

Abstract
Respiration produces rhythmic activity in the entire olfactory system, driving neurons in the olfactory epithelium, olfactory bulb (OB), and cortex. The rhythmic nature of this activity is believed to be a critical component of sensory processing. OB projection neurons, mitral and tufted cells exhibit both spiking and subthreshold membrane potential oscillations rhythmically coupled to respiration. However, the network and synaptic mechanisms that produce respiration-coupled activity, and the effects of respiration on lateral inhibition, a major component of sensory processing in OB circuits, are not known. Is respiration-coupled activity in mitral and tufted cells produced by sensory synaptic inputs from nasal airflow alone, cortico-bulbar feedback, or intrinsic membrane properties of the projection neurons? Does respiration facilitate or modulate the activity of inhibitory lateral circuits in the OB? Here, in vivo intracellular recordings from identified mitral and tufted cells in anesthetized rats demonstrate that nasal airflow provides excitatory synaptic inputs to both cell types and drives respiration-coupled spiking. Lateral inhibition, inhibitory postsynaptic potentials evoked by intrabulbar microstimulation, was modulated by respiration. In individual mitral and tufted cells, inhibition was larger at specific respiratory phases. However, lateral inhibition was not uniformly larger during a particular respiratory phase in either cell type. Removing nasal airflow abolished respiration-coupled spiking in both cell types and nearly eliminated spiking in mitral, but not tufted, cells. In the absence of nasal airflow, lateral inhibition was weaker in mitral cells and less modulated in tufted cells. Thus, respiration drives distinct network activities that functionally modulate sensory processing in the OB.
Introduction
The perception of odors depends critically on respiration-coupled synaptic and spiking activities. In the absence of odorants, rhythmic activity in the olfactory bulb (OB) is coupled to the respiratory motion of air, which may induce an oscillatory, mechanical activation of the olfactory receptor neurons (ORNs) (Grosmaitre et al., 2007). In the presence of odorants, respiratory-coupled activity of OB projection neurons is thought to establish specific temporal windows to encode, relay, and process olfactory information (Chaput et al., 1992; Philpot et al., 1997; Kepecs et al., 2006).
Respiration-coupled activity in the OB was first described by local field potential and extracellular recording methods (Adrian, 1950; Walsh, 1956). These and other studies showed that OB projection neurons, identified by recording depth, were coupled to respiration (Macrides and Chorover, 1972; Buonviso et al., 2003). Subsets of these neurons fired action potentials during inspiration, others during expiration, and still others showed no evidence of respiration-coupled spiking activity (Onoda and Mori, 1980). Removing airflow over the nasal epithelium abolished respiration-coupled spiking and local field potential activity in the OB and olfactory cortex (Sobel and Tank, 1993; Fontanini et al., 2003). Whether respiration-coupled spiking activity in both mitral and tufted cells is produced by excitatory synaptic inputs from nasal airflow alone or whether respiration facilitates or modulates the activity of inhibitory lateral circuits in the OB is not known.
The two types of bulbar projection neurons, the mitral and tufted cells, are believed to code different aspects, such as detection or discrimination, of odor information (Scott, 1981; Nagayama et al., 2004). Morphologically, mitral cells are larger and have longer apical dendrites than tufted cells (Macrides and Schneider, 1982). Anatomically, these two classes of neurons receive similar inputs. Both receive direct excitatory input from ORNs, inhibitory input from local granule cells, but project in distinct patterns to different olfactory cortical regions (Price and Powell, 1970; Nagayama et al., 2010; Najac et al., 2011). Physiologically, the mitral and tufted neurons are different: tufted cells have larger responses to odorants, respond at lower threshold to antidromic inputs, have higher input resistances, and show a smaller extent of lateral inhibition (Schneider and Scott, 1983; Nagayama et al., 2004; Griff et al., 2008). Mitral and tufted cells also have distinct intrinsic membrane properties: tufted cells can fire rhythmically in vitro and continue firing even in the absence of synaptic inputs; mitral cells are not intrinsically active (Hayar et al., 2004b; De Saint Jan et al., 2009). At the present time, however, it is not known whether respiration couples spiking and depolarizes mitral and tufted cell to the same extent or whether the action of local inhibitory circuits is modulated by respiration.
Here, we used intracellular in vivo recording methods in conjunction with intrabulbar electrical microstimulation in anesthetized rats to assess the role of respiration-coupled synaptic inputs in shaping spontaneous activity and modulating synaptic and network processing in the OB. We show that nasal airflow produces respiration-coupled spiking in mitral and tufted cells and strengthens lateral inhibition in mitral cells.
Materials and Methods
Surgical preparation.
All of the procedures in this study conformed to the Guide for the Care and Use of Laboratory Animals (1996, National Academy of Sciences) and were approved by the Yale Institutional Animal Care and Use Committee. Male Sprague Dawley rats weighing 250–370 g (8–15 weeks old) were anesthetized with urethane (1000 mg/kg, i.p.) and maintained with supplemental doses delivered once every hour or as needed. Atropine sulfate (0.06 mg/kg, s.c.) was used to reduce the secretions along the respiratory tract, and lidocaine was applied topically at the incision. Throughout the experiment, the animal's temperature was maintained at 37°C via a feedback system. Animals were prepared for recording by exposing the dorsal surface of the OB with a craniotomy, removing the dura mater, and placing four stimulating electrodes across the olfactory nerve layer (ONL) (Ezeh et al., 1993) caudal to the recording site. Caudal placement of stimulation electrodes allowed us to activate local inhibitory circuitry without direct presynaptic stimulation of impaled neurons (Scott, 1981). The locations of the stimulating electrodes were marked with DiI to facilitate visualization, and the OB was covered with 4% agar in 0.9% saline to minimize pulsations.
In vivo intracellular and extracellular recordings of mitral and tufted cells.
Intracellular recordings were made with high-impedance glass micropipettes (40–80 MΩ). Extracellular recordings were made with lower-impedance (10–30 MΩ) glass electrodes filled with 2% biocytin dissolved in 1 m potassium acetate and 0.05 m Tris buffer (Ezeh et al., 1993). The recording electrode was advanced with a hydraulic microdrive. Recordings were made using an intracellular amplifier (Molecular Devices) in current-clamp mode with a bridge circuit. Before the impalement, the direct current offset was corrected, and the bridge was balanced. During impalement, the bridge was balanced as necessary, and, at the end of the recording, an offset trace was recorded. All membrane potential measurements were corrected offline to account for the measured pipette offset.
Criteria for acceptable recording.
Neurons were only included in the analysis described below if they met the following criteria: neurons had to (1) exhibit stable membrane potentials for the course of the recording (>20 min with 0 holding current), (2) show negligible motion artifacts, (3) have a resting membrane potential more hyperpolarized than approximately −45 mV, and (4) generate action potentials that crossed 0 mV.
Identification of cell classes.
Mitral and tufted cells were recovered after intracellular recording. Neurons were identified by type after recordings by morphology after histological recovery using intracellular injections of biotin. If the soma was located in the mitral cell body layer and the dendrites showed a primary apical tufted in a glomerulus as well as lateral processes, the filled neuron was classified as a mitral cell. If the soma was located in the external plexiform layer (EPL) or deep glomerular layer, the dendrites showed a glomerular tuft, lateral processes, and a putative axon, the neuron was classified as a tufted cell (Scott, 1981).
Histology.
After recordings, animals were transcardially perfused with 4% paraformaldehyde (PFA), and the brain was dissected and postfixed in PFA overnight. Free-floating sections of OB tissues (70 μm) were cut on a vibrating microtome. Sections were labeled with mouse streptavidin–Cy3 overnight at room temperature with gentle shaking (1:500; Ros et al., 2009). Tissue sections were wet mounted in mounting medium containing DAPI. Fluorescent neurons were located in single sections, aligned to recording penetrations, and imaged using a laser scanning confocal microscope.
Artificial nasal breathing.
In 19 of 34 preparations, a double tracheotomy was performed using previously established methods (Onoda and Mori, 1980). A vacuum and a filtered airline were used to simulate inspiration and expiration by directing airflow in and out of the nasal cavity. Flow rates were controlled using two airflow meters and matched to physiological conditions [500 ml/min for both inspiration and expiration (Ravel and Pager, 1990)]. A two-way pinch valve (Cole-Palmer) was triggered by custom software written in Spike 2 (Cambridge Electronic Design) to allow switching between inspiration (vacuum line open, airline closed) and expiration (vacuum line closed, airline open). During periods of artificial nasal breathing, the frequency and phase of nasal airflow were matched to the animal's natural respiration using a piezoelectric stretch sensor to monitor the chest expansion and trigger the pinch valve. Nasal airflow frequency and phase were monitored using a thermistor implanted in the ipsilateral naris.
Lateral olfactory tract lesion.
In 5 of 19 tracheotomized preparations, OB mitral and tufted cell axons were lesioned, the anteriormost portion of the ipsilateral lateral olfactory tract (LOT) was exposed through the orbit, the dura was carefully dissected, and a bent 30 gauge needle was used to sever the most superficial tissue (∼500 μm). These procedures effectively extinguish OB output to the ipsilateral olfactory cortices and some, but not all, cortical backprojections to the OB (Shepherd and Haberly, 1970). LOT targeting was confirmed histologically (see Fig. 3A).
Figure 3.

Respiration-coupled spiking and membrane potential oscillations in mitral cells are independent of cortico-bulbar feedback. A, Mitral cells were recorded and identified in double-tracheotomized animals in which the LOT was transected [green in Ai, coronal view, bregma +5.20 mm (Paxinos and Watson, 2004); Aii, histologically identified at the dotted line and arrows]. B, An identified mitral cell showed respiration-coupled membrane potential depolarizations and spiking during artificial nasal airflow (Bi) but not without nasal airflow (Bii, unfiltered membrane potentials in black, chest and nasal respiration traces below). Biii, Respiration-triggered average of the membrane potential (median filtered with 1 SD confidence intervals) and spiking PSTH during artificial nasal airflow (red) and without nasal airflow (blue). C, D, Mitral cells recorded in LOT-lesioned animals (red) showed respiratory-coupled spiking (C) and membrane potential depolarizations (D) with artificial nasal airflow as observed in mitral cells recorded in non-lesioned animals (gray). E, Nasal airflow was necessary for respiration-coupled spiking. F, However, respiration-coupled membrane potential depolarizations were not significantly altered in the absence of nasal airflow. G, Vector strength decreased significantly without nasal airflow (*p < 0.01, two-sided paired Students t test; n = 3). Scale bar: Aii, 750 μm.
ONL microstimulations.
Electrical microstimulations of the ONL were applied with concentric bipolar electrodes (platinum–iridium 150 μm outer diameter) to evoke IPSPs. Stimulation parameters (0.2–1 ms in duration and 0.1–10 mA in amplitude) were chosen to evoke long-lasting, complex responses in individual mitral and tufted cells (Ezeh et al., 1993). Each stimulation trial was separated by at least one breathing cycle. Inspiration and expiration stimulation epochs were staggered and triggered using the respiration trace monitored from the chest expansion. At least 10 trials were collected from the ONL microstimulations evoking observable IPSP response form a 10 Hz stimulus train at 1 mA. Evoked potentials were averaged. For reversal potential estimates, averages were smoothed and median filtered to remove spikes. The respiratory modulation of evoked IPSPs was measured as the difference of IPSP magnitudes between the preferred and nonpreferred phases.
GABAA receptor pharmacology.
Mitral/tufted (MT) cells (i.e., all mitral, tufted, and external tufted cells) were extracellularly recorded in response to ONL microstimulation before and 30 min after application of 5 μl of 5 mm bicuculline methiodide (BMI) to the OB surface (Yu et al., 1993). Cells were recorded extracellularly with glass pipettes and identified as MT cells if LOT shock elicited an antidromic spike or with a collision test (Schneider and Scott, 1983). Significance of spiking suppression was assessed with an unpaired Student's t test across all trials between pre-BMI and post-BMI responses within the time window defined by the epoch of baseline suppression in spiking.
Respiration phase of coupled spiking and membrane potentials.
The mean vector measured the degree of spike coupling with the respiration cycle. The mean spiking vector is a measure of the average phase (angle) and strength of correlation (vector strength: 0, uncorrelated; 1, perfect correlation) of the spontaneous spiking to the respiration cycle (Roux et al., 2006). The mean vector was defined by the best fit of the respiration-triggered peristimulus time histogram (PSTH) of all spontaneous spikes at 0 holding current (0°, 360° = peak of inspiration; 180° = peak of expiration). The active respiration phase was defined as the respiratory phase of peak membrane depolarization. The inactive phase was defined as +180° of phase after peak depolarization. The respiratory phase of greatest spiking was estimated by the mean vector direction using the circular statistics toolbox (Berens, 2009) in Matlab (version 2009a; MathWorks). The degree of coupling was estimated by the mean vector length (vector strength). Significant respiration coupling was defined by p < 0.05 for Rayleigh's test of non-uniformity. The rate of spiking (SR) modulation (Fig. 1C,D) was measured by the following:
The percentage change in respiratory modulation under conditions of artificial nasal airflow (n) and no nasal airflow (nn) was measured by
 |
for spiking and
 |
for membrane potential change. Significance of membrane potential oscillations in the absence of nasal airflow was assessed by a Student's t test between the SD and maximum depolarization of the smoothed (to remove spikes) and respiration-triggered (p < 0.01) membrane potential.
Figure 1.

Respiration-coupled spiking and membrane potential depolarizations in OB mitral and tufted cells. A, B, Identified mitral (Ai) and tufted (Bi) cells showed respiration-coupled spiking and membrane potential (Vm) depolarizations with respiration [Aii, Bii, unfiltered membrane potential (black), median-filtered membrane potential (blue), respiration trace at bottom: rising phase is inspiration, falling phase is expiration]. Aiii, Biii, The respiration-triggered spike raster (black) and PSTHs of these neurons showed a peak in spiking and a membrane potential depolarization during expiration (spiking in red, Vm in blue, mean with 1 SD confidence intervals from n = 50 respiration cycles). C, D, Highly respiration-coupled mitral and tufted cells, respectively (p < 0.05; see Results), showed maximal spiking (red) and membrane depolarization (blue) during the early expiratory phase, in which mitral cells showed a smaller peak firing rate than tufted cells. Note the Vm trace for tufted cells shows multiple peaks because there are neurons that were coupled to both inspiration and expiration. E, Distribution of vector strength with coupled respiratory phase showed similar vector strengths and coupled phases for mitral and tufted cells (mitral, n = 3; tufted, n = 5). Scale bars: Ai, Bi, 200 μm.
Statistical tests.
The Rayleigh's test for non-uniformity of circular data was used to asses significant respiration coupling of spiking at the p < 0.05 confidence level unless otherwise noted. This test rejects the null hypothesis that the respiratory-triggered spiking histogram is uniformly distributed for any non-uniform distribution. Pearson's correlation was used to quantify the significance of ONL distance with the largest observable IPSP at the p < 0.05 confidence level. Paired and unpaired one-sided and two-sided Student's t tests were used to test significance at the p < 0.05 confidence level unless otherwise noted.
Reversal potential estimates.
The reversal potential of both spontaneous postsynaptic potentials (PSPs) with respiration and evoked IPSPs from ONL microstimulation were measured by injecting at least three values (in addition to the zero-current resting condition) of hyperpolarizing direct current into the recorded neuron. The effect of hyperpolarization on the membrane potential and PSP magnitudes was measured (Sachdev et al., 2004). A linear regression of the PSP magnitude (ΔV = Vpeak − Vrest) as a function of resting membrane potential (Vmbaseline) was used to estimate the reversal potential of the PSPs by extrapolating the fit to 0 mV. Vmbaseline was measured 100 ms before either the peak depolarization (for the active phase) for spontaneous activity or 100 ms before the stimulus artifact for evoked responses (similar results were obtained using the average membrane potential across the respiration cycle; data not shown). The errors of the fit parameters of the ΔV versus Vmbaseline line were plotted at 2 SDs from the mean of the fit to estimate significance by the t statistics at the 95% confidence level (p < 0.05). We considered four hypothetical network mechanisms that could account for respiration-coupled spiking and membrane potential oscillations. (1) If respiration-coupled activity is caused by excitatory synaptic inputs from ORNs during the active phase and no synaptic input during the inactive phase, hyperpolarizing the membrane potential (Vmbaseline) should increase the magnitude of depolarizing PSPs (ΔV) during the active phase and the slope of the ΔV/Vmbaseline regression line should be negative in the active phase and 0, or negligible, in the inactive phase. (2) Respiration-coupled activity could also be produced by inhibition in the inactive phase generating rebound spiking in the active phase (Balu and Strowbridge, 2007). Hyperpolarizing the membrane potential below the reversal potential for inhibition should reveal a depolarizing PSP during the inactive phase (Buonviso et al., 2003; Fontanini et al., 2003) and should not affect the depolarization in the active phase; the slope of the ΔV/Vmbaseline regression should be negative in the inactive phase and zero in the active phase. (3) Respiration-coupled activity may also be produced by inhibition in the inactive phase suppressing uniform excitatory synaptic inputs throughout the respiration cycle. The ΔV/Vm baseline regression should show a negative slope in both the inactive and active phases, and the reversal potential in the inactive phase should indicate a mixture of excitation and inhibition. (4) Finally, respiration-coupled activity may be produced by inhibition in the inactive phase and excitation in the active phase. The ΔV/Vmbaseline regression line should have a negative slope in both the inactive and active phases; the reversal potential in the inactive phase should be more hyperpolarized than mechanism 3, indicating synaptic inhibition.
Driving force estimates.
To test whether IPSP differences with respiration were attributable to driving force changes from the baseline membrane potential with respiration, a linear conductance model estimated the expected IPSP differences as they varied with respiration phase. Zero millivolts was assumed as the reversal potential for excitation and −77 mV for inhibition. The difference in the baseline membrane potentials between respiration phases were compared with the PSP magnitude differences between respiration phases. If the PSP difference was larger than the baseline membrane potential difference, passive driving force changes could not explain the phenomenon.
Results
Spontaneous respiratory activity of mitral and tufted cells
To examine respiration-coupled spiking and membrane potential activity, intracellular recordings were obtained from 49 OB neurons in 34 adult rats. In 15 intact animals, we recorded from 25 neurons and identified three mitral and five tufted cells that showed highly significant respiration-coupled spiking (p < 0.01, Rayleigh's criterion). To examine the effect of nasal airflow on synaptic input to mitral and tufted cells, the remaining 19 animals were doubly tracheotomized. We recorded from 12 mitral and four tufted cells in these animals. The LOT was lesioned in five of the tracheotomized animals. We recorded from three mitral cells in these animals. To examine the excitatory and inhibitory synaptic components associated with membrane potential depolarizations during respiration, current was injected to hyperpolarize the membrane potential of 10 mitral and 7 tufted cells.
Cell types were determined by morphological reconstruction from intracellular injections of biotin (see Materials and Methods). Mitral cell somas were located in the mitral cell layer; they also had apical dendrites with a glomerular tuft (the glomerular tuft for the mitral cell from Fig. 1Ai was located in a preceding section), axons, and long lateral dendrites extending in the EPL. Tufted cells somas were located in the deep glomerular and EPLs and had apical glomerular dendritic tufts, lateral dendrites, and axons (Fig. 1Bi).
Respiration-coupled spiking was characterized by computing the mean spiking vector, a measure of the average phase (angle) and correlation (length) of spontaneous spikes to the respiration cycle. The length, or vector strength, ranged from 0 (maximally uncorrelated) to 1 (maximally correlated). This analysis categorizes neurons as respiration coupled or uncoupled and thus combines into one category the previously described single-peak simple coupled, excitatory synchronized, and suppressed synchronized categories (Buonviso et al., 1992). The majority of recorded and histologically identified mitral and tufted cells had a significant coupling of spontaneous spiking with the respiration cycle (7 of 14 mitral, 6 of 9 tufted) (Rayleigh's criterion, p < 0.05).
During normal nasal respiration, the spiking of mitral and tufted cells typically exhibited a preferred respiratory phase. For example, the individual mitral cell in Figure 1A fired spikes at the transition between expiration and inspiration with a vector strength of 0.15 and a preferred phase of 0 ± 50° (Fig. 1Aii,Aiii, p < 1e-12, Rayleigh's criterion, mean ± SD, expiration occurred from 0 to 180° and inspiration from 180 to 360°). The tufted cell in Figure 1B fired spikes during expiration, had a vector strength of 0.78, and had a preferred phase of 38 ± 13° (Fig. 1Bii,Biii, p < 1e-12, Rayleigh's criterion). Note that, in this example, respiration coupling in the tufted cell was stronger than in the mitral cell, because the tufted cell firing was heavily biased toward expiration. Significantly respiration-coupled neurons showed increases in spiking during expiration (n = 3 mitral, 5 tufted) or inspiration (n = 4 mitral, 1 tufted; p < 0.05, Rayleigh's criterion).
Highly significant respiration-coupled mitral and tufted cells showed maximal spiking and largest membrane depolarization during the early expiratory phase (Fig. 1C–E, p < 0.01, Rayleigh's criterion). Although the phase coupling of these strongly coupled cells was essentially identical, tufted cells achieved significantly higher peak spiking rates than mitral cells as measured by the maximum value of the average population firing rate (Table 1; p < 0.05 two-sided unpaired Student's t test). However, tufted and mitral cells showed similar average spiking rates and were similarly modulated with respiration (Table 1).
Table 1.
Summary of membrane potential and spiking activity results from mitral and tufted cells
| Avg [Vm] (mV) | ΔVm (mV) | Avg [Sp. rate] (Hz) | Max [Sp. rate] (Hz) | ΔSp. rate (Hz) | Vector strength | |
|---|---|---|---|---|---|---|
| Mitral intact | −51.3 ± 4.9 | 0.88 ± 1.08 | 15.9 ± 5.4 | 31.5 ± 10.6 | 15.6 ± 5.3 | 0.26 ± 0.11 |
| Tufted intact | −47.8 ± 5.3 | 0.71 ± 2.56 | 22.2 ± 14.2 | 57.8 ± 16.9 | 28.5 ± 30.6 | 0.47 ± 0.40 |
| Mitral trach. nasal | −54.6 ± 4.7 | 0.56 ± 0.54 | 12.2 ± 5.5 | 22.0 ± 91.9 | 59.7 ± 5.2 | 0.36 ± 0.25 |
| Mitral trach. nonasal | −54.2 ± 4.7 | 0.21 ± 0.37 | 1.5 ± 3.3* | 2.4 ± 4.8 | 20.4 ± 14.9* | 0.06 ± 0.09 |
| Tufted trach. nasal | −56.3 ± 9.2 | 0.20 ± 0.84 | 18.6 ± 4.2 | 24.6 ± 2.0 | 23.2 ± 2.2 | 0.10 ± 0.08 |
| Tufted trach. nonasal | −56.1 ± 6.9 | 0.08 ± 0.10 | 15.2 ± 8.6 | 12.6 ± 7.2 | 33.1 ± 15.8 | 0.01 ± 0.01 |
| Mitral LOT trach. nasal | −60.4 ± 1.3 | 0.81 ± 1.96 | 9.8 ± 4.9 | 21.9 ± 36.5 | 42.9 ± 33.2 | 0.35 ± 0.37 |
| Mitral LOT trach. nonasal | −57.8 ± 3.9 | 0.51 ± 0.89 | 9.5 ± 9.4 | 11.9 ± 22.1 | 15.1 ± 13.2 | 0.05 ± 0.05 |
* Indicates a significant decrease in the spiking rate observed during nasal airflow (p < 0.05, two-sided paired Student's t test, n = 5, see Results).
To compare the phasic activity between neurons, we divided the respiratory cycle into active and inactive phases: the active phase was the fraction of the respiration cycle associated with the membrane depolarization, and the inactive phase was the remaining part of the cycle (this combines inspiration and expiration-coupled cells into a normalized phase group; see Materials and Methods). We measured the duration of the active and inactive phases in strongly coupled mitral cells (Rayleigh's criterion, p < 0.05, n = 3). On average, these neurons were depolarized and increased their spiking rate for 200 ± 64 ms, followed by 300 ± 64 ms during which the neurons did not fire and were not depolarized. These epochs of activity and inactivity corresponded to a respiratory rate of two breaths per second. The respiration rate for our recordings ranged between 0.9 and 3 Hz on average, with a respiration cycle period from 0.3 to 1.1 s. Using an absolute time basis for averaging, the expiration- or inspiration-triggered activity generates similar results: the activity of mitral and tufted neurons showed respiration-related modulation (Cury and Uchida, 2010). However, the variability in respiration period across animals smears the average phasic relationship between spiking and the respiration rhythm.
The magnitudes of membrane potential depolarization from baseline associated with the active respiratory phase and the baseline membrane potentials themselves were not significantly different between mitral and tufted cells (Table 1).
Effects of nasal airflow on spontaneous activity
To determine whether respiration-coupled mitral and tufted cell activity is driven by nasal airflow, we recorded from mitral and tufted cells in animals that had undergone a double tracheotomy in which the artificial respiration was locked to the timing of intrinsic respiration (see Materials and Methods).
Artificial nasal breathing resulted in rhythmic depolarizations and spiking activities coupled to the cyclic pattern of the airflow in both mitral (Fig. 2A) and tufted (Fig. 2B) cells. During artificial nasal breathing, the spiking activity of these neurons remain phased locked to the respiration cycle. These example mitral and tufted cells had a vector strength of 0.78 and 0.15, respectively (p < 1e-12/p < 0.05, Rayleigh's criterion). When nasal airflow was removed and the animal continued to breathe without generating nasal airflow, phase locking was abolished (Fig. 2Aiii,Aiv, Biii,Biv). In the absence of nasal airflow, these example mitral and tufted cells had a vector strength of 0.09 and 0.04, respectively (p > 0.68/p > 0.95, Rayleigh's criterion).
Figure 2.
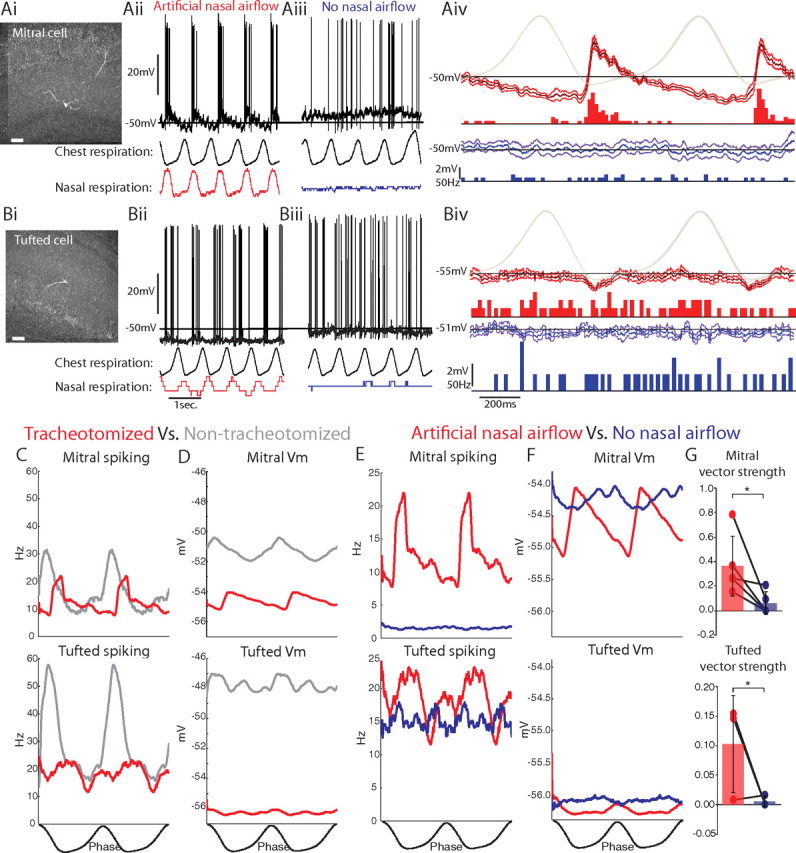
Spiking and membrane potential activities of mitral and tufted cells during artificial nasal airflow and without nasal airflow. Ai, Bi, Mitral and tufted cells, respectively, recorded and identified in double-tracheotomized animals, showed respiration-coupled membrane potential depolarizations and spiking during artificial nasal airflow (Aii, Bii) but not without nasal airflow (Aiii, Biii, unfiltered membrane potentials in black, chest and nasal respiration traces below). Aiv, Biv, Respiration-triggered average of the membrane potential (median filtered with 1 SD confidence intervals) and spiking PSTH during artificial nasal airflow (red) and without nasal airflow (blue, chest respiration shown in light gray). C, D, Populations of mitral and tufted cells recorded in tracheotomized animals (red) showed respiratory-coupled spiking (C) and membrane potential depolarizations (D) with artificial nasal airflow as observed in populations of non-tracheotomized animals (gray). E, Nasal airflow was necessary for respiration-coupled spiking in mitral and tufted cell populations. F, However, respiration-coupled membrane potential depolarizations were not significantly altered in the absence of nasal airflow. G, Vector strength decreased significantly without nasal airflow for both mitral and tufted cells (*p < 0.05 two-sided paired Student's t test; n = 11 mitral, n = 4 tufted). Scale bars: Ai, Bi, 100 μm.
Artificial nasal breathing reproduced the phenomenon of respiration-coupled spiking and membrane potential depolarizations as seen in the intact animal (p < 0.01, Rayleigh's criterion, nmitral = 5, ntufted = 3). Tracheotomies alone did not significantly alter the average spiking rate, membrane potential depolarization, or average resting membrane potential (Fig. 2C,D, Table 1). However, during artificial nasal breathing, tufted cells did not achieve the maximum spiking rates observed in nontracheotomized animals (Fig. 2C, p < 0.05, two-sided unpaired Student's t test, n = 3). Thus, artificial nasal breathing in the normal physiological range [500 ml/min (Ravel and Pager, 1990)] may not drive the spontaneous activity of mitral and tufted cells to the extent observed in the non-tracheotomized animals.
Removing nasal airflow completely abolished the respiration-coupled spiking observed during artificial nasal breathing in populations of both mitral and tufted cells [mitral: p < 1e-14 (nasal airflow), p > 0.77 (no nasal airflow), n = 5; tufted: p < 2e-6 (nasal airflow), p > 0.97 (without nasal airflow), n = 3, Rayleigh's test for non-uniformity] and significantly reduced the average vector strength of respiration-coupled spiking (mitral: −81 ± 36%, tufted: −95 ± 9%; Fig. 2G, p < 0.05, two-sided paired Student's t test). Thus, nasal airflow alone is critical for rhythmic, respiration-coupled spiking of mitral and tufted cells.
Interestingly, in the tracheotomized animals, removing nasal airflow did not significantly affect the magnitude of the average membrane potential depolarization (mitral: 0.56 ± 0.30 mV nasal airflow vs 0.21 ± 0.37 mV without nasal airflow, n = 11; tufted: 0.20 ± 0.84 vs 0.08 ± 0.10 mV, n = 5) or the resting membrane potentials in either cell type (Table 1). Respiration-coupled (6.18 ± 4.29 mV nasal airflow vs 2.87 ± 1.02 mV without nasal airflow, n = 4) and uncoupled (1.76 ± 0.91 mV nasal airflow vs 2.13 ± 0.74 mV without nasal airflow, n = 5) mitral cells both showed membrane potential oscillations in the absence of nasal airflow. Note, however, that these membrane potential depolarizations were significant in only three of nine mitral cells and in one of three tufted cells in double-tracheotomized animals in the absence of nasal airflow (p < 0.01, one-sided paired Student's t test). In contrast, in non-tracheotomized animals, respiration-coupled membrane potential depolarizations were significant in all strongly coupled neurons (Fig. 1, p < 0.01, one-sided paired Student's t test, n = 6). This suggests that cortical inputs or intrinsic membrane properties may also contribute to these membrane potential depolarizations.
In the double-tracheotomized animals, removing nasal airflow decreased the peak spiking rates for both cell types, nearly abolished average spiking in mitral but not tufted cells (Table 1, p < 0.05, two-sided paired Student's t test, n = 5), and reduced the spike rate modulation in mitral but not tufted cells (Table 1, p < 0.05, two-sided paired Student's t test, n = 5). Overall, removing nasal airflow had a smaller effect on the modulation of tufted cell spiking (+30 ± 159%) than on mitral cells (−66 ± 38%). Compared with mitral cells, tufted cells maintained similar average spiking rates after nasal airflow was removed (12 ± 73 and 82 ± 253% of spiking observed during nasal airflow, respectively).
Thus, removing nasal airflow over the olfactory epithelium abolished rhythmic spiking in both mitral and tufted cells, indicating that this receptor input is necessary for inducing respiration-coupled spiking in OB mitral and tufted cells. These results also indicate that intrinsic membrane properties and rhythmic synaptic input from olfactory cortical areas were not sufficient to generate rhythmic spiking of either cell type, although they may play a role in generating rhythmic membrane potential oscillations (Fontanini et al., 2003; Hayar et al., 2004b).
Effects of cortico-bulbar feedback on spontaneous activity
To assess the contributions of the cortico-bulbar feedback loop circuit on rhythmic activity in the OB, mitral cells were recorded after lesioning the LOT in double-tracheotomized animals, allowing us to compare spontaneous activities with and without nasal airflow in the absence of cortico-bulbar feedback (Fig. 3A) (Haberly and Bower, 1984). The absence of cortico-bulbar feedback did not abolish respiration-coupled spiking and membrane potential depolarizations in mitral cells during artificial nasal breathing (Fig. 3B, p < 0.01, Rayleigh's criterion, n = 3). As in the non-LOT lesioned mitral cells, removing nasal airflow abolished respiration-coupled spiking. For example, the mitral cell in Figure 3B had a vector strength of 0.75 (p < 1e-12, Rayleigh's criterion) during artificial nasal breathing and 0.0 (p = 1.0, Rayleigh's criterion) without nasal airflow attributable to a total absence of spiking. Mitral cells recorded with and without lesioned LOTs had similar peak spiking rates, average spiking rates, and membrane potential depolarizations with respiration (Fig. 3C,D, Table 1).
Although the LOT lesion did not affect the spiking properties of mitral cells, the average membrane potential of mitral cells in LOT-lesioned animals was more hyperpolarized (Table 1, p < 0.05, unpaired Student's t test, nLOT-lesion = 3, nnon-lesion = 8). One potential explanation for this effect is that the cortico-bulbar feedback circuit maintains these neurons at a constant depolarized membrane potential. Together, these results suggest that cortico-bulbar feedback does not generate respiration-coupled spiking or membrane potential depolarizations in mitral cells.
Next, we compared mitral cell activity in the LOT lesioned experiments when artificial nasal airflow was present or abolished. When artificial nasal airflow was abolished in LOT-lesioned animals (Fig. 3E,F, in blue), there was a significant reduction in the normalized vector strength of spiking of mitral cells (−86 ± 40%; Fig. 3G, p < 0.01, two-sided paired Student's t test, n = 3). Spiking was also no longer coupled to respiration in the absence of nasal airflow (p < 0.01, Rayleigh's test for non-uniformity, n = 3). When nasal airflow was abolished, the changes observed in the peak spiking rate, average spiking rate, and the average change in respiratory spiking rate modulation were not significantly different between LOT-lesioned and non-LOT-lesioned mitral cells (Table 1). As with non-LOT lesioned mitral cells, the membrane potential depolarizations did not change significantly, and the average membrane potentials were slightly more depolarized without nasal airflow (Fig. 3F, Table 1).
Synaptic mechanisms of respiration-coupled membrane potential depolarizations
To examine the mechanisms producing respiration-coupled spiking and membrane potential oscillations, we used current injection to hyperpolarize the membrane potential of mitral (Fig. 4A) and tufted (Fig. 4B) cells recorded in intact animals. These methods can be used to estimate the excitatory and inhibitory contributions to the spontaneous PSPs seen at rest.
Figure 4.

Excitatory and inhibitory synaptic inputs contribute to spontaneous respiration-coupled membrane potential oscillations. A, B, Mitral and tufted cells (respectively, from Fig. 1) showed spontaneously depolarizing PSPs in the respiration-triggered membrane potential without holding current (top trace in black, averaged over 50, 22 cycles and median filtered), which increased in size (ΔV) at hyperpolarized holding potentials (bottom traces) during the phase of maximal spiking seen in the PSTH (top black, respiration trace in gray, active phase: magenta/cyan; inactive phase: red/blue; T, respiration cycle period). C, A comparison of the slopes and estimated reversal potentials of ΔV versus Vmbaseline fits shows evidence for synaptic excitation and inhibition (see Materials and Methods). D, ΔV values in the active phase were strongly correlated with the vector strength of spontaneous respiration-coupled spiking in mitral cells. PSP sizes (ΔV) were measured as the difference between the peak and baseline membrane potentials (for the active phase, baseline measured 100 ms before the peak depolarization) or the difference between the membrane potential half a respiration cycle after the peak and the baseline membrane potential (for the inactive phase).
Neurons were selected for these analyses if their spiking rates were coupled to one phase of respiration, the resting membrane potential was stable and approximately −45 mV or more hyperpolarized, spikes crossed 0 mV and were related to a measurable depolarizing PSP, and, finally, the neuron remained stable even when it was held at three or more different hyperpolarized membrane potentials for at least 10 respiration cycles (six neurons satisfied this criteria, three mitral and three tufted cells). Hyperpolarization increased the magnitude of the depolarizing PSPs in the active phase of both cell types (Fig. 4A,B) but did not do so consistently for the mitral cells, possibly the result of active conductances. Hyperpolarization did not reveal evidence of reversed IPSPs during the inactive phases of mitral cells but did reveal a depolarizing tail in the inactive phase for tufted cells (Fig. 4B), suggesting that IPSPs occur during the inactive phase in tufted cells.
Our analysis of the slope and zero crossing of the PSPs revealed that depolarizing PSPs in the active phase of the mitral and tufted cells reversed at −3.5 ± 3.7 and −14.4 ± 10.5 mV, respectively (Fig. 4C), and both had significantly negative regression line slopes (p < 0.01, two-sided unpaired Student's t test). These reversal potentials correspond to EPSPs, which reverse at 0 mV, rather than IPSPs, which reverse at approximately −80 mV. Thus, spontaneous synaptic inputs to tufted cells in the active respiration phase were primarily excitatory (see Materials and Methods; p < 0.05, two-sided unpaired Student's t test, n = 3). In the inactive phase, hyperpolarization revealed PSPs in tufted cells only that reversed at −30.5 ± 34.5 mV (Fig. 4C) and had a significantly negative regression line slope, suggesting the presence of both excitatory and inhibitory inputs (p < 0.05, two-sided unpaired Student's t test, n = 2 of 3). Reversal potentials in the inactive phase could not be determined for the mitral cells likely because they received little synaptic input during the inactive phase. These data indicate that, in the four projection neurons in which reversal potentials were estimated, the spontaneous PSP in the active phase (during a depolarization) was excitatory. In the inactive phase, there was a mixture of both inhibitory and excitatory input to the tufted cells.
To further test for differences between mitral and tufted cells, the magnitudes of the depolarizations in the active phase (ΔV) were plotted against the strength of spontaneous respiration-coupled spiking (vector strength) for significantly coupled neurons (p < 0.05, Rayleigh's criterion). Mitral cells had larger active phase depolarizations for a given vector strength than tufted cells (Fig. 4D, p < 0.001, regression t statistic, nmitral = 9, ntufted = 9). Thus, when the membrane potential of mitral cells was depolarized in the active phase of respiration, their spiking became more correlated with respiration. Tufted cells did not show as strong a relationship with depolarization and respiratory-spike coupling. These results support a mechanism of excitatory synaptic inputs during the active phase driving respiration-coupled spiking in mitral cells.
Together, these results demonstrate that excitatory synaptic inputs resulting from nasal airflow at the periphery couple mitral and tufted cells spiking with respiration and cause most of the spontaneous spikes in mitral but not tufted cells. Although cortico-bulbar feedback is in a position to augment respiration-coupled spiking, it did not. However, whether this rhythmic input from the nasal peripheral affects sensory processing in the OB is not known. To understand whether respiration-coupled synaptic activity affects local synaptic circuit processing, we investigated whether the circuits producing lateral inhibition in the OB were modulated by respiration.
Evoked IPSP responses in mitral and tufted cells
To test for a respiratory modulation of lateral inhibition, mitral (n = 14) and tufted (n = 8) cells were recorded intracellularly, whereas IPSPs were evoked by electrical microstimulation of the ONL. Four stimulating electrodes were placed on the surface of the ONL ∼0.3–1.5 mm from the recording site (Fig. 5A) to stimulate spatially distinct, surround neuronal pools (Willhite et al., 2006; Kim et al., 2011). The specific threshold for evoking IPSPs (100 μA to several milliamps) varied greatly across neurons and within the same neuron from different ONL microstimulation electrodes. There was no significant difference between mitral and tufted peak IPSP latencies (mitral, 25.9 ± 23.9 ms; tufted, 19.0 ± 18.4 ms).
Figure 5.

ONL microstimulations evoke IPSPs in mitral and tufted cells. A, Dorsal view of the OB with ONL microelectrodes 1 to 4 (1 is the most proximal to the approximate recording locations, colored X symbols of the mitral and tufted cells in C and D). B, Putative neuronal circuits mediating IPSPs in mitral (red) and tufted (blue) cells. C, D, Evoked IPSPs in mitral and tufted cells from ONL stimulus 1 (traces are averages from 10 trials, the tufted cell was held with −80 pA of direct current to suppress spontaneous spiking). C, D, Insets, Histograms of the best ONL stimuli in 14 identified mitral and eight identified tufted cells showed a significant distance-dependent decrease in mitral cells only (p < 0.01 Pearson's correlation; see Results). E, The ionotropic GABAA receptors mediated the suppression of spiking after ONL stimulations in extracellularly recorded MT cells. Ei, PSTH after ONL microstimulation showed a suppression in spontaneous spiking (arrow). Eii, After application of 5 mm BMI, the suppressive response to ONL microstimulation was significantly reduced (arrow, n = 3). A, Anterior; P, posterior; M, medial; L, lateral; GL, glomerular layer; GrL, granule cell layer; MCL, mitral cell layer.
The canonical polysynaptic circuit for lateral inhibition between mitral cells is from inhibitory granule cells via dendrodendritic synapses, indicated in red in Figure 5B (Mori and Takagi, 1978; Jahr and Nicoll, 1980). Electrical microstimulation of olfactory receptor axons in the ONL (Bi) drives mitral cells (Bii) to spike, and action potentials backpropagate into the lateral dendrites, activating distal granule cells (Biii) (Xiong and Chen, 2002). Granule cells release GABA onto the recorded mitral cell (Biv) evoking an IPSP.
The response of a mitral cell to ONL microstimulation (Fig. 5C) was composed of an initial, fast depolarization (leading to spike initiation), followed by a long-lasting hyperpolarization produced by IPSPs (Getchell and Shepherd, 1975; Ezeh et al., 1993). IPSP magnitudes were measured as the difference between the membrane potential before stimulus and the membrane potential at the largest hyperpolarization (Vmbaseline, −56.2 mV; ΔV, 8.1 ± 0.3 mV; latency, 19.4 ms).
In tufted cells, in addition to the canonical circuit described above, lateral inhibition may be provided by the short axon SA cell or other inhibitory cell types in the glomerular layer and EPL (Fig. 5B, blue circuit) (Christie et al., 2001; Shao et al., 2009). Electrical stimulation of olfactory receptor axons in the ONL (Bi) drives tufted cells (Bii) to spike, the action potentials propagate into axon collaterals, driving distal short axon cells (Biii) (Hayar et al., 2004a), and the short axon cells inhibit the recorded tufted cell (Biv) (Kiyokage et al., 2010).
In a tufted cell (Fig. 5D), electrical stimulation evoked a small initial depolarization followed by a characteristic hyperpolarization after the stimulus artifact. The other electrodes did not evoke a significant IPSP (Vmbaseline, −50.7 mV; ΔV, 1.7 ± 0.1 mV; latency, 14.9 ms). The IPSPs evoked by ONL stimulation were distance dependent in mitral cells but not tufted cells (Fig. 5C,D, insets, p < 0.01, Pearson's correlation, nmitral = 14).
The effect of ONL microstimulation in producing inhibition was also assessed pharmacologically with extracellular recordings from MT cells. The activity of the same unit was followed during and after a 5 μl perfusion of 5 mm BMI (a competitive GABAA receptor antagonist) to the surface of the OB. As has been reported previously, BMI application had no effect on spontaneous spiking of the MT cells (Yu et al., 1993). ONL microstimulation evoked an artifact that lasted for several milliseconds (Fig. 5E), followed by a suppression of spontaneous spiking (arrow; 3–17 ms). This suppression was followed by an increase in spiking, corresponding temporally to the previously reported rebound spiking, from 17 to 35 ms (Balu and Strowbridge, 2007). The stimulus-evoked suppression of spiking in the 3–20 ms after the stimulus was significantly reduced after BMI application (p < 0.0015, two-sided paired Student's t test, n = 3). These results suggest that the suppression in spiking recorded extracellularly, and the corresponding IPSPs recorded intracellularly, are mediated by GABAA receptors, a known pathway for intrabulbar lateral inhibition (Jahr and Nicoll, 1980).
Respiratory modulation of evoked IPSP magnitudes
To test whether respiration modulated lateral inhibition, we synchronized each ONL microstimulation evoking a significant IPSP to inspiration (Fig. 6A, light gray) and expiration (dark gray; see Materials and Methods). Timing ONL microstimulation to respiratory phase modulated the magnitude of the evoked IPSP. The example mitral cell showed a 66% larger hyperpolarization during expiration compared with the stimulus-evoked hyperpolarization during inspiration (Fig. 6B: ΔVexpiration, 2.5 ± 0.3 mV; ΔVinspiration, 1.5 ± 0.2 mV; Vmbaseline inspiration, −54.4 ± 0.3 mV; expiration, −54.1 ± 0.3 mV; latency, 18.5 ms; 20, 20 trials at 5 mA stimulation). The respiratory modulation observed for individual ONL stimuli was not uniform across all ONL stimuli locations. For example, the mitral cell in Figure 6B showed a larger IPSP during expiration stimulation of ONL1 but showed a larger IPSP during inspiration stimulation of ONL3 (Fig. 6C: ΔVinspiration, 4.3 ± 0.5 mV; ΔVexpiration, 2.6 ± 0.2 mV; Vmbaseline: inspiration, −57.6 ± 0.3 mV and expiration, −57.6 ± 0.3 mV; latency, 12.5 ms; 20, 20 trials at 5 mA stimulation). In four neurons in which the respiratory phase significantly modulated evoked IPSP magnitudes from at least two ONL stimulation locations, three showed a significantly different phase preferences between electrode locations (p < 0.05, two-sided paired Student's t test). Thus, IPSPs evoked from different ONL stimulation locations did not preserve a global IPSP phase preference within the same neuron. In other words, different ONL microstimulation locations evoked maximal IPSPs at different phases of respiration. These results imply that specific lateral connections within the bulb are modulated differently with respiration.
Figure 6.

The respiratory phase of ONL microstimulations modulates evoked IPSP magnitudes in mitral and tufted cells. A, IPSPs were evoked by ONL microstimulation at two phases of respiration (inspiration, light gray; expiration, dark gray). Calibration bar, 100 ms. B, C, The same mitral cell showed a larger IPSP during expiration from ONL microstimulus 1 and a larger IPSP during inspiration from ONL microstimulus 3 (both stimuli, 5 mA). The IPSP was measured as the difference in the membrane potential before stimulus and at the largest evoked hyperpolarization (green and red vertical lines indicate preferred and nonpreferred IPSP measurements, respectively; averages are unfiltered) Note that this cell showed a higher spontaneous activity during stimulation with ONL3. D, Respiratory-modulated mitral and tufted cells showed a significant decrease in the normalized IPSP magnitude during the nonpreferred phase (mitral, 50.2% decrease, n = 7; tufted, 72.7% decrease, n = 7; p < 0.05, paired Student's t test, SEM). E, In tracheotomized animals, evoked IPSPs in mitral cells during the preferred phase showed a significant decrease in magnitude when artificial nasal airflow was removed (41.9%, *p < 0.05, paired Student's t test, SEM, nmitral = 11, ntufted = 4).
Respiration phase significantly modulated the evoked IPSP magnitude in 7 of 14 (50%) mitral cells and in 7 of 8 (88%) tufted cells (p < 0.01, two-sided paired Student's t test). In neurons that showed a significant respiratory modulation of evoked IPSPs, the stimulus evoked a twofold larger IPSP in mitral cells and a threefold to fourfold larger IPSP in tufted cells (preferred vs nonpreferred phase; Fig. 6D). However, there was no uniform preferred phase for larger IPSPs. Some were larger during inspiration (38% of mitral and 50% of tufted cells) and some during expiration (62% of mitral and 50% of tufted cells). Similarly, there was no significant relationship with the spontaneous activity: some neurons showed larger IPSPs during their active phase (38% of mitral and 50% of tufted cells); some showed larger IPSPs during their inactive phase (62% of mitral and 50% of tufted cells). Thus, just as there is no single active phase of respiration for neurons in the bulb (i.e., neurons can be active during inspiration or expiration), there is also no single phase of respiration in which ONL stimulation evokes larger IPSPs.
The variation in magnitudes of evoked IPSPs with respiration phase could have arisen from differences in sensory input, i.e., from differences in nasal airflow between the inspiration and expiration phases at the ORNs (Wilson and Sullivan, 1999). Conversely, the respiratory modulation of IPSPs could have arisen from centrifugal input, i.e., respiratory corollary discharge signals. Abolishing nasal airflow could help distinguish between these possibilities. Abolishing nasal airflow should eliminate the phase modulation effect if sensory input modulates the level of inhibition but should have no effect if centrifugal input modulates levels of inhibition. To test this possibility, IPSPs were evoked in mitral and tufted cells recorded in double-tracheotomized animals during both artificial nasal airflow and without nasal airflow. Just as in non-tracheotomized animals, in tracheotomized animals during artificial nasal breathing, the phase of respiration significantly modulated the magnitude of evoked IPSPs in mitral and tufted cells (p < 0.05, two-sided paired Student's t test, n = 7 of 11 mitral, n = 4 of 4 tufted). Even in animals with lesioned LOTs, the phase of respiration modulated the magnitude of evoked IPSPs in mitral cells during artificial nasal breathing (p < 0.05, two-sided paired Student's t test, n = 3).
To test whether the respiratory modulation of IPSPs arose from the activation of peripheral sensory inputs, we abolished nasal airflow. This significantly reduced the magnitude of the preferred IPSPs measured during artificial nasal airflow in mitral but not tufted cells (Fig. 6E, p < 0.05, one-sided paired Student's t test, nmitral = 11). Removing airflow also significantly reduced the respiratory modulation of IPSPs in tufted but not mitral cells (preferred − nonpreferred, p < 0.05, two-sided paired Student's t test, ntufted = 4). Together, these results suggest that respiration modulates lateral inhibition in mitral and tufted cells through functionally distinct neuronal circuits.
This modulation of lateral inhibition may result from differences in the amount of inhibition evoked in the two phases of respiration (i.e., fewer inhibitory or more excitatory inputs were activated during one phase), or the modulation may be related to the magnitude of the total inhibitory conductance (i.e., fewer inhibitory inputs were activated by the stimulus during one phase). The reversal potentials of the IPSPs evoked during both phases of respiration were used to distinguish between these mechanisms. The magnitude of the hyperpolarization evoked by ONL stimuli in both mitral and tufted cells, during both inspiration and expiration, decreased when the membrane potential was hyperpolarized relative to the resting potential (Fig. 7A). By holding each neuron at three to six membrane potentials during ONL stimulation, we estimated the reversal potentials of ONL evoked PSPs at their peak magnitude (typically 8–25 ms after the stimulus). Evoked PSPs in the example mitral cell in Figure 7A were estimated to reverse at −89.6 ± 21.8 mV during inspiration and −88.5 ± 20.5 mV during expiration, suggesting that the hyperpolarization induced by ONL stimulation results from synaptic inhibition (Fig. 7B). On average, there were no significant differences in the evoked PSP reversal potential between the two phases for individual neurons or populations (Fig. 7C; mitral, −78.5 ± 68.9 mV inspiration, −77.0 ± 71.8 mV expiration, n = 4; tufted: −77.4 ± 49.5 mV inspiration, −56.7 ± 39.9 mV expiration, n = 3).
Figure 7.

ONL-evoked responses show similar reversal potentials during inspiration and expiration. A, A mitral cell showed a larger evoked IPSP during expiration (dark trace; inspiration is light trace), which decreased in magnitude (ΔV) as the neuron was progressively hyperpolarized (below) (ΔV: expiration, 10.7 ± 0.2 mV; inspiration, 9.5 ± 0.2 mV; Vmbaseline: inspiration, −49.8 mV; expiration, −49.2 mV; latency, 17.0 ms, from 10 trials each with unfiltered traces). B, No significant differences in reversal potentials could be distinguished between the two phases within single cells (inspiration, −89.6 ± 21.8 mV; expiration, −88.5 ± 20.5 mV). C, Population data showed no significant difference in reversal potential (mitral cells inspiration: magenta, −78.5 ± 68.9 mV; expiration: red, −77.0 ± 71.8 mV; tufted cells inspiration: cyan, −77.4 ± 49.5 mV; expiration: blue, −56.7 ± 39.9 mV). D, The evoked IPSPs in the mitral cell in A, combining inspiration and expiration trials, reversed at −89 ± 12.4 mV. E, Population data showed reversals of −78 ± 37.8 mV (mitral) and −60 ± 50.1 mV (tufted) when combining inspiration and expiration trials. All traces were offset corrected.
To examine whether the ONL stimulation evoked more or less synaptic inhibition in mitral or tufted cells, data from both phases were combined and the reversal potentials determined for each cell class (Fig. 7D,E). The example mitral cell had a reversal potential of −89 ± 12.4 mV (Fig. 7D). The average reversal potential was −78 ± 37.8 mV for all mitral cells and −60 ± 50.1 mV for tufted cells (Fig. 7D, red, n = 4; Fig. 7E, blue, n = 3).
These results suggest that ONL stimulation evoked similar mixtures of excitatory and inhibitory conductances during both phases of respiration and that as seen in the slope of the ΔV versus Vmbaseline curves (Fig. 7B,C); the stimulus likely evokes more inhibition during the phase of respiration in which the larger IPSPs were observed. Alternatively, evoked IPSP magnitude differences with respiratory phase could have resulted from a difference in the driving force of inhibition, produced by the small membrane potential depolarization related to respiration. However, the small differences in membrane potentials between inspiration and expiration (|Vminspiration − Vmexpiration|) in mitral and tufted cells could not account for the measured IPSP differences (|ΔVinspiration − ΔVexpiration|) based on driving force alone in 11 of 31 neurons.
Discussion
The respiration cycle is emerging as the key context for functional processing in the olfactory system. Here, we provide new evidence that respiration drives mitral and tufted cell activity differentially and increases and modulates the efficacy of lateral inhibition. Although nasal airflow synchronized the spiking of identified mitral and tufted cells to respiratory-coupled excitatory synaptic input from the sensory epithelium, nasal airflow selectively induced spontaneous spiking in mitral but not tufted cells. Finally, direct measurements of IPSPs, evoked within the OB, show that respiration produced a modulation of lateral inhibition in mitral and tufted cells and increased the magnitude of lateral inhibition in mitral cells. Thus, respiration drives rhythmic spontaneous activity, modulates specific bulbar circuits, and establishes a structure for olfactory information processing.
Respiration-coupled activity in mitral and tufted cells
The passage of air over the olfactory epithelium modifies activity in the entire olfactory system. Nasal airflow is hypothesized to activate mechanical receptors of ORNs and modulate patterns of excitatory synaptic inputs to OB glomeruli (Spors and Grinvald, 2002; Grosmaitre et al., 2007; Carey et al., 2009; Oka et al., 2009). Previous extracellular work has also shown that nasal airflow couples spontaneous and odor-evoked spiking with respiration (Onoda and Mori, 1980; Sobel and Tank, 1993; Courtiol et al., 2011) and synchronizes odor responses (Adrian, 1950). Respiration-coupled bulbar activity is powerful enough to generate slow-wave oscillations in olfactory cortex neurons (Fontanini et al., 2003). Thus, nasal airflow generates activity that supports the encoding and processing of sensory information (Chaput et al., 1992; Philpot et al., 1997; Kepecs et al., 2006).
Our results show that nasal airflow produces respiration-coupled spiking in mitral and tufted cells through excitatory synaptic inputs from the nasal epithelium in anesthetized rats with slow and stable breathing rates. The reversal potential for the associated spontaneous PSPs was 0–15 mV for both mitral and tufted cells, consistent with the reversal potential for excitatory synaptic inputs. Abolishing nasal airflow shut down mitral cell spiking and abolished rhythmic spiking in tufted cells, also consistent with an excitatory synaptic drive to these cells. The different effects of nasal airflow on mitral and tufted cell spiking may arise from differences in the intrinsic membrane properties, local inputs, or cortical/modulatory inputs to these neurons.
Because there are no known differences in the sensory, local, cortical, or modulatory inputs to the mitral and tufted cells, differences in the intrinsic membrane properties may account for the effects of removing nasal airflow on mitral and tufted cell spiking. There are well-known differences in the mitral and tufted cells: tufted cells are smaller, genetically distinct, have shorter apical dendrites, have higher input resistances, have a lower threshold for action potentials, and are intrinsically active (Price and Powell, 1970; Greer and Shepherd, 1982; Macrides and Schneider, 1982; Hayar et al., 2004b; De Saint Jan et al., 2009). The differences in the intrinsic membrane properties of mitral and tufted cells could account for the differential effects of removing nasal airflow on their activity especially because tufted cells are intrinsically active in the absence of synaptic inputs (Hayar et al., 2004b).
Consistent with the idea that intrinsic membrane properties account for the effects of nasal airflow on mitral cells activity, when we removed cortico-bulbar feedback by lesioning the LOT, the respiration coupling of spiking and membrane potential depolarizations in mitral cells were not dramatically altered. Thus, synaptic inputs from cortico-bulbar feedback do not produce respiration-coupled spiking.
In a small set of neurons (4 of 12), rhythmic membrane potential depolarizations persisted even in the absence of nasal airflow and cortico-bulbar feedback. These respiration-coupled membrane potential depolarizations may arise from intrinsic subthreshold oscillations very near the respiration frequency (Desmaisons et al., 1999; Heyward et al., 2001). Although these membrane potential depolarizations were not reflected in the spiking output of the neurons, this subset of neurons may represent a subclass of the mitral and tufted cells that are strongly modulated by cortical, centrifugal, or other modulatory inputs (Fontanini et al., 2003; Laaris et al., 2007; Tsuno et al., 2008; Petzold et al., 2009).
Respiration modulates lateral inhibitory synaptic and network processing
The passage of air over the olfactory epithelium is thought to drive activity of inhibitory granule cells in-phase with the respiration cycle (Young and Wilson, 1999; Cang and Isaacson, 2003; Margrie and Schaefer, 2003). Lateral inhibition, via granule cell activity, is hypothesized to functionally synchronize mitral cell spiking to specific phases of the respiration cycle (Schaefer et al., 2006; Schoppa, 2006; Arevian et al., 2008; Giridhar et al., 2011), or ongoing gamma activity (Cenier et al., 2009; David et al., 2009), that results in sharpened odor responses (Tan et al., 2010) during active sniffing or during odor-guided behaviors (Verhagen et al., 2007; Carey et al., 2009). Lateral inhibition also differentially modulates the odor-evoked spiking phases of mitral cells within a single glomerulus. This effect of lateral inhibition may create both a rate and a phase code for olfactory information (Dhawale et al., 2010).
Our results in the anesthetized animal demonstrate that the magnitude of lateral inhibition evoked by ONL microstimulation is modulated during the respiration cycle in the majority of both mitral and tufted cells. In mitral cells, this respiratory modulation did not depend on airflow across the nasal epithelium or on an intact cortico-bulbar feedback circuit. However, in tufted cells, the respiratory modulation was greater in the presence of nasal airflow. Removing nasal airflow significantly decreased the magnitude of lateral inhibition at the preferred phase in mitral but not tufted cells. These results suggest that the neuronal circuits providing the respiratory modulation of lateral inhibition are functionally distinct between mitral and tufted cells. The respiratory modulation in tufted cells may arise from sensory inputs at the periphery resulting from nasal airflow, whereas the respiratory modulation of lateral inhibition in mitral cells may arise from cortical, centrifugal, or brainstem (respiratory corollary discharge) inputs arising through the olfactory peduncle.
Lateral inhibition was not universally stronger during inspiration or expiration; it was also not stronger when neurons were spontaneously active or more depolarized. These results are consistent with the observation that individual OB neurons are active during inspiration or expiration or may be unmodulated. Thus, although the respiration rhythm modulates lateral inhibition within single neurons, there is no single preferred phase for lateral inhibition in the OB. This modulation may depend on the animals' behavioral state, as the magnitude and the efficiency of various inputs change. Also note that, in a subset of experiments, a fast (<5 ms) stimulus-evoked spike could activate granule cells and cause reciprocal inhibition. The subsequent IPSP that occurred after the spike could consist of both a reciprocal and a lateral IPSP (Fig. 5C). Consistent with this, our results indicate that the evoked IPSPs were GABAA receptor-mediated and reversed near the chloride equilibrium potential (−78 mV; Schoppa, 2006) during both respiratory phases. These results indicate that the evoked hyperpolarization is inhibitory and that the stimulus evokes larger inhibitory conductances during the preferred phase of respiration.
Our results also suggest that olfactory stimuli could activate a single inhibitory circuit differently during inspiration and expiration, the phases in which orthonasal and retronasal processing occurs (Shepherd, 2006; Masaoka et al., 2010; Haehner et al., 2011). Functionally, this could be relevant for modulating contrast gain control, selectivity tuning, temporal tuning, and temporal decorrelation (Lei et al., 2002; Vucinic et al., 2006; Linster and Cleland, 2010; Tan et al., 2010; Giridhar et al., 2011).
Functional implications
Respiration drives and synchronizes the activity of the entire olfactory system. Odor-evoked responses, odor perception, and discrimination occur in the presence of respiration-coupled activities. Intentionally, or coincidently, sniffing and respiration are also thought to drive mechanical receptors of the ORNs and provide the olfactory system with continuously modulating levels of baseline sensory inputs (Ma, 2007). Respiration and sniffing are actively modulated in the awake animal; each breath alters the physical contact between the volatile odorants and the olfactory epithelium. By modulating the strength of lateral inhibition within OB circuits, respiration and sniffing could enhance the detection or discrimination of odors and selectively promote the synchrony of mitral or tufted cells through distinct functional circuits. Thus, respiration is not just a simple driver of baseline activity, but it also modulates specific bulbar circuits in a context-dependent manner and establishes a framework for the processing of olfactory information.
Footnotes
This work was supported by NIH/NIDCD Grants DC000086 (G.M.S.), DC008874 (D.C.W.), F31DC009921, and 5T32NS007224 (M.E.P.). We thank S. Gautam and J. Verhagen for technical assistance with the double tracheotomies, M. Fletcher, C. Greer, M. Krause, J. Mazer, M. McGinley, S. Nagayama, and E. Zagha for helpful comments, and C. Greer for use of the laser scanning confocal microscope.
The authors declare no competing financial interests.
References
- Adrian ED. The electrical activity of the mammalian olfactory bulb. Electroencephalogr Clin Neurophysiol. 1950;2:377–388. doi: 10.1016/0013-4694(50)90075-7. [DOI] [PubMed] [Google Scholar]
- Arevian AC, Kapoor V, Urban NN. Activity-dependent gating of lateral inhibition in the mouse olfactory bulb. Nat Neurosci. 2008;11:80–87. doi: 10.1038/nn2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu R, Strowbridge BW. Opposing inward and outward conductances regulate rebound discharges in olfactory mitral cells. J Neurophysiol. 2007;97:1959–1968. doi: 10.1152/jn.01115.2006. [DOI] [PubMed] [Google Scholar]
- Berens P. CircStat: a MATLAB toolbox for circular statistics. J Stat Software. 2009;31:1–21. [Google Scholar]
- Buonviso N, Chaput MA, Berthommier F. Temporal pattern analyses in pairs of neighboring mitral cells. J Neurophysiol. 1992;68:417–424. doi: 10.1152/jn.1992.68.2.417. [DOI] [PubMed] [Google Scholar]
- Buonviso N, Amat C, Litaudon P, Roux S, Royet JP, Farget V, Sicard G. Rhythm sequence through the olfactory bulb layers during the time window of a respiratory cycle. Eur J Neurosci. 2003;17:1811–1819. doi: 10.1046/j.1460-9568.2003.02619.x. [DOI] [PubMed] [Google Scholar]
- Cang J, Isaacson JS. In vivo whole-cell recording of odor-evoked synaptic transmission in the rat olfactory bulb. J Neurosci. 2003;23:4108–4116. doi: 10.1523/JNEUROSCI.23-10-04108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey RM, Verhagen JV, Wesson DW, Pírez N, Wachowiak M. Temporal structure of receptor neuron input to the olfactory bulb imaged in behaving rats. J Neurophysiol. 2009;101:1073–1088. doi: 10.1152/jn.90902.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenier T, David F, Litaudon P, Garcia S, Amat C, Buonviso N. Respiration-gated formation of gamma and beta neural assemblies in the mammalian olfactory bulb. Eur J Neurosci. 2009;29:921–930. doi: 10.1111/j.1460-9568.2009.06651.x. [DOI] [PubMed] [Google Scholar]
- Chaput MA, Buonviso N, Berthommier F. Temporal patterns in spontaneous and odour-evoked mitral cell discharges recorded in anaesthetized freely breathing animals. Eur J Neurosci. 1992;4:813–822. doi: 10.1111/j.1460-9568.1992.tb00191.x. [DOI] [PubMed] [Google Scholar]
- Christie JM, Schoppa NE, Westbrook GL. Tufted cell dendrodendritic inhibition in the olfactory bulb is dependent on NMDA receptor activity. J Neurophysiol. 2001;85:169–173. doi: 10.1152/jn.2001.85.1.169. [DOI] [PubMed] [Google Scholar]
- Courtiol E, Amat C, Thévenet M, Messaoudi B, Garcia S, Buonviso N. Reshaping of bulbar odor response by nasal flow rate in the rat. PLoS One. 2011;6:e16445. doi: 10.1371/journal.pone.0016445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cury KM, Uchida N. Robust odor coding via inhalation-coupled transient activity in the mammalian olfactory bulb. Neuron. 2010;68:570–585. doi: 10.1016/j.neuron.2010.09.040. [DOI] [PubMed] [Google Scholar]
- David FO, Hugues E, Cenier T, Fourcaud-Trocmé N, Buonviso N. Specific entrainment of mitral cells during gamma oscillation in the rat olfactory bulb. PLoS Comput Biol. 2009;5:e1000551. doi: 10.1371/journal.pcbi.1000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Saint Jan D, Hirnet D, Westbrook GL, Charpak S. External tufted cells drive the output of olfactory bulb glomeruli. J Neurosci. 2009;29:2043–2052. doi: 10.1523/JNEUROSCI.5317-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmaisons D, Vincent JD, Lledo PM. Control of action potential timing by intrinsic subthreshold oscillations in olfactory bulb output neurons. J Neurosci. 1999;19:10727–10737. doi: 10.1523/JNEUROSCI.19-24-10727.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawale AK, Hagiwara A, Bhalla US, Murthy VN, Albeanu DF. Non-redundant odor coding by sister mitral cells revealed by light addressable glomeruli in the mouse. Nat Neurosci. 2010;13:1404–1412. doi: 10.1038/nn.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezeh PI, Wellis DP, Scott JW. Organization of inhibition in the rat olfactory bulb external plexiform layer. J Neurophysiol. 1993;70:263–274. doi: 10.1152/jn.1993.70.1.263. [DOI] [PubMed] [Google Scholar]
- Fontanini A, Spano P, Bower JM. Ketamine-xylazine-induced slow (<1.5 Hz) oscillations in the rat piriform (olfactory) cortex are functionally correlated with respiration. J Neurosci. 2003;23:7993–8001. doi: 10.1523/JNEUROSCI.23-22-07993.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getchell TV, Shepherd GM. Synaptic actions on mitral and tufted cells elicited by olfactory nerve volleys in the rabbit. J Physiol. 1975;251:497–522. doi: 10.1113/jphysiol.1975.sp011105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giridhar S, Doiron B, Urban NN. Timescale-dependent shaping of correlation by olfactory bulb lateral inhibition. Proc Natl Acad Sci U S A. 2011;108:5843–5848. doi: 10.1073/pnas.1015165108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer CA, Shepherd GM. (1982). Mitral cell degeneration and sensory function in the neurological mutant mouse Purkinje cell degeneration (PCD) Brain Res. 1982;235:156–161. doi: 10.1016/0006-8993(82)90206-2. [DOI] [PubMed] [Google Scholar]
- Griff ER, Mafhouz M, Chaput MA. Comparison of identified mitral and tufted cells in freely breathing rats. II. Odor-evoked responses. Chem Senses. 2008;33:793–802. doi: 10.1093/chemse/bjn040. [DOI] [PubMed] [Google Scholar]
- Grosmaitre X, Santarelli LC, Tan J, Luo M, Ma M. Dual functions of mammalian olfactory sensory neurons as odor detectors and mechanical sensors. Nat Neurosci. 2007;10:348–354. doi: 10.1038/nn1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberly LB, Bower JM. Analysis of association fiber system in piriform cortex with intracellular recording and staining techniques. J Neurophysiol. 1984;51:90–112. doi: 10.1152/jn.1984.51.1.90. [DOI] [PubMed] [Google Scholar]
- Haehner A, Gruenewald G, Dibenedetto M, Hummel T. Responses to olfactory and intranasal trigeminal stimuli: relation to the respiratory cycle. Neuroscience. 2011;175:178–183. doi: 10.1016/j.neuroscience.2010.11.038. [DOI] [PubMed] [Google Scholar]
- Hayar A, Karnup S, Ennis M, Shipley MT. External tufted cells: a major excitatory element that coordinates glomerular activity. J Neurosci. 2004a;24:6676–6685. doi: 10.1523/JNEUROSCI.1367-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayar A, Karnup S, Shipley MT, Ennis M. Olfactory bulb glomeruli: external tufted cells intrinsically burst at theta frequency and are entrained by patterned olfactory input. J Neurosci. 2004b;24:1190–1199. doi: 10.1523/JNEUROSCI.4714-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyward P, Ennis M, Keller A, Shipley MT. Membrane bistability in olfactory bulb mitral cells. J Neurosci. 2001;21:5311–5320. doi: 10.1523/JNEUROSCI.21-14-05311.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahr CE, Nicoll RA. Dendrodendritic inhibition: demonstration with intracellular recording. Science. 1980;207:1473–1475. doi: 10.1126/science.7361098. [DOI] [PubMed] [Google Scholar]
- Kepecs A, Uchida N, Mainen ZF. The sniff as a unit of olfactory processing. Chem Senses. 2006;31:167–179. doi: 10.1093/chemse/bjj016. [DOI] [PubMed] [Google Scholar]
- Kim DH, Phillips ME, Chang AY, Patel HK, Nguyen KT, Willhite DC. Lateral connectivity in the olfactory bulb is sparse and segregated. Front Neural Circuits. 2011;5:5. doi: 10.3389/fncir.2011.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyokage E, Pan YZ, Shao Z, Kobayashi K, Szabo G, Yanagawa Y, Obata K, Okano H, Toida K, Puche AC, Shipley MT. Molecular identity of periglomerular and short axon cells. J Neurosci. 2010;30:1185–1196. doi: 10.1523/JNEUROSCI.3497-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laaris N, Puche A, Ennis M. Complementary postsynaptic activity patterns elicited in olfactory bulb by stimulation of mitral/tufted and centrifugal fiber inputs to granule cells. J Neurophysiol. 2007;97:296–306. doi: 10.1152/jn.00823.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei H, Christensen TA, Hildebrand JG. Local inhibition modulates odor-evoked synchronization of glomerulus-specific output neurons. Nat Neurosci. 2002;5:557–565. doi: 10.1038/nn0602-859. [DOI] [PubMed] [Google Scholar]
- Linster C, Cleland TA. Decorrelation of odor representations via spike timing-dependent plasticity. Front Comput Neurosci. 2010;4:157. doi: 10.3389/fncom.2010.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M. Encoding olfactory signals via multiple chemosensory systems. Crit Rev Biochem Mol Biol. 2007;42:463–480. doi: 10.1080/10409230701693359. [DOI] [PubMed] [Google Scholar]
- Macrides F, Chorover SL. Olfactory bulb units: activity correlated with inhalation cycles and odor quality. Science. 1972;175:84–87. doi: 10.1126/science.175.4017.84. [DOI] [PubMed] [Google Scholar]
- Macrides F, Schneider SP. Laminar organization of mitral and tufted cells in the main olfactory bulb of the adult hamster. J Comp Neurol. 1982;208:419–430. doi: 10.1002/cne.902080410. [DOI] [PubMed] [Google Scholar]
- Margrie TW, Schaefer AT. Theta oscillation coupled spike latencies yield computational vigour in a mammalian sensory system. J Physiol. 2003;546:363–374. doi: 10.1113/jphysiol.2002.031245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaoka Y, Satoh H, Akai L, Homma I. Expiration: the moment we experience retronasal olfaction in flavor. Neurosci Lett. 2010;473:92–96. doi: 10.1016/j.neulet.2010.02.024. [DOI] [PubMed] [Google Scholar]
- Mori K, Takagi SF. An intracellular study of dendrodendritic inhibitory synapses on mitral cells in the rabbit olfactory bulb. J Physiol. 1978;279:569–588. doi: 10.1113/jphysiol.1978.sp012362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama S, Takahashi YK, Yoshihara Y, Mori K. Mitral and tufted cells differ in the decoding manner of odor maps in the rat olfactory bulb. J Neurophysiol. 2004;91:2532–2540. doi: 10.1152/jn.01266.2003. [DOI] [PubMed] [Google Scholar]
- Nagayama S, Enerva A, Fletcher ML, Masurkar AV, Igarashi KM, Mori K, Chen WR. Differential axonal projection of mitral and tufted cells in the mouse main olfactory system. Front Neural Circuits. 2010;4:pii, 120. doi: 10.3389/fncir.2010.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najac M, De Saint Jan D, Reguero L, Grandes P, Charpak S. Monosynaptic and polysynaptic feed-forward inputs to mitral cells from olfactory sensory neurons. J Neurosci. 2011;31:8722–8729. doi: 10.1523/JNEUROSCI.0527-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka Y, Takai Y, Touhara K. Nasal airflow rate affects the sensitivity and pattern of glomerular odorant responses in the mouse olfactory bulb. J Neurosci. 2009;29:12070–12078. doi: 10.1523/JNEUROSCI.1415-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoda N, Mori K. Depth distribution of temporal firing patterns in olfactory bulb related to air-intake cycles. J Neurophysiol. 1980;44:29–39. doi: 10.1152/jn.1980.44.1.29. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 5. San Diego: Elsevier Academic; 2004. [Google Scholar]
- Petzold GC, Hagiwara A, Murthy VN. Serotonergic modulation of odor input to the mammalian olfactory bulb. Nat Neurosci. 2009;12:784–791. doi: 10.1038/nn.2335. [DOI] [PubMed] [Google Scholar]
- Philpot BD, Foster TC, Brunjes PC. Mitral/tufted cell activity is attenuated and becomes uncoupled from respiration following naris closure. J Neurobiol. 1997;33:374–386. doi: 10.1002/(sici)1097-4695(199710)33:4<374::aid-neu3>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Price JL, Powell TP. The synaptology of the granule cells of the olfactory bulb. J Cell Sci. 1970;7:125–155. doi: 10.1242/jcs.7.1.125. [DOI] [PubMed] [Google Scholar]
- Ravel N, Pager J. Respiratory patterning of the rat olfactory bulb unit activity: nasal versus tracheal breathing. Neurosci Lett. 1990;115:213–218. doi: 10.1016/0304-3940(90)90457-k. [DOI] [PubMed] [Google Scholar]
- Ros H, Sachdev RN, Yu Y, Sestan N, McCormick DA. Neocortical networks entrain neuronal circuits in cerebellar cortex. J Neurosci. 2009;29:10309–10320. doi: 10.1523/JNEUROSCI.2327-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux SG, Garcia S, Bertrand B, Cenier T, Vigouroux M, Buonviso N, Litaudon P. Respiratory cycle as time basis: an improved method for averaging olfactory neural events. J Neurosci Methods. 2006;152:173–178. doi: 10.1016/j.jneumeth.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Sachdev RN, Ebner FF, Wilson CJ. Effect of subthreshold up and down states on the whisker-evoked response in somatosensory cortex. J Neurophysiol. 2004;92:3511–3521. doi: 10.1152/jn.00347.2004. [DOI] [PubMed] [Google Scholar]
- Schaefer AT, Angelo K, Spors H, Margrie TW. Neuronal oscillations enhance stimulus discrimination by ensuring action potential precision. PLoS Biol. 2006;4:e163. doi: 10.1371/journal.pbio.0040163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider SP, Scott JW. Orthodromic response properties of rat olfactory bulb mitral and tufted cells correlate with their projection patterns. J Neurophysiol. 1983;50:358–378. doi: 10.1152/jn.1983.50.2.358. [DOI] [PubMed] [Google Scholar]
- Schoppa NE. Synchronization of olfactory bulb mitral cells by precisely timed inhibitory inputs. Neuron. 2006;49:271–283. doi: 10.1016/j.neuron.2005.11.038. [DOI] [PubMed] [Google Scholar]
- Scott JW. Electrophysiological identification of mitral and tufted cells and distributions of their axons in olfactory system of the rat. J Neurophysiol. 1981;46:918–931. doi: 10.1152/jn.1981.46.5.918. [DOI] [PubMed] [Google Scholar]
- Shao Z, Puche AC, Kiyokage E, Szabo G, Shipley MT. Two GABAergic intraglomerular circuits differentially regulate tonic and phasic presynaptic inhibition of olfactory nerve terminals. J Neurophysiol. 2009;101:1988–2001. doi: 10.1152/jn.91116.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM. Smell images and the flavour system in the human brain. Nature. 2006;444:316–321. doi: 10.1038/nature05405. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Haberly LB. Partial activation of olfactory bulb: analysis of field potentials and topographical relation between bulb and lateral olfactory tract. J Neurophysiol. 1970;33:643–653. doi: 10.1152/jn.1970.33.5.643. [DOI] [PubMed] [Google Scholar]
- Sobel EC, Tank DW. Timing of odor stimulation does not alter patterning of olfactory bulb unit activity in freely breathing rats. J Neurophysiol. 1993;69:1331–1337. doi: 10.1152/jn.1993.69.4.1331. [DOI] [PubMed] [Google Scholar]
- Spors H, Grinvald A. Spatio-temporal dynamics of odor representations in the mammalian olfactory bulb. Neuron. 2002;34:301–315. doi: 10.1016/s0896-6273(02)00644-x. [DOI] [PubMed] [Google Scholar]
- Tan J, Savigner A, Ma M, Luo M. Odor information processing by the olfactory bulb analyzed in gene-targeted mice. Neuron. 2010;65:912–926. doi: 10.1016/j.neuron.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuno Y, Kashiwadani H, Mori K. Behavioral state regulation of dendrodendritic synaptic inhibition in the olfactory bulb. J Neurosci. 2008;28:9227–9238. doi: 10.1523/JNEUROSCI.1576-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen JV, Wesson DW, Netoff TI, White JA, Wachowiak M. Sniffing controls an adaptive filter of sensory input to the olfactory bulb. Nat Neurosci. 2007;10:631–639. doi: 10.1038/nn1892. [DOI] [PubMed] [Google Scholar]
- Vucinić D, Cohen LB, Kosmidis EK. Interglomerular center-surround inhibition shapes odorant-evoked input to the mouse olfactory bulb in vivo. J Neurophysiol. 2006;95:1881–1887. doi: 10.1152/jn.00918.2005. [DOI] [PubMed] [Google Scholar]
- Walsh RR. Single spike activity in the olfactory bulb. Am J Physiol. 1956;186:255–257. doi: 10.1152/ajplegacy.1956.186.2.255. [DOI] [PubMed] [Google Scholar]
- Willhite DC, Nguyen KT, Masurkar AV, Greer CA, Shepherd GM, Chen WR. Viral tracing identifies distributed columnar organization in the olfactory bulb. Proc Natl Acad Sci U S A. 2006;103:12592–12597. doi: 10.1073/pnas.0602032103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DA, Sullivan RM. Respiratory airflow pattern at the rat's snout and an hypothesis regarding its role in olfaction. Physiol Behav. 1999;66:41–44. doi: 10.1016/s0031-9384(98)00269-8. [DOI] [PubMed] [Google Scholar]
- Xiong W, Chen WR. Dynamic gating of spike propagation in the mitral cell lateral dendrites. Neuron. 2002;34:115–126. doi: 10.1016/s0896-6273(02)00628-1. [DOI] [PubMed] [Google Scholar]
- Young TA, Wilson DA. Frequency-dependent modulation of inhibition in the rat olfactory bulb. Neurosci Lett. 1999;276:65–67. doi: 10.1016/s0304-3940(99)00781-8. [DOI] [PubMed] [Google Scholar]
- Yu GZ, Kaba H, Saito H, Seto K. Heterogeneous characteristics of mitral cells in the rat olfactory bulb. Brain Res Bull. 1993;31:701–706. doi: 10.1016/0361-9230(93)90144-z. [DOI] [PubMed] [Google Scholar]