoming a worldwide health problem: epidemiology, etiopathology, treatment, pathogenesis and laboratory medicine. Heart Fail Rev. 2012;17:45–64. doi: 10.1007/s10741-010-9211-5. [DOI] [PubMed] [Google Scholar]
- 4.Bern C, Kjos S, Yabsley MJ, Montgomery SP. Trypanosoma cruzi and Chagas’ Disease in the United States. Clin Microbiol Rev. 2011;24:655–681. doi: 10.1128/CMR.00005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gutteridge WE. Existing chemotherapy and its limitations. Br Med Bull. 1985;41:162–168. doi: 10.1093/oxfordjournals.bmb.a072044. [DOI] [PubMed] [Google Scholar]
- 6.Kerr ID, Lee JH, Farady CJ, Marion R, Rickert M, Sajid M, Pandey KC, Caffrey CR, Legac J, Hansell E, McKerrow JH, Craik CS, Rosenthal PJ, Brinen LS. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J Biol Chem. 2009;284:25697–25703. doi: 10.1074/jbc.M109.014340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamphuis IG, Kalk KH, Swarte MB, Drenth J. Structure of papain refined at 1.65 A resolution. J Mol Biol. 1984;179:233–256. doi: 10.1016/0022-2836(84)90467-4. [DOI] [PubMed] [Google Scholar]
- 8.McGrath ME. The lysosomal cysteine proteases. Annu Rev Biophys Biomol Struct. 1999;28:181–204. doi: 10.1146/annurev.biophys.28.1.181. [DOI] [PubMed] [Google Scholar]
- 9.Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim Biophys Acta. 2012;1824:68–88. doi: 10.1016/j.bbapap.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doyle PS, Zhou YM, Engel JC, McKerrow JH. A Cysteine Protease Inhibitor Cures Chagas’ Disease in an Immunodeficient-Mouse Model of Infection. Antimicrob. Agents Chemother. 2007;51:3932–3939. doi: 10.1128/AAC.00436-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKerrow JH, Doyle PS, Engel JC, Podust LM, Robertson SA, Ferreira R, Saxton T, Arkin M, Kerr ID, Brinen LS, Craik CS. Two approaches to discovering and developing new drugs for Chagas disease. Mem Inst Oswaldo Cruz. 2009;104(Suppl 1):263–269. doi: 10.1590/s0074-02762009000900034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferreira RS, Bryant C, Ang KK, McKerrow JH, Shoichet BK, Renslo AR. Divergent modes of enzyme inhibition in a homologous structure-activity series. J Med Chem. 2009;52:5005–5008. doi: 10.1021/jm9009229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferreira RS, Simeonov A, Jadhav A, Eidam O, Mott BT, Keiser MJ, McKerrow JH, Maloney DJ, Irwin JJ, Shoichet BK. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J Med Chem. 2010;53:4891–4905. doi: 10.1021/jm100488w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eakin AE, Mills AA, Harth G, McKerrow JH, Craik CS. The sequence, organization, and expression of the major cysteine protease (cruzain) from Trypanosoma cruzi. J Biol Chem. 1992;267:7411–7420. [PubMed] [Google Scholar]
- 15.Eakin AE, McGrath ME, McKerrow JH, Fletterick RJ, Craik CS. Production of crystallizable cruzain, the major cysteine protease from Trypanosoma cruzi. J Biol Chem. 1993;268:6115–6118. [PubMed] [Google Scholar]
- 16.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 17.Shahian T, Lee GM, Lazic A, Arnold LA, Velusamy P, Roels CM, Guy RK, Craik CS. Inhibition of a viral enzyme by a small-molecule dimer disruptor. Nat Chem Biol. 2009;5:640–646. doi: 10.1038/nchembio.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 19.Goddard TD, Kneller DG. Sparky. Vol. 3. University of California; San Francisco: 1999. [Google Scholar]
- 20.Cavanagh J, Fairbrother WE, Palmer AG, III, Skelton NJ. Protein NMR Spectroscopy Principles and Practice. Academic Press; San Diego: 1996. [Google Scholar]
- 21.Lee GM, Shahian T, Baharuddin A, Gable JE, Craik CS. Enzyme Inhibition by Allosteric Capture of an Inactive Conformation. J Mol Biol. 2011;411:999–1016. doi: 10.1016/j.jmb.2011.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardner KH, Kay LE. The use of 2H, 13C, 15N multidimensional NMR to study the structure and dynamics of proteins. Ann Rev Biophys Biomol Struct. 1998;27:357–406. doi: 10.1146/annurev.biophys.27.1.357. [DOI] [PubMed] [Google Scholar]
- 23.Lecaille F, Authie E, Moreau T, Serveau C, Gauthier F, Lalmanach G. Subsite specificity of trypanosomal cathepsin L-like cysteine proteases. Probing the S2 pocket with phenylalanine-derived amino acids. Eur J Biochem. 2001;268:2733–2741. doi: 10.1046/j.1432-1327.2001.02172.x. [DOI] [PubMed] [Google Scholar]
- 24.Mott BT, Ferreira RS, Simeonov A, Jadhav A, Ang KK, Leister W, Shen M, Silveira JT, Doyle PS, Arkin MR, McKerrow JH, Inglese J, Austin CP, Thomas CJ, Shoichet BK, Maloney DJ. Identification and optimization of inhibitors of Trypanosomal cysteine proteases: cruzain, rhodesain, and TbCatB. J Med Chem. 2010;53:52–60. doi: 10.1021/jm901069a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kautz RA, Goetzinger WK, Karger BL. High-Throughput Microcoil NMR of Compound Libraries Using Zero-Dispersion Segmented Flow Analysis. J Comb Chem. 2004;7:14–20. doi: 10.1021/cc0498940. [DOI] [PubMed] [Google Scholar]
- 26.Durrant JD, Keranen H, Wilson BA, McCammon JA. Computational identification of uncharacterized cruzain binding sites. PLoS Negl Trop Dis. 2010;4:e676. doi: 10.1371/journal.pntd.0000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vasiljeva O, Reinheckel T, Peters C, Turk D, Turk V, Turk B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr Pharm Des. 2007;13:387–403. doi: 10.2174/138161207780162962. [DOI] [PubMed] [Google Scholar]
- 28.Reiser J, Adair B, Reinheckel T. Specialized roles for cysteine cathepsins in health and disease. J Clin Invest. 2010;120:3421–3431. doi: 10.1172/JCI42918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson FA, Lewis SD, Shafer JA. Determination of a low pK for histidine-159 in the S-methylthio derivative of papain by proton nuclear magnetic resonance spectroscopy. Biochemistry. 1981;20:44–48. doi: 10.1021/bi00504a008. [DOI] [PubMed] [Google Scholar]
- 30.Jerala R, Zerovnik E, Kidric J, Turk V. pH-induced conformational transitions of the propeptide of human cathepsin L. A role for a molten globule state in zymogen activation. J Biol Chem. 1998;273:11498–11504. doi: 10.1074/jbc.273.19.11498. [DOI] [PubMed] [Google Scholar]
- 31.Salmon D, do Aido-Machado R, Diehl A, Leidert M, Schmetzer O, de ALAP, Scharfstein J, Oschkinat H, Pires JR. Solution structure and backbone dynamics of the Trypanosoma cruzi cysteine protease inhibitor chagasin. J Mol Biol. 2006;357:1511–1521. doi: 10.1016/j.jmb.2006.01.064. [DOI] [PubMed] [Google Scholar]
- 32.Chiva C, Barthe P, Codina A, Gairi M, Molina F, Granier C, Pugniere M, Inui T, Nishio H, Nishiuchi Y, Kimura T, Sakakibara S, Albericio F, Giralt E. Synthesis and NMR structure of p41icf, a potent inhibitor of human cathepsin L. J Am Chem Soc. 2003;125:1508–1517. doi: 10.1021/ja0207908. [DOI] [PubMed] [Google Scholar]
- 33.Luo S-C, Chen C-Y, Lin Y-S, Jeng W-Y, Chuang W-J. Letter to the Editor: Backbone 1H, 15N and 13C resonance assignments of the 28 kDa mature form of streptopain*. J Biomol NMR. 2003;25:165–166. doi: 10.1023/a:1022291604936. [DOI] [PubMed] [Google Scholar]
- 34.Wang CC, Houng HC, Chen CL, Wang PJ, Kuo CF, Lin YS, Wu JJ, Lin MT, Liu CC, Huang W, Chuang WJ. Solution structure and backbone dynamics of streptopain: insight into diverse substrate specificity. J Biol Chem. 2009;284:10957–10967. doi: 10.1074/jbc.M807624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cencic R, Mayer C, Juliano MA, Juliano L, Konrat R, Kontaxis G, Skern T. Investigating the substrate specificity and oligomerisation of the leader protease of foot and mouth disease virus using NMR. J Mol Biol. 2007;373:1071–1087. doi: 10.1016/j.jmb.2007.08.061. [DOI] [PubMed] [Google Scholar]
- 36.Harris R, Eidhoff U, Vinzenz D, Renatus M, Gerhartz B, Hommel U, Driscoll PC. Backbone 1H, 13C, and 15N resonance assignments for the 26-kD human de-ubiquitinating enzyme UCH-L3. Biomol NMR Assign. 2007;1:51–53. doi: 10.1007/s12104-007-9012-5. [DOI] [PubMed] [Google Scholar]
- 37.Andersson FI, Jackson SE, Hsu ST. Backbone assignments of the 26 kDa neuron-specific ubiquitin carboxyl-terminal hydrolase L1 (UCH-L1) Biomol NMR Assign. 2010;4:41–43. doi: 10.1007/s12104-009-9203-3. [DOI] [PubMed] [Google Scholar]
- 38.Zhang S, Zhong N, Ren X, Jin C, Xia B. 1H, 13C and 15N resonance assignments of SARS-CoV main protease N-terminal domain. Biomol NMR Assign. 2011;5:143–145. doi: 10.1007/s12104-010-9287-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicastro G, Masino L, Frenkiel TA, Kelly G, McCormick J, Menon RP, Pastore A. Letter to the Editor: Assignment of the 1H, 13C, and 15N resonances of the Josephin domain of human ataxin-3. J Biomol NMR. 2004;30:457–458. doi: 10.1007/s10858-004-4343-3. [DOI] [PubMed] [Google Scholar]
- 40.Nicastro G, Menon RP, Masino L, Knowles PP, McDonald NQ, Pastore A. The solution structure of the Josephin domain of ataxin-3: structural determinants for molecular recognition. Proc Natl Acad Sci U S A. 2005;102:10493–10498. doi: 10.1073/pnas.0501732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicastro G, Todi SV, Karaca E, Bonvin AM, Paulson HL, Pastore A. Understanding the role of the Josephin domain in the PolyUb binding and cleavage properties of ataxin-3. PLoS One. 2010;5:e12430. doi: 10.1371/journal.pone.0012430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aramini JM, Rossi P, Huang YJ, Zhao L, Jiang M, Maglaqui M, Xiao R, Locke J, Nair R, Rost B, Acton TB, Inouye M, Montelione GT. Solution NMR structure of the NlpC/P60 domain of lipoprotein Spr from Escherichia coli: structural evidence for a novel cysteine peptidase catalytic triad. Biochemistry. 2008;47:9715–9717. doi: 10.1021/bi8010779. [DOI] [PubMed] [Google Scholar]
- 43.Ilangovan U, Ton-That H, Iwahara J, Schneewind O, Clubb RT. Structure of sortase, the transpeptidase that anchors proteins to the cell wall of Staphylococcus aureus. Proc Natl Acad Sci U S A. 2001;98:6056–6061. doi: 10.1073/pnas.101064198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coulombe R, Grochulski P, Sivaraman J, Menard R, Mort JS, Cygler M. Structure of human procathepsin L reveals the molecular basis of inhibition by the prosegment. EMBO J. 1996;15:5492–5503. [PMC free article] [PubMed] [Google Scholar]
- 45.Lyo V, Cattaruzza F, Kim TN, Walker AW, Paulick M, Cox D, Cloyd J, Buxbaum J, Ostroff J, Bogyo M, Grady EF, Bunnett NW, Kirkwood KS. Active Cathepsins B, L and S in Murine and Human Pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2012;303:G894–903. doi: 10.1152/ajpgi.00073.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.