

Abstract
INTRODUCTION
Cervical paragangliomas are slow-growing tumours that eventually cause lower cranial nerve palsies and infiltrate the skull base. Surgical treatment may cause the same deficits and, in some, risks more serious neurological deficits. We describe a classification used to guide investigation, consent and management of cervical paragangliomas based on extensive experience.
METHODS
The case notes of patients managed by the senior author at a tertiary referral skull base unit between 1987 and 2010 were reviewed retrospectively. A total of 87 cervical paragangliomas were identified in 70 patients (mean age: 46 years, range: 13–77 years). Of these, 35 patients had 36 vagal paragangliomas, 43 patients had 50 carotid body paragangliomas and 8 had both. One cervical paraganglioma arose from neither the carotid body nor the nodose ganglion. The main outcome measures were death, stroke, gastrostomy and tracheotomy.
RESULTS
All tumours were classified pre-operatively based on their relationship to the carotid artery, skull base and lower cranial nerves. Type 1 tumours were excised with a transcervical approach, type 2 with a transcervical-parotid approach and type 3 with a combined transcervical-parotid and infratemporal fossa approach. Type 4 patients underwent careful assessment and genetic counselling before any treatment was undertaken. There were no peri-operative deaths; two patients had strokes, one required a long-term feeding gastrostomy and none required a tracheotomy.
CONCLUSIONS
The use of a pre-operative classification system guides management and surgical approach, improves accuracy of consent, facilitates audit and clarifies which patients should be referred to specialised centres.
Keywords: Paraganglioma, Classification, Glomus tumour, Carotid artery
Paragangliomas are rare tumours that arise from paraganglia of neural crest origin. They have an incidence of approximately 1:300,000 and a third arise in the head and neck. The majority of the remainder are either adrenal or extra-adrenal tumours.1 A significant number of cervical paragangliomas are relatively asymptomatic, causing little more than a lump in the neck. The rest present with lower cranial nerve deficits that may result in a husky voice, dysphagia, mild dysarthria, Eustachian tube dysfunction and wasting of the trapezius.1
Although they synthesise and store catecholamines, only about 1% of head and neck paragangliomas are functional whereas most of those within the thorax, abdomen and pelvis secrete vasoactive amines and cause hypertension.1,2
Surgery has been considered the treatment of choice for cervical paragangliomas in selected patients but, in some parts of the world, radiotherapy or a watchful waiting policy is preferred. Indeed, there is mounting evidence that radiotherapy, whether conventionally fractionated or as a single treatment from the ‘gamma knife’ or ‘cyber knife’, will prevent further tumour growth at least within the temporal bone.3,4
If untreated, these slow-growing tumours are likely to cause a gradual loss of lower cranial nerve function with associated hoarseness and difficulty in swallowing. Certainly, morbidity increases with growth and management is extremely difficult once they have infiltrated the temporal bone. Intracranial extension is rare but has potentially devastating consequences.
That said, the life expectancy for patients with paragangliomas remains good, lower cranial neuropathies of gradual onset are generally well tolerated and the growth rate of tumours may be so slow as to appear negligible for years, in some cases well beyond the professional lifespan of the diagnosing clinician. However, the life expectancy of patients with surgically treated carotid body tumours also matches that of the normal population.5
The challenge for advocates of surgical management is to demonstrate morbidity and mortality as low as that associated with radiation therapy, with the advantageous outcome of tumour removal as opposed to simply the cessation or slowing of tumour growth. The key to this lies in both accurate pre-operative appraisal and minimising the risks associated with surgery.
The goals of any classification are to guide surgical approach and management, allow comparison of outcomes and facilitate consent informed by the risks associated with a particular tumour. Head and neck paragangliomas may be divided into two main groups: cervical and temporal. Cervical paragangliomas include carotid and vagal paragangliomas (carotid body tumour and glomus vagale), and temporal comprise jugular and tympanic paragangliomas (glomus jugulare and tympanicum). The most common paraganglioma is that of the carotid body.6
In 1979 Oldring and Fisch described a classification system for temporal paragangliomas that fulfilled these goals7 but no similar pre-operative classification system exists for cervical paragangliomas. We have developed a classification system that has been used in our department to guide the investigation and management of these tumours. We present it here illustrated by our case series of cervical paragangliomas.
Methods
Patients
A prospectively maintained database of skull base patients managed by the senior author from 1987 to 2010 was consulted to identify patients with paragangliomas arising in the neck. Paragangliomas with their epicentre in the temporal bone and extending into the neck were excluded from the analysis.
A total of 70 patients were identified with 87 cervical paragangliomas. Of these, 35 patients had 36 vagal paragangliomas and 42 patients had 50 carotid body paragangliomas. Eight patients had both tumours. One cervical paraganglioma arose from neither the carotid body nor the nodose ganglion. There were 27 men and 43 women in the series with an age range of 13–77 years and a mean age of 46 years (Table 1). Those with multiple or familial tumours and those with malignant tumours tended to be younger than those with benign sporadic tumours (Fig 1).
Table 1.
Patient demographics
| Sex | |
| Female | 43 (61%) |
| Male | 27 (39%) |
| Age at diagnosis (years) | |
| Mean | 46 |
| Standard deviation | 17 |
| Paraganglioma site of origin | |
| Carotid | 50 |
| Vagal | 36 |
| Other | 1 |
| Classification (patients) | |
| 1 | 11 |
| 2 | 14 |
| 3 | 24 |
| 4 | 21 |
| Modality of treatment (tumours)* | |
| Surgery | 57 |
| Observation | 20 |
| Radiation | 11 |
| Embolisation alone | 1 |
Tumours may have multiple modalities of treatment.
Figure 1.
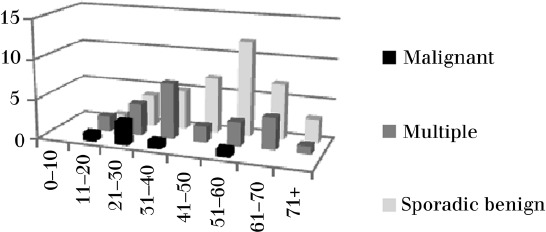
Age distribution of cervical paragangliomas
The patient pathway
Our patients typically present with a cervical or parapharyngeal mass and are referred to us with preliminary imaging data, either ultrasonography or computed tomography (CT). When seen in clinic, a comprehensive history is taken and a clinical examination performed. The history includes an exhaustive search for a genetic pedigree. Existing image data are reviewed and additional scans are arranged as necessary. Those patients who have a positive family history or present with multiple tumours are referred for genetic assessment.
All patients have fludeoxyglucose positron emission tomography (FDG PET) to exclude multiple lesions, 24-hour urinary catecholamine assessment and magnetic resonance imaging (MRI) of the head and neck to assess the degree to which the internal carotid artery (ICA) is encased by the tumour and its distance from the skull base. The MRI also adds specificity to the pre-operative diagnosis of a paraganglioma with the identification of a ‘salt and pepper’ pattern of T1w and T2w hyperintensity and hypointensity.
Tumours are classified as per Table 2. All patients with type 2 and 3 tumours have a CT angiogram and those with type 3 tumours undergo carotid angiography with cross-flow studies if the CT angiogram is equivocal. Trial balloon occlusion is undertaken on those patients when the carotid artery is likely to be lost and an interposition graft deemed impossible.
Table 2.
Classification of cervical paragangliomas. Each tumour is classified as type 1–4, with the suffix ‘c’ or ‘v’ to indicate whether the origin is from the carotid body or ganglion nodosum (vagale). Note that multiple tumours are type 4 regardless of their size
| Type 1 | Confined to within 2cm of carotid bifurcation and encases <50% of internal carotid artery and no cranial nerve deficits |
| Type 2 | Extends beyond 2cm from carotid bifurcation or encases >50% of internal carotid artery or associated cranial nerve deficits and rostral margin >2cm from skull base |
| Type 3 | Extending to within 2cm of or through skull base |
| Type 4 | Bilateral/multiple tumours |
A decision is then made on whether to advise tumour resection based on: patient choice; biological age; co-morbidities; tumour size and number (solitary or multiple); associated cranial nerve deficits; stage of disease; cerebral circulation; and potential for cross-flow.
Surgical approach
Type 1 tumours were excised using a transcervical approach (Fig 2) and type 2 tumours were resected using an extended transcervical-parotid approach. Removal or reflection of the lower pole of the parotid gland, together with mobilisation of the attachment of the sternomastoid to the mastoid tip, provides significant additional exposure of the ICA that facilitates superior vascular control (Fig 3). For type 2 and type 3 lesions, the potential need for arterial resection and replacement should be considered.
Figure 2.
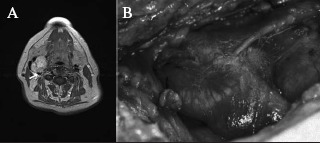
Type 1 tumour: T2 weighted axial image (A) with an arrow indicating the tumour abutting <50% of the internal carotid artery perimeter (note the characteristic tumoural internal vascular flow void) and operative photograph of the tumour (B) encasing <50% of the internal carotid artery
Figure 3.
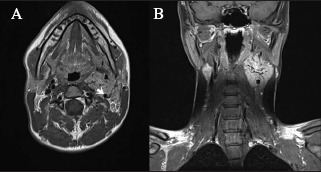
Type 2 tumour: T1 weighted axial (A) and coronal (B) magnetic resonance imaging of the tumour encasing >50% of internal carotid artery but >2cm from skull base. Arrow indicates internal carotid artery.
Type 3 tumours may sometimes be excised completely through a transcervical-parotid approach. However, consideration should also be given to infratemporal, far lateral or juxtacondylar approaches.8–11 It is a mistake to approach a tumour that approximates but does not enter the skull base in a manner that does not allow adequate distal control of the ICA (Fig 4). The choice between these approaches is dictated by the relationship of the upper limit of the tumour to the temporal bone and the functional status of the ear.
Figure 4.
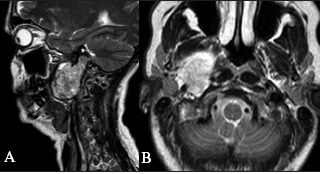
Type 3 tumour: sagittal (A) and axial (B) T2 weighted images of the tumour abutting the skull base and extending into the jugular foramen
Pre-operative embolisation is considered and employed only with very large tumours. In the event of an abnormal cerebral circulation and an extensive tumour that needs to be removed, a high flow bypass may be required.
The full complement of paragangliomas should be sought in all type 4 patients as the implications of surgical treatment can be far reaching. The removal of these tumours should not be rushed; preservation of at least one vagus and hearing should be the priority (Fig 5). The surgical approach for type 4 patients is dictated by the subcategory of each individual tumour.
Figure 5.
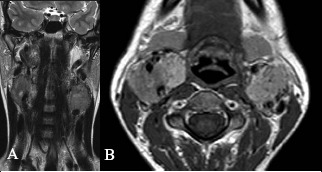
Type 4 tumour: T2 weighted coronal (A) and post gadolinium T1 weighted axial (B) images showing bilateral carotid body paragangliomas and a right vagal paraganglioma
In the event of a tumour proving impossible to dissect free of the carotid artery, our preference is to leave small amounts of tumour attached to the artery wall rather than resect the artery at that stage. On the other hand, gross disease inseparable from the artery does dictate that an arterial resection and graft are required. Small volumes of residual tumour are monitored by interval MRI or ultrasonography.
The external carotid artery is commonly sacrificed if encased by the tumour; its peripheral branches are then ligated as they emerge from the tumour. Particular care must be taken when attempting to ligate the external carotid close to the carotid bifurcation. Attention must be given to preserving the vagus, in particular avoiding traction, compression and heat injury. Similarly, the superior laryngeal nerve, which runs medial to the internal carotid artery, is also vulnerable. Damage to this nerve increases the likelihood of aspiration. Releasing the head of the sternomastoid from the mastoid tip may facilitate dissection and reduces the risk of traction-induced neuropathy, particularly of the accessory nerve.
Results
Vascular events/mortality
Table 3summarises the management and complications involving the ICA. There were no deaths in the immediate post-operative period. Five of the tumours were malignant; one patient with malignant disease died and another died of unrelated causes. Our impression is that patients with malignant tumours presented at a younger age and the disease followed a more aggressive course but the limited number of patients in our series means that this difference was not statistically significant (p=0.15).
Table 3.
Internal carotid artery interventions and outcomes
| Age (years) | Tumour classification | Intervention | Planned? | Outcome | Note |
|---|---|---|---|---|---|
| 60 | 3v | Internal carotid artery resection with primary anastomosis | No | No morbidity | |
| 52 | 3c | Interposition graft | Yes | No morbidity | |
| 70 | 2c | Interposition graft | Yes | Stroke | Thrombosed day 2 |
| 22 | 2v | Interposition graft | No | No morbidity | |
| 54 | 3c | Radiological occlusion and resection | Yes | Stroke | Incomplete pre-operative occlusion |
| 14 | 3v | Radiological occlusion and resection | No | No morbidity |
v = vagal paraganglioma; c = carotid paraganglioma
Morbidity/neural deficits
Those patients with vagal paragangliomas who did not have a pre-operative vagal palsy lost ipsilateral vagal function at the time of tumour resection.
Of eleven patients with type 1 tumours, nine underwent excision with no recurrence or acquired neural deficit. The tumours in the other two patients were managed conservatively by clinical observation with serial scans.
Eleven patients with type 2 tumours underwent surgery, with one recurrence (in a patient with a malignant tumour), and three were managed conservatively. One patient suffered from first bite pain and two patients acquired vagal palsies (one vagal paraganglioma 2v, one carotid 2c). One patient who had a planned carotid interposition graft developed a stroke on the second post-operative day as a result of graft thrombosis.
Twenty-four patients had type 3 tumours, of which nineteen were excised. Five patients were treated with radiotherapy, which followed surgery in three cases where residual disease demonstrated growth. Eight patients developed new lower cranial nerve palsies following surgery. In two cases this was limited to the loss of a previously working vagus nerve following the resection of a vagal tumour. One patient with an extremely large type 3 vagal paraganglioma required the insertion of a percutaneous gastrostomy tube for long-term feeding. He found swallowing impossible with ipsilateral vagal and hypoglossal palsies and also tended to aspirate. One patient developed a stroke in the peri-operative period despite pre-operative carotid balloon occlusion.
Of 21 patients with 39 type 4 tumours, 22 tumours were excised, 15 were managed conservatively, 1 was treated primarily with gamma knife radiotherapy and 1 with embolisation alone. Our caution in managing this group meant that the only new cranial neuropathy caused by resection was an accessory nerve palsy.
First bite syndrome
First bite pain was experienced by the majority of patients with type 3 tumours. This generally settled over time and was eased by pregabalin or gabapentin prescribed for a period of up to 12 months. Dietary adjustments, avoiding sharp tasting foods or citrus drinks were also found to be helpful by some. Persistent first bite pain was an issue for five patients, all of whom had type 3 tumours, one of which originated from the carotid body.
Horner's syndrome
Disruption of cervical sympathetic fibres lead to the development of Horner's syndrome in eight patients but was not problematic for anyone.
Discussion
The 2004 World Health Organization classification of endocrine tumours defined head and neck paragangliomas as:
‘[…] tumours arising from the paraganglia distributed along the parasympathetic nerves in the head, neck and mediastinum. These tumours are histologically identical and are named by anatomical site of origin.’12
Classification of paragangliomas based on site of origin groups cervical paragangliomas arising from the carotid body in one group and those arising from the ganglion nodosum, known as glomus vagale or vagal paragangliomas, in another. In practice, the risks of surgery for both groups depend on their relationship to the ICA, the lower cranial nerves and the skull base. It is therefore rational to use one classification system for both of these groups of cervical paragangliomas, maximising the number of patients in each subset and hence the utility of the classification. We believe our classification addresses these shortcomings. We have found it a useful guide to pre-operative investigation, surgical approach and informed consent.
The morbidity and mortality associated with surgery has reduced dramatically over the last 50 years. In Elder's 1962 review of paragangliomas treated in Holland, 6% of the patients who underwent attempted resection of carotid body paragangliomas died. Furthermore, 35% of patients developed central neurological symptoms that only resolved in a quarter of those affected.13
The mortality associated with resection of cervical paragangliomas is almost entirely due to their intimate relationship to the ICA. Management of ICA injury is particularly difficult should the injury be sustained at the skull base. It is essential to assess this risk pre-operatively and plan the surgical approach carefully to ensure adequate exposure of the ICA lest reconstruction is required. This may necessitate a transtemporal approach so that the ascending or horizontal carotid artery can be exposed within the skull base.
In the series presented here, there was no peri-operative mortality and a stroke rate of 4.5% (two patients). It is important to note that both of these strokes developed in patients with large, rapidly growing tumours for whom the risk was identified prior to surgery. The potential risk was discussed with both patients and their carers very carefully beforehand. In order to do this, it is essential to have a reliable method of pre-operative assessment. We advise the use of cross-sectional imaging, CT and conventional angiography as well as trial balloon occlusion if necessary. Definitive balloon occlusion following a successful trial should allow safe resection of the ICA but this cannot be guaranteed. The stroke in our series following a suboptimal balloon occlusion demonstrated that this is not always the case.
Vascular duplex ultrasonography has been advocated by some and provides excellent imaging of the cervical carotid.14 It is less useful intracranially and does not provide information regarding the patency of the circle of Willis, which may influence intraoperative decisions.
Shamblin et al proposed a classification of carotid body tumours that relied on intraoperative and histopathological findings.15 They analysed 90 carotid body tumours and advocated a 3-point scheme in which low-grade tumours were easily dissected from the carotid artery while high-grade tumours infiltrated the arterial wall and could not be removed completely without resecting the artery. This scheme allowed for a comparison of outcomes but failed to address the risk of cranial nerve involvement or proximity to the skull base.9 Its retrospective nature meant it could not guide surgical approach or inform consent and hence its utility is limited. Despite this disadvantage, it is commonly employed but used incorrectly.
A pre-operative, radiological ‘Shamblin’ classification has been proposed by Arya et al.16 Their classification only applies to carotid paragangliomas and their relationship to the carotid artery. In other words, it only addresses one aspect of the surgery. Our classification scheme accurately identified those at increased risk of stroke and neural deficits.
The morbidity of surgery is largely caused by acquired lower cranial nerve palsies. The associated acute speech and swallowing problems have a very significant impact on quality of life. Of particular note is the intimate relationship of the vagus nerve with vagal paragangliomas, which develop within the nerve. Although there are reports of preservation of vagal integrity following resection of these tumours, most of the authors of such reports (eg Kollert et al)17 concede that function is inevitably lost. In our experience, the nerve is grossly infiltrated as it enters and exits the tumour, and no plane for dissection exists between it and the tumour. We therefore counsel patients and plan surgery on the assumption that vagal function, if present pre-operatively, will be lost if these tumours are resected. This is of particular relevance for patients with bilateral tumours.
Although sporadic cases account for the majority, in recent years it has been appreciated that up to 50% of paragangliomas may be familial, linked to a number of gene mutations that encode subunits of the succinate dehydrogenase enzymes.1,6,18,19 In familial cases, bilateral and multiple tumours are often present.
The use of either octreotide or iodine-131-metaiodobenzylguanidine has been advocated in identifying synchronous paragangliomas. We have found the uptake of these isotopes relatively inconsistent in cervical paragangliomas.20,21 Our preference is therefore to identify synchronous tumours using FDG PET.
Many familial cases can go undetected for generations owing to difficulties in diagnosis, missed tumours, incomplete gene penetrance and genomic imprinting.18 Familial cases tend to present at a younger age and are not subject to the female preponderance evident in sporadic paragangliomas.18 The most common mutation is to the succinate dehydrogenase subunit D (SDHD). While mutations of this gene may be transmitted from either parent, the disease phenotype is only expressed when the gene is paternal in origin. The SDHD gene is therefore subject to maternal imprinting. Mutations to subunits B and C are not subject to imprinting and hence are expressed when inherited from either parent. Patients with this latter mutation tend to have more aggressive disease and are liable to develop abdominal tumours. This must be considered when taking a family history in order to allow accurate assessment of risk.
The association between the development of paragangliomas and von Hippel–Lindau syndrome, multiple endocrine neoplasia type 2 (a or b) and neurofibromatosis type 1 should also be considered. If in doubt, the patient should be managed in an appropriate multidisciplinary clinic.22,23
Even solitary tumours with a family history need to be managed with caution as the consequences of cranial neuropathy may be compounded by the subsequent development of additional tumours. A number of our patients have been referred after a carotid body paraganglioma has been removed and a vagal palsy acquired. Some have contralateral vagal paragangliomas that are then extremely difficult to manage. Surgical resection would render them unable to speak or swallow, and in need of a tracheotomy and percutaneous gastrostomy. The only options for management are observation, awaiting the inevitable and radiosurgery, which may or may not hasten the natural course of the disease in terms of neural function.
In order to emphasise the caution required in managing these patients, we choose to classify those with multiple tumours as a separate subgroup rather than just considering and classifying the presenting tumour.
It should not be forgotten that some of these tumours are malignant. It is usually impossible to differentiate between benign and malignant paragangliomas on the basis of histology. The diagnosis is almost always made by detecting metastases, which in our experience are most often pulmonary or vertebral. Metastases grow slowly and it is possible for patients to survive a very long time with a significant tumour load. It is therefore important to detect metastases, particularly in the vertebral column as the consequences of collapse or cord compression are to some extent preventable. It is possible that some of the tumours removed may have been malignant without this becoming apparent and concern about potential malignancy remains an argument in favour of resection over radiation treatment.3
Conclusions
Our classification groups tumours that present similar treatment challenges, complications and benefits of surgery. When dealing with relatively rare tumours, it is imperative that the groups are not unnecessarily small so that meaningful comparison can be made. The classification should be undertaken pre-operatively in order to plan therapy and facilitate informed consent. It is important to identify patients at increased risk of complications who would benefit by referral to a centre with specific expertise. We recommend that all patients with types 2, 3 and 4 tumours should be referred to an experienced specialist centre for the management of these tumours.
References
- 1.Baysal BE. Hereditary paraganglioma targets diverse paraganglia. J Med Genet. 2002;39:617–622. doi: 10.1136/jmg.39.9.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zak FG, Lawson W. The Paraganglionic Chemoreceptor System: Physiology, Pathology and Clinical Medicine. New York: Springer; 1982. [Google Scholar]
- 3.Hinerman RW, Amdur RJ, et al. Definitive radiotherapy in the management of paragangliomas arising in the head and neck: a 35-year experience. Head Neck. 2008;30:1,431–1,438. doi: 10.1002/hed.20885. [DOI] [PubMed] [Google Scholar]
- 4.Navarro Martín A, Maitz A, et al. Successful treatment of glomus jugulare tumours with gamma knife radiosurgery: clinical and physical aspects of management and review of the literature. Clin Transl Oncol. 2010;12:55–62. doi: 10.1007/s12094-010-0467-y. [DOI] [PubMed] [Google Scholar]
- 5.Nora JD, Hallett JW, et al. Surgical resection of carotid body tumors: long-term survival, recurrence, and metastasis. Mayo Clin Proc. 1988;63:348–352. doi: 10.1016/s0025-6196(12)64856-3. [DOI] [PubMed] [Google Scholar]
- 6.van der Mey AG, Maaswinkel-Mooy PD, et al. Genomic imprinting in hereditary glomus tumours: evidence for new genetic theory. Lancet. 1989;2:1,291–1,294. doi: 10.1016/s0140-6736(89)91908-9. [DOI] [PubMed] [Google Scholar]
- 7.Oldring D, Fisch U. Glomus tumors of the temporal region: surgical therapy. Am J Otol. 1979;1:7–18. [PubMed] [Google Scholar]
- 8.Fisch U, Fagan P, Valavanis A. the infratemporal fossa approach for the lateral skull base. Otolaryngol Clin North Am. 1984;17:513–552. [PubMed] [Google Scholar]
- 9.Wen HT, Rhoton AL, Katsuta T, de Oliveira E. Microsurgical anatomy of the transcondylar, supracondylar, and paracondylar extensions of the far-lateral approach. J Neurosurg. 1997;87:555–585. doi: 10.3171/jns.1997.87.4.0555. [DOI] [PubMed] [Google Scholar]
- 10.Spektor S, Anderson GJ, et al. Quantitative description of the far-lateral transcondylar transtubercular approach to the foramen magnum and clivus. J Neurosurg. 2000;92:824–831. doi: 10.3171/jns.2000.92.5.0824. [DOI] [PubMed] [Google Scholar]
- 11.Schipper J, Spetzger U, et al. Juxtacondylar approach in temporal paraganglioma surgery: when and why? Skull Base. 2009;19:43–47. doi: 10.1055/s-0028-1103129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.World Health Organization. Pathology and Genetics: Tumours of Endocrine Organs. Lyon, France: IARC; 2004. [Google Scholar]
- 13.Elders RA. Paraganglioma. Groningen, Netherlands: University of Groningen; 1962. [PhD dissertation] [Google Scholar]
- 14.Stoeckli SJ, Schuknecht B, Alkadhi H, Fisch U. Evaluation of paragangliomas presenting as a cervical mass on color-coded Doppler sonography. Laryngoscope. 2002;112:143–146. doi: 10.1097/00005537-200201000-00025. [DOI] [PubMed] [Google Scholar]
- 15.Shamblin WR, ReMine WH, Sheps SG, Harrison EG. Carotid body tumor (chemodectoma). Clinicopathologic analysis of ninety cases. Am J Surg. 1971;122:732–739. doi: 10.1016/0002-9610(71)90436-3. [DOI] [PubMed] [Google Scholar]
- 16.Arya S, Rao V, Juvekar S, Dcruz AK. Carotid body tumors: objective criteria to predict the Shamblin group on MR imaging. Am J Neuroradiol. 2008;29:1,349–1,354. doi: 10.3174/ajnr.A1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kollert M, Minovi AA, Draf W, Bockmühl U. Cervical paragangliomas-tumor control and long-term functional results after surgery. Skull Base. 2006;16:185–191. doi: 10.1055/s-2006-950386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drovdlic CM, Myers EN, et al. Proportion of heritable paraganglioma cases and associated clinical characteristics. Laryngoscope. 2001;111:1,822–1,827. doi: 10.1097/00005537-200110000-00029. [DOI] [PubMed] [Google Scholar]
- 19.Neumayer C, Moritz A, et al. Novel SDHD germ-line mutations in pheochromocytoma patients. Eur J Clin Invest. 2007;37:544–551. doi: 10.1111/j.1365-2362.2007.01822.x. [DOI] [PubMed] [Google Scholar]
- 20.Telischi FF, Bustillo A, et al. Octreotide scintigraphy for the detection of paragangliomas. Otolaryngol Head Neck Surg. 2000;122:358–362. doi: 10.1016/S0194-5998(00)70048-9. [DOI] [PubMed] [Google Scholar]
- 21.Taïeb D, Sebag F, et al. Does iodine-131 meta-iodobenzylguanidine (MIBG) scintigraphy have an impact on the management of sporadic and familial phaeochromocytoma? Clin Endocrinol. 2004;61:102–108. doi: 10.1111/j.1365-2265.2004.02077.x. [DOI] [PubMed] [Google Scholar]
- 22.Neumann HP, Berger DP, et al. Phaeochromocytomas, multiple endocrine neoplasia type 2, and von Hippel–Lindau disease. N Engl J Med. 1993;329:1,531–1,538. doi: 10.1056/NEJM199311183292103. [DOI] [PubMed] [Google Scholar]
- 23.DeAngelis LM, Kelleher MB, Post KD, Fetell MR. Multiple paragangliomas in neurofibromatosis: a new neuroendocrine neoplasia. Neurology. 1987;37:129–133. doi: 10.1212/wnl.37.1.129. [DOI] [PubMed] [Google Scholar]