

Abstract
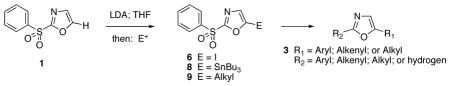
Deprotonation of 2-(phenylsulfonyl)-1,3-oxazole (1) readily provides a useful C-5 carbanion which is reactive with a variety of electrophiles. Aldehydes and ketones are useful substrates, and the formation of 5-iodo- and 5-tri-n-butylstannyl oxazoles affords access to cross-coupling reactions. Subsequent nucleophilic displacement of the 2-phenylsulfonyl group provides a general route for the synthesis of 2,5-disubstituted-1,3-oxazoles.
Oxazoles represent an important class of five-membered heterocycles.1 Continuing interest in the chemistry of 1,3-oxazoles has been undoubtedly stimulated by the incorporation of this ring system in a variety of biologically significant secondary metabolites, such as hennoxazole A,2 telomestatin,3 phorboxazoles A and B,4 the ulapualides,5 diazonamides A and B,6 and rhizopodin.7 Depsipeptides often contain 1,3-oxazoles as a result of oxidative cyclodehydrations of serine or threonine residues resulting in 2,4-disubstitution of the heterocycle.8 Additional examples of naturally occurring 1,3-oxazoles feature 2,5-disubstitution and 5-monosubstitution, and these substances may also exhibit significant biological activity.9
The proliferation of interesting structures within this family has inspired the development of methods to address the synthesis of specific substitution patterns. Williams and Wipf have devised oxidative cyclodehydration strategies to provide a general route for the de novo preparation of 2,4-disubstituted oxazoles.10 Several laboratories have recently described cross-coupling reactions for alkenylation and arylation at C-2 of the oxazole nucleus11 as well as studies of Stille reactions of 2-phenyl-1,3-oxazoles leading to C-4 and C-5 substitution.12 Direct metalations via deprotonations of the 1,3-oxazole ring have been studied in some cases.13 Removal of the C-2 hydrogen leads to an equilibrium of the ring-closed carbanion and the isomeric ring-opened isonitrile, and acylation reactions produce 4,5-disubstituted oxazoles derived from the Cornforth rearrangement.14 Examples of ring metalations via complex-induced proximity effects15 (CIPE) have been recorded in [2,4]bisoxazoles16 and for 2-methyloxazole-4-carboxylic acid.17 Very recently, Stambuli and coworkers have described the selective C-5 deprotonation of 2-methylthio-1,3-oxazole with tert-butyllithium leading to the production of 2,5-disubstituted oxazoles.18 These studies advance the earlier precedents of Shafer and Molinski and reports of the regioselective copper-mediated allylation of 2-(n-butylthio)-1,3-oxazole.19 In this letter, we disclose a compilation of our findings for the site selective deprotonation and the synthetic utility of 2-(phenylsulfonyl)-1,3-oxazole in processes of alkylation, alkenylation and arylation to afford a general pathway for the synthesis of 2,5-disubstituted oxazoles.
Direct incorporation of five-membered heterocycles into complex molecules is an important and fundamental strategy in medicinal chemsitry. Thus, we have explored the kinetic deprotonation of 2-(phenylsulfonyl)-1,3-oxazole (1) and the utility of carbanion 2. Reactions of 2 with a variety of electrophiles and the subsequent replacement of the 2-phenylsulfonyl group provide a general route for the preparation of 2,5-disubstituted oxazoles 3. Our studies are precedented by the selective (C-4) deprotonation of 1-[2′-(trimethylsilyl)ethoxymethyl]-2-(phenylsulfonyl)imidazole (4) and reactions with common electrophiles followed by mild removal of SEM as illustrated in the preparation of 5.20 Reductive desulfonylation with 2% Na(Hg) in methanol leads to the introduction of the intact imidazole nucleus.
The preparation of 1 began with the C-2 deprotonation of oxazole using n-butyllithium at −78 °C (THF). Diphenyldisulfide was introduced and reactions were allowed to stir at room temperature (24 h) to give 2-phenylthio-1,3-oxazole (91%) for oxidation with ammonium molybdate tetrahydrate (2.2 equiv) in ethanol containing 30% aqueous H2O2 at 22 °C (98% yield).
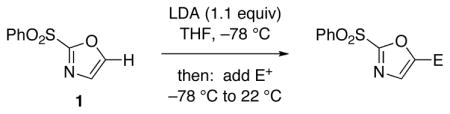
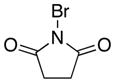
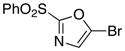
The selective deprotonation of 2-(phenylsulfonyl)-1,3-oxazole (1) with LDA (1.1 equiv) in THF at −78 °C afforded a well-behaved C-5 carbanion 2 which reacted efficiently with a number of electrophiles (Table 1). Under these reaction conditions, alkylation of 2 with allyl bromide proceeded poorly. This problem was overcome by transmetalation of 2 to give the corresponding organozinc species for a Negishi cross-coupling to yield the desired alkylation product 14 (90%).
Table 1.
Reactions of 2-(Phenylsulfonyl)-1,3-oxazole 1
| |||
|---|---|---|---|
| entry | electrophile | producta,b | yieldc (%) |
| 1 |
|
 6
6
|
87 |
| 2 |
|
 7
7
|
81 |
| 3 | n-Bu3SnCl |
|
86 |
| 4 | CH3I |
|
91 |
| 5 |
|
 10
10
|
72 |
| 6 |
|
 11
11
|
75 |
| 7 |
|
 12
12
|
87 |
| 8 |
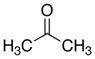
|
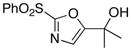 13
13
|
81 |
Reaction conditions: A THF solution of sulfone 1 (1.0 equiv; 2.0 mmol) was added into a solution of LDA (1.1 equiv) in THF at −78 °C. After stirring under N2 for 1 h, electrophile in THF solution was added with subsequent warming to 22 °C.
Products were purified by flash silica gel chromatography.
Yields are reported for isolated and purified product.
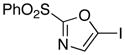
The efficient production of the iodide 6 (Table 1, entry 1) facilitates studies of Suzuki cross-coupling reactions toward the preparation of 2,5-disubstituted oxazoles. For example, the arylation reaction of 6 with commercially available 4-methoxyphenyl boronic acid using 10 mol% Pd(PPh3)4 at 70 °C gave 15 in 94% isolated yield. Similarly, our expectations that the 5-stannyl derivative 8 (Table 1, entry 3) would be useful for Stille processes have been confirmed by the effective cross-coupling with (Z)-iodide 16 21 using 10 mol% Pd(PPh3)4 in the presence of LiCl (6 equiv) and CuCl (5 equiv) in DMSO at 60 °C to yield oxazole 17 (90% yield).
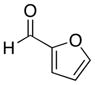
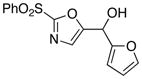
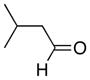
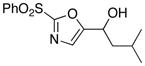
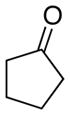
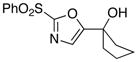
With the successful introduction of selective substitution at the C-5 position of 1, our subsequent studies focused on opportunities for the replacement of the 2-phenylsulfonyl group as a general scheme for the synthesis of 2,5-disubstituted-1,3-oxazoles. In this regard, we have found that organolithium species react via an addition-elimination pathway leading to the effective nucleophilic displacement of the C-2 sulfonyl group. Examples are compiled in Table 2. To our knowledge, this preparation of 1,3-oxazoles describes the first report for replacement of C-2 sulfonyl functionality utilizing aryl, alkenyl, and alkyllithium reagents. Small-scale reactions (approximately 1 molar) demonstrate complete consumption of starting 2-phenylsulfonyl oxazoles upon slow introduction of the organolithium species (1.2 equiv) and warming from 0 °C to room temperature over 5–10 minutes. We have briefly examined reactions of our 2-phenylsulfonyl oxazoles with the analogous cuprates and have observed an expected lack of reactivity at low temperatures whereas warming to 22 °C led to uncharacterized products which do not contain the 1,3-oxazole nucleus. Similar results were observed in our limited attempts to use allylmagnesium chloride. Overall, the replacement of the 2-sulfonyl substituent upon treatment with reactive organolithium reagents appears to be a general process which may be utilized for the introduction of additional functionalization as evident by reactions with acyl carbanion equivalents affording oxazoles 22, 23, and 28 (Table 3, entries 4, 5 and 10).
Table 2.
Preparation of 2,5-Disubstituted-1,3-oxazoles 3 via Displacement of the 2-Phenylsulfonyl Substituent
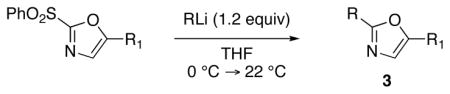
| ||||
|---|---|---|---|---|
| entry | lithium reagent | sulfone | oxazole producta,b | yieldc (%) |
| 1 |
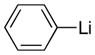
|
|
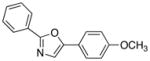 19
19
|
90 |
| 2 |
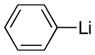
|
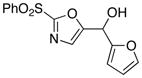 10
10
|
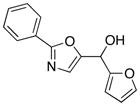 20
20
|
85 |
| 3 |
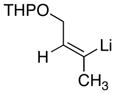
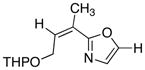
|
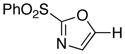 1
1
|
 21
21
|
78 |
| 4 |
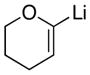
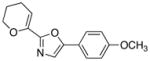
|
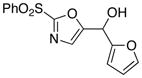 10
10
|
 22
22
|
71 |
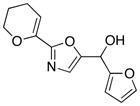
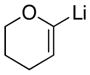
| 5 |
|
|
 23
23
|
83 |
| 6 |
|
|
|
69 |
| 7 |
|
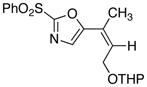 17
17
|
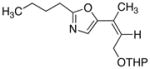 25
25
|
76 |
| 8 |
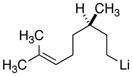
|
|
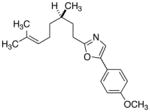 26
26
|
79 |
| 9 |
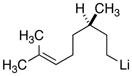
|
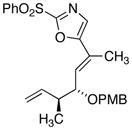 18d
18d
|
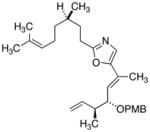 27
27
|
81 |
| 10 |
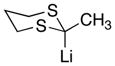
|
|
 28
28
|
82 |
Reaction conditions: Lithium reagents (1.2 equiv or 2.2 equiv in entries 2 and 4) were added under N2 to a THF solution of sulfones (0.053 mmol) at 0 °C. Reactions were generally complete within 5 minutes and warmed to 22 °C prior to addition of aq. NH4Cl.
Products were purified by flash silica gel chromatography.
All yields are reported for isolated and purified products.
Compound 18 was prepared via the Stille cross-coupling reaction as described for 17.
In summary, an efficient preparation of 2-(phenylsulfonyl)-1,3-oxazole facilitates a convenient, site-selective deprotonation to produce a reactive carbanion for halogenation, stannylation, and alkylation reactions. Negishi, Suzuki, and Stille cross-coupling processes are demonstrated to afford high yielding arylations and alkenylations leading to the preparation of a variety of 2,5-disubstituted oxazoles. The replacement of the 2-phenylsulfonyl substituent upon reaction with aryl, alkenyl, and akyllithium reagents provides a versatile pathway for the synthesis of a wide variety of complex 2,5-disubstituted oxazoles.
Supplementary Material
Scheme 1.
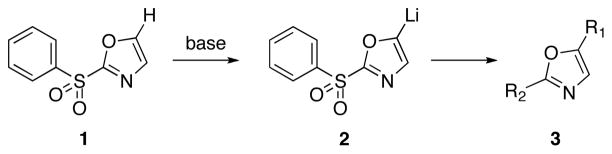
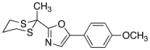
Site Selective Substitutions of 1,3-Oxazole
Scheme 2.
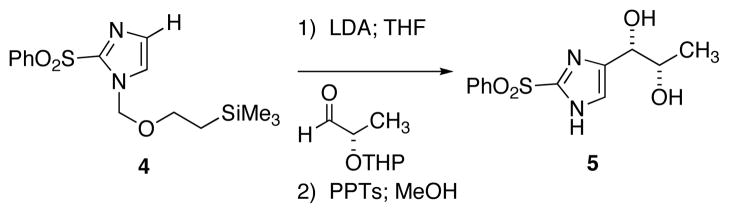
Selective C-4 Deprotonation of Imidaxzole 4
Scheme 3.
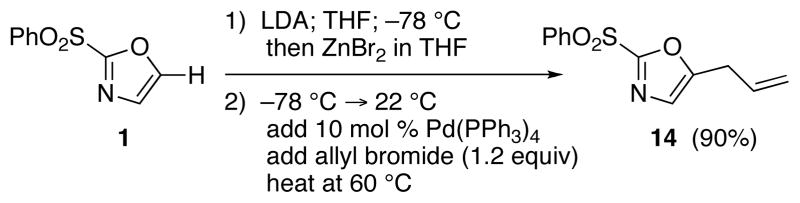
Negishi Coupling for C-5 Allylation of 1
Scheme 4.

Stille Cross Coupling Reactions
Acknowledgments
We acknowledge the support of Indiana University and partial support of the National Institutes of Health (GM042897). We acknowledge the assistance of Fese Mokube (Indiana University) for the preparation of compound 18.
Footnotes
Supporting Information Available Experimental procedures and characterization data, including proton and carbon NMR spectra for all products (64 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Taylor EC, Wipf P. Oxazoles: Synthesis, Reactions, and Spectroscopy. John Wiley & Sons; Hoboken: 2003. [Google Scholar]
- 2.Ichiba T, Yoshida WY, Scheuer PJ, Higa T, Gravalos DG. J Am Chem Soc. 1991;113:3173–3174. [Google Scholar]
- 3.Shin-ya K, Wierzba K, Matsuo K, Ohtani T, Yamada Y, Furihata K, Hayakawa Y, Seto H. J Am Chem Soc. 2001;123:1262–1263. doi: 10.1021/ja005780q. [DOI] [PubMed] [Google Scholar]
- 4.Searle PA, Molinski TF. J Am Chem Soc. 1995;117:8126–8131. [Google Scholar]
- 5.(a) Rosener JA, Scheuer PJ. J Am Chem Soc. 1986;108:846–847. [Google Scholar]; (b) Dalisay DS, Rogers EW, Edison AS, Molinski TF. J Nat Prod. 2009;72:732–738. doi: 10.1021/np8007649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For structure revision of diazonamide A: Li J, Burgett AWG, Esser L, Amezcua C, Harran PG. Angew Chem Int Ed. 2001;40:4770–4773. doi: 10.1002/1521-3773(20011217)40:24<4770::aid-anie4770>3.0.co;2-t.
- 7.Hagelueken G, Albrecht SC, Steinmetz H, Jansen R, Heinz DW, Kalesse M, Schubert WD. Angew Chem Int Ed. 2008;48:595–598. doi: 10.1002/anie.200802915. [DOI] [PubMed] [Google Scholar]
- 8.For examples: Kanoh K, Matsuo Y, Adachi K, Imagawa H, Nishizawa M, Shizuri Y. J Antibiot. 2005;58:289–292. doi: 10.1038/ja.2005.36.Perez LJ, Faulkner DJ. J Nat Prod. 2003;66:247–250. doi: 10.1021/np0204601.Kohno J, Kameda N, Nisho M, Kinumaki A, Komatsubara S. J Antibiot. 1996;49:1063–1065. doi: 10.7164/antibiotics.49.1063.
- 9.(a) Giddens AC, Boshoff HIM, Franzblau SG, Barry CE, III, Copp BR. Tetrahedron Lett. 2005;46:7355–7357. [Google Scholar]; (b) Fresneda PM, Castañeda M, Blug M, Molina P. Synlett. 2007:324–326. [Google Scholar]; (c) Shiomi K, Arai N, Shinose M, Takahashi Y, Yoshida H, Iwabuchi J, Tanaka Y, Omura S. J Antibiot. 1995;48:714–719. doi: 10.7164/antibiotics.48.714. [DOI] [PubMed] [Google Scholar]
- 10.Phillips AJ, Uto Y, Wipf P, Reno MJ, Williams DR. Org Lett. 2000;2:1165–1168. doi: 10.1021/ol005777b.For initial application of this methodology in the synthesis of hennoxazole A: Williams DR, Brooks DA, Berliner MA. J Am Chem Soc. 1999;121:1303–1305.
- 11.(a) Besselièvre F, Piguel S, Mahuteau-Betzer F, Grierson DS. Org Lett. 2008;10:4029–4032. doi: 10.1021/ol801512q. [DOI] [PubMed] [Google Scholar]; (b) Flegeau EF, Popkin ME, Greaney MF. Org Lett. 2008;10:2717–2720. doi: 10.1021/ol800869g. [DOI] [PubMed] [Google Scholar]; (c) Hodgetts KJ, Kershaw MT. Org Lett. 2002;4:2905–2907. doi: 10.1021/ol0262800. [DOI] [PubMed] [Google Scholar]; (d) Smith AB, III, Minbiole KP, Freeze S. Synlett. 2001:1739–1742. [Google Scholar]
- 12.Hämmerle J, Spina M, Schnürch M, Mihovilovic MD, Stanetty P. Synthesis. 2008:3099–3107. [Google Scholar]
- 13.For leading references: Vedejs E, Luchetta LM. J Org Chem. 1999;64:1011–1014. doi: 10.1021/jo981367d.Whitney SE, Rickborn B. J Org Chem. 1991;56:3058–3063.
- 14.Williams DR, McClymont EL. Tetrahedron Lett. 1993;34:7705–7708. [Google Scholar]
- 15.Whisler MC, MacNeil S, Sneickus V, Beak P. Angew Chem Int Ed. 2004;43:2206–2225. doi: 10.1002/anie.200300590. [DOI] [PubMed] [Google Scholar]
- 16.Williams DR, Brooks DA, Meyer KG. Tetrahedron Lett. 1998;39:8023–8026. [Google Scholar]
- 17.Meyers AI, Lawson JP. Tetrahedron Lett. 1981;22:3163–3166. [Google Scholar]
- 18.Lee K, Counceller CM, Stambuli JP. Org Lett. 2009;11:1457–1459. doi: 10.1021/ol900260g. [DOI] [PubMed] [Google Scholar]
- 19.(a) Shafer CM, Molinski TF. J Org Chem. 1998;65:551–555. doi: 10.1021/jo971410h. [DOI] [PubMed] [Google Scholar]; (b) Marino JP, Nguyn HN. Tetrahedron Lett. 2003;44:7395–7398. [Google Scholar]
- 20.Phillips JG, Fadnis L, Williams DR. Tetrahedron Lett. 1997;38:7835–7838. [Google Scholar]
- 21.Denmark SE, Pan W. J Organomet Chem. 2002;653:98–104. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.