

Abstract
Autism spectrum disorder (ASD) has been associated with mitochondrial disease (MD). Interestingly, most individuals with ASD and MD do not have a specific genetic mutation to explain the MD, raising the possibility of that MD may be acquired, at least in a subgroup of children with ASD. Acquired MD has been demonstrated in a rodent ASD model in which propionic acid (PPA), an enteric bacterial fermentation product of ASD-associated gut bacteria, is infused intracerebroventricularly. This animal model shows validity as it demonstrates many behavioral, metabolic, neuropathologic and neurophysiologic abnormalities associated with ASD. This animal model also demonstrates a unique pattern of elevations in short-chain and long-chain acyl-carnitines suggesting abnormalities in fatty-acid metabolism. To determine if the same pattern of biomarkers of abnormal fatty-acid metabolism are present in children with ASD, the laboratory results from a large cohort of children with ASD (n=213) who underwent screening for metabolic disorders, including mitochondrial and fatty-acid oxidation disorders, in a medically based autism clinic were reviewed. Acyl-carnitine panels were determined to be abnormal if three or more individual acyl-carnitine species were abnormal in the panel and these abnormalities were verified by repeated testing. Overall, 17% of individuals with ASD demonstrated consistently abnormal acyl-carnitine panels. Next, it was determined if specific acyl-carnitine species were consistently elevated across the individuals with consistently abnormal acyl-carnitine panels. Significant elevations in short-chain and long-chain, but not medium-chain, acyl-carnitines were found in the ASD individuals with consistently abnormal acyl-carnitine panels—a pattern consistent with the PPA rodent ASD model. Examination of electron transport chain function in muscle and fibroblast culture, histological and electron microscopy examination of muscle and other biomarkers of mitochondrial metabolism revealed a pattern consistent with the notion that PPA could be interfering with mitochondrial metabolism at the level of the tricarboxylic-acid cycle (TCAC). The function of the fatty-acid oxidation pathway in fibroblast cultures and biomarkers for abnormalities in non-mitochondrial fatty-acid metabolism were not consistently abnormal across the subgroup of ASD children, consistent with the notion that the abnormalities in fatty-acid metabolism found in this subgroup of children with ASD were secondary to TCAC abnormalities. Glutathione metabolism was abnormal in the subset of ASD individuals with consistent acyl-carnitine panel abnormalities in a pattern similar to glutathione abnormalities found in the PPA rodent model of ASD. These data suggest that there are similar pathological processes between a subset of ASD children and an animal model of ASD with acquired mitochondrial dysfunction. Future studies need to identify additional parallels between the PPA rodent model of ASD and this subset of ASD individuals with this unique pattern of acyl-carnitine abnormalities. A better understanding of this animal model and subset of children with ASD should lead to better insight in mechanisms behind environmentally induced ASD pathophysiology and should provide guidance for developing preventive and symptomatic treatments.
Keywords: acyl-carnitines, autism spectrum disorder, clostridia, microbiome, mitochondrial disease, propionic acid
Introduction
Autism spectrum disorders (ASD) are a heterogeneous group of neurodevelopmental disorders that are characterized by impairments in social interaction and communication along with restrictive and repetitive behaviors.1 Many of the cognitive and behavioral features of ASD are believed to arise from central nervous system dysfunction, but abnormalities in many non-central nervous system tissues have been associated with ASD.2, 3 Recent studies have implicated abnormalities in systemic physiology that transcend organ specific dysfunction, at least in some children with ASD.2, 3, 4 Thus, it is possible that organs other than the brain and/or systemic abnormalities could be the source of the primary pathophysiological that manifest, in part, with secondary brain dysfunction.
A recent meta-analysis found that 5% of children with ASD meet criteria for a classic mitochondrial disease (MD) and suggest that this subgroup has distinct clinical characteristics that distinguish it from the general ASD population.3, 5 This study also found that about 30% of the general ASD population exhibited biomarkers consistent with MD.3 The high prevalence of abnormal mitochondrial biomarkers in ASD has been suggested to be due to mitochondrial dysfunction that is more prevalent and distinct from classic MD. Such a notion is supported by a recent study that found that 80% of the children with ASD demonstrated below normal function of the electron transport chain (ETC) in lymphocytes.6
The reason for mitochondrial dysfunction in ASD is unknown, but the fact that only 23% of children with ASD and MD have a known mitochondrial deoxyribonucleic acid (mtDNA) abnormality suggests that MD may be acquired rather than genetic in many ASD cases.3 Indeed, some have suggested that the systemic abnormalities in ASD such as mitochondrial dysfunction may arise from environmental triggers7 in genetically sensitive subpopulations.8, 9 Enteric short-chain fatty-acids, such as propionic acid (PPA),10, 11, 12, 13, 14, 15, 16, 17 which are fermentation by-products of ASD-associated enteric bacteria (that is, Clostridia, Desulfovibrio, Sutterella and Bacteroidetes), have been suggested as a possible environmental triggers in ASD.18, 19 Interestingly, humans with impairments in PPA metabolism20, 21, 22 exhibit neurodevelopmental conditions with ASD features.23
Recently, a rodent model has been development in which reversible (30 min) bouts of ASD-type (that is, stereotyped, perseverative and impaired social) behaviors are produced by brief intracerebroventricular infusions of PPA (http://www.psychology.uwo.ca/autism/autism6.htm for behavioral video). This animal model demonstrates several characteristics that have been reported in ASD such as tics, electrographic seizures, innate neuroinflammation and redox, lipid, phosphatidylethanolamine, mitochondrial, acyl-carnitine and carnitine abnormalities.10, 11, 12, 14, 15, 16, 17 This animal model provides an understanding of how exogenous agents, such as PPA, can cause reversible behavioral, metabolic, neuropathological and neurophysiological changes associated with ASD. Most importantly, this animal model has predictive value as it demonstrates biomarkers of abnormal mitochondrial fatty-acid metabolism (that is, acyl-carnitine elevations) that could be used as routine biomarkers if found in children with ASD.
Several lines of evidence suggest that mitochondrial fatty-acid oxidation could be abnormal in a subset of children with ASD. First, free carnitine, the cofactor used to transport long-chain and very-long-chain fatty-acids into the mitochondrial matrix, has been shown to be depleted in children with ASD.24 Free carnitine can be depleted if it remains bound to unprocessed fatty-acids due to a reduction in mitochondrial fatty-acid beta-oxidation.25 Second, elevations in long-chain and very-long-chain fatty-acids have been reported in children with ASD as compared with controls, suggesting excess unprocessed fatty-acid in the serum of children with ASD.26 Third, a case study and case series of patients with ASD have reported elevations in acyl-carnitines, the standard biomarker for mitochondrial fatty-acid oxidation deficits.27, 28 Thus, there is ample evidence to suggest that abnormalities in fatty-acid metabolism (that is, acyl-carnitine elevations) may be found in children with ASD.
Figure 1 demonstrates the acyl-carnitine elevations in brain homogenates found in rats exposed to intracerebroventricular infusions of PPA as compared with those exposed to phosphate buffered saline vehicle control. These abnormalities included short-chain (2–5 carbon length) and long-chain (13–18 carbon length) acyl-carnitines but not medium-chain (6–12 carbon length) acyl-carnitines.13 We hypothesize that a subset of children with ASD manifest biomarkers of abnormal mitochondrial fatty-acid metabolism that are similar to those reported in the PPA rodent model of ASD. Here we review the charts of consecutive patients seen in a medically based autism clinic who underwent a systematic workup for mitochondrial disorders per recently published guidelines, which included screening for fatty-acid metabolism disorders.3 Overall, 17% of children with ASD were found to demonstrate a unique pattern of acyl-carnitine abnormalities that were similar to the acyl-carnitine abnormalities found in the rodent PPA model of ASD. The potential causes of these abnormalities and their possible relation to ASD pathogenesis is discussed.
Figure 1.

Acyl-carnitine elevations in the brain of rats treated intracerebroventricularly with propionic acid. Notice that the majority of fatty-acid elevations were in short-chain (2–5 carbon length) and long-chain (13–18 carbon length) fatty-acids as compared with the medium-chain (6–12 carbon length) fatty-acids. This is adapted from Thomas et al.13 where it was presented as a table.
Materials and methods
Subject population
Parents of patients seen from 2008–2011 in a medically based autism clinic were requested to consent to allow their child's medical information to be anonymously abstracted into a clinical database that contained medical history, physical examination findings and the results of neurological and metabolic testing. Approximately 98% of parents (326 total patients) signed the consent.
Metabolic evaluation
A standardized metabolic workup for mitochondrial metabolism disorders was conducted on most patients.29, 30 The algorithm for this evaluation is depicted in Figure 2. Initial testing included laboratory tests to identify abnormalities in the respiratory chain, tricarboxylic-acid cycle (TCAC) and fatty-acid oxidation pathways. Abnormalities detected in initial testing were confirmed with repeat testing. If abnormalities could not be replicated, laboratories tests were reconsidered during metabolic stress or illness if a high index of suspicion remained for the patient.
Figure 2.

Algorithm for metabolic workup of autistic spectrum disease patients evaluated in the medically based autism clinic. Patients are screened with biomarkers of abnormal mitochondrial function in the fasting state. Abnormalities are verified with repeat fasting biomarker testing. For patients with biomarkers for a fatty-acid oxidation defect, other disorders of fatty-acid metabolism are ruled-out before further workup for a mitochondrial disorder. Patients with consistent biomarkers for mitochondrial dysfunction are first investigated for genetic causes of their mitochondrial disorder before considering a muscle and/or skin biopsy. mtDNA, mitochondrial deoxyribonucleic acid; RBC, red-blood cell.
An acyl-carnitine panel, which measures short-chain, medium-chain and long-chain acyl-carnitines, was used as the primarily laboratory test to detect defects in the fatty-acid oxidation pathway. An acyl-carnitine panel was measured at initial testing in 213 of the consented patients. The acyl-carnitine panel was considered abnormal if three or more acyl-carnitines were elevated in the panel. An abnormal acyl-carnitine panel was confirmed by repeat testing.
If acyl-carnitine abnormalities were confirmed, non-mitochondrial disorders of fatty-acid metabolism were ruled-out before a MD workup was initiated. Disorders ruled-out included generalized hyperlipidemia, hypercholesterolemia, multiple carboxylase deficiencies (that is, biotinidase deficiency), zinc deficiency, abnormal copper metabolism and hypoglycemia. After such disorders were ruled-out, a MD workup was pursued. The initial step in the MD workup was examination for mtDNA gene abnormalities by either a targeted analysis for common mutations and/or deletions and/or sequencing of the entire mtDNA genome (Baylor Medical Genetics Laboratory, Houston, TX, USA).31, 32
When a conclusive mtDNA abnormality could not be identified, nuclear mitochondrial gene testing and/or a muscle and/or skin biopsy was recommended. Nuclear mitochondrial gene abnormalities were ruled-out using an oligonucleotide array with comparative genomic hybridization analysis that examines ∼180 nuclear genes involved in mitochondria function, including genes involved in fatty-acid oxidation, carnitine metabolism, mitochondrial biogenesis, mtDNA maintenance, transcription and translation, and ETC complex assembly (MitoMet, Baylor Medical Genetics Laboratory).
In some patients, the quadricep muscle was biopsied and analyzed with light and electron microscopy, as well as for mtDNA content.33, 34 In some patients, fibroblasts obtained from a skin biopsy were cultured. ETC function was examined on frozen muscle and cultured fibroblasts (Baylor Medical Genetics Laboratory).35 Both uncorrected ETC function and ETC function correcting for citrate synthase are presented. Fibroblasts were incubated with d3-palmitate and ℒ-carnitine in duplicate for 72 h to determine function of the fatty-acid oxidation pathway (Baylor Institute of Metabolic Disease, Dallas, TX).36
Determination of acyl-carnitine abnormalities
To calculate the prevalence of having an abnormal acyl-carnitine panel in the ASD sample, the prevalence of having an acyl-carnitine panel with the first laboratory test was multiplied by the percent of patients confirmed to have an abnormal acyl-carnitine panel on repeat laboratory testing. This was done to account for the fact that some patients did not repeat the acyl-carnitine panel even though it was abnormal. To determine the specific acyl-carnitine species that were consistently elevated across the subgroup of patients with consistently abnormal acyl-carnitine panels, we examined the first two acyl-carnitine panels measured for each patient. Values for each individual acyl-carnitine species (for each patient) were transformed to a percent of the upper limit of normal for the specific acyl-carnitine species. The mean and s.e. were then calculated for each acyl-carnitine species to summarize the group data. Statistical significance was calculated as the significance of the difference between the upper limit of normal and the group mean for each acyl-carnitine specific using a z-distribution derived from the group mean and s.e.
Measurement of glutathione metabolism
Glutathione metabolism was evaluated in four participants. These patients were compared with normative values established in a previous study on redox metabolism.37 These controls included 42 healthy children ranged from 2–7 years of age with no history of developmental delay or neurological symptoms. Independent sample t-tests were used for comparison. Fasting blood samples were collected into ethylenediaminetetraacetic acid vacutainer tubes and were immediately chilled on ice before centrifuging at 4000 g for 10 min at 4 °C. To prevent metabolite inter-conversion the ice-cold samples were centrifuged within 15 min of the blood collection and the plasma stored at −80 °C until analysis within 2 weeks. Details of the methodology for high-pressure liquid chromatography with electrochemical detection and metabolite quantitation have been previously described.38 Total and free-reduced glutathione, oxidized glutathione (GSSG) and the total-reduced glutathione/GSSG and free-reduced glutathione/GSSG ratios were measured.
Results
Prevalence and patterns of abnormal acyl-carnitines
Seventy-four (35%) of the 213 patients tested demonstrated an increase in three or more acyl-carnitines when initially measured. Forty-two (57%) of the 74 underwent repeat acyl-carnitine testing. Three or more acyl-carnitines were abnormal a second time in 20 (48%) of the 42 patients, resulting in a prevalence of 17% of ASD children who manifested consistent acyl-carnitine panel abnormalities. Figure 3 demonstrates the mean values of each acyl-carnitine species relative of the upper limit of normal. C4OH, C14 and C16:1 were significantly elevated as compared with the upper limit of normal (z=2.18, P=0.01; z=5.71, P<0.0001; z=2.85,P=0.02, respectively), and were 186%, 226% and 131% of the upper limit of normal, respectively.
Figure 3.

Average acyl-carnitine values (with s.e. bars) from 20 patients with consistent abnormal elevations in multiple acyl-carnitines. Acyl-carnitine values are represented as percent upper limit of normal for each acyl-carnitine species. Notice that C4OH, C14 and C16:1 are significantly elevated as compared with the maximum upper limit of normal.
Clinical characteristics
Clinical characteristics of the 20 patients with consistent elevations in the acyl-carnitine panel are given in Supplementary Table 1. The average age was 8.7 years (s.d. 2.25) with a male to female ratio of 3:1. Autistic disorder was diagnosed in 70% of the participants while 25% had a diagnosis of pervasive developmental disorder-not otherwise specified and 5% were diagnosed with Asperger syndrome. Developmental regression was reported in 45% of patients.
Glutathione metabolism
The subset of children in which glutathione metabolism was examined demonstrated significantly lower total-reduced glutathione (t=12.75, P<0.0001) and free-reduced glutathione (t=10.04, P<0.0001) values and total-reduced glutathione/GSSG (t=9.07, P<0.001) and free-reduced glutathione/GSSG (t=4.69, P<0.0001) ratios as well as higher GSSG (t=2.61, P=0.01) values as compared with typically developing controls (Figure 4), suggesting both a reduction in the production of gluthathione and increase in gluthathione utilization by reactive oxygen species.
Figure 4.
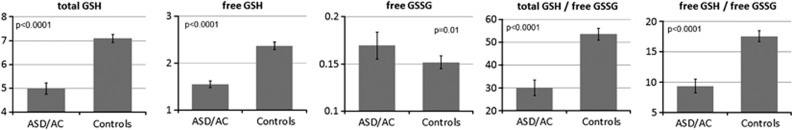
Gluthathione abnormalities in four children with consistent elevations in multiple acyl-carnitine species. Notice that the patients have lower total (tGSH, μM) and free (fGSH, μM) reduced gluthathione, as well as lower tGSH/fGSSG (free-oxidized gluthathione, μM) and fGSH/fGSSG ratios and higher fGSSG as compared with typically developing controls, suggesting both a reduction in the production of gluthathione and increase in gluthathione utilization by reactive species.
Genetic characteristics
Nuclear DNA examinations were normal in the great majority of patient (94%) in which such examinations were conducted. mtDNA was normal in 85% of the patients in which it was examined. The two mtDNA abnormalities that were identified involved novel maternally inherited homoplasmic cytochrome B gene mutations (15533A>G and 15404T>C), which altered evolutionary conserved amino acids. mtDNA content in muscle from four patients ranged from 109–189% of normal with a mean of 160.5% (s.d.±27%).
Neurological and biochemical testing
Neurological and biochemical characteristics of the patients are given in Supplementary Table 2. No abnormalities were found in the majority of patients that underwent an extended 23 h video electroencephalogram. Acyl-glycine panel, amino acids, glucose, insulin, Co-Q10, biotin, cholesterol and triglyceride levels were unremarkable in all patients in which they were tested. Urine organic acids were abnormal in the majority in which it was tested with elevations in TCAC metabolites, specifically elevations in citrate and/or isocitrate representing the majority of the abnormalities. Lactate was elevated in about half of the patients. Carnitine panel was abnormal in half of the patients in whom it was measured with 33% of the patients having high-esterified carnitine and 17% having low free carnitine. Creatine kinase and pyruvate was elevated in a minority of patients in which it was measured. Interestingly, red-blood cell zinc was borderline low in 70% of the patients in which it was measured and red-blood cell copper was slightly elevated in 35% of the patients in which it was measured. The great majority of individuals (90%) in which red-blood cell zinc and red-blood cell copper were both measured demonstrated an abnormality in at least one.
Muscle histology
All five of the patients that underwent muscle biopsy demonstrated abnormal histological and electron microscopy findings. Four demonstrated fiber type 1 predominance with two also demonstrating fiber type 2 atrophy. The fifth demonstrated myofiber size irregularity, increased sarcoplasmic lipid and scattered succinate dehydrogenase hyper-reactive fibers. Electron microscopy demonstrated an increased number of mitochondria in the subsarcolemmal region in all and also in the intermyofibrillar region in two. Mitochondria were maloriented in two patients and degeneration of membranous organelles was seen in three cases.
ETC and fatty-acid oxidation function
ETC function was testing on all five muscles biopsies. Corrected and uncorrected ETC activity is shown in Figures 5a and b, respectively, and demonstrate a partial defect in complexes I/III and I/III rotenone sensitive (RS). ETC and fatty-acid oxidation testing was conducted on fibroblast cultures from eight patients. Overall complex II/III activity was deficient across patients, and complex I/III RS demonstrated variable and elevated (>200% of the mean) activity, which was also seen to a lesser extent in complex IV (Figures 5c and d). Functional fatty-acid oxidation testing demonstrated elevations in the short-chain fatty-acid D3-C4 (Figures 5e and f). This effect was due to three patients with high D4-C4. Only one patient had a high enough elevation for a short-chain acyl-CoA dehydrogenase defect to be considered. However, sequencing of exons 1–10 of the short-chain acyl-CoA dehydrogenase gene (GeneDx, Gaithersburg, MD, USA)39 for the patient was normal.
Figure 5.

Electron transport chain (ETC) function of muscle (a, b) and fibroblast culture (c, d), as well as function of the fatty-acid oxidation pathway in fibroblast cultures (e, f). Graph values represent percent of normal ETC function, uncorrected (a, c) or corrected for citrate synthase (b, d). Muscle ETC results suggest a partial defect in complexes I/III and I/III rotenone sensitive (RS) while fibroblast culture ETC function suggests a partial defect in complex II/III activity. In fibroblast culture ETC studies complexes I/III RS and IV demonstrate considerable variability due to overactivity (>200% of the mean) in complex I/III RS in three patients and complex IV in one case. Fatty-acid oxidation values represent mean of specific acyl-carnitine species (higher is worse) uncorrected (e) and corrected for citrate synthase (f). Elevation in the short-chain fatty-acid D3-C4 was due to three patients demonstrating high D4-C4 values. The one patient with a significantly elevated D4-C4 value was found not to have a mutation in ;the short-chain acyl-CoA dehydrogenase gene suggesting that the abnormalities in fatty-acids in fibroblast culture were due to other mitochondrial metabolism abnormalities.
MD diagnosis criteria
Using the Morava et al.40 criteria for the five patients that underwent muscle biopsy found that three patients were rated as having a definite MD and two patients were rated as having probable MD.
Discussion
In this report, 17% of a large cohort of children with ASD demonstrated consistent elevations in short-chain and long-chain, but not medium-chain, acyl-carnitines. This pattern of acyl-carnitine abnormalities is similar to elevations in brain acyl-carnitines seen in the PPA rodent model of ASD.13 Other metabolic abnormalities, specifically mitochondrial dysfunction and glutathione abnormalities, were identified in the patient cohort that are similar to the PPA rodent model of ASD. Such abnormalities are discussed below in detail.
Mitochondrial abnormalities is ASD patients are consistent with PPA toxicity
ETC function testing in muscle demonstrated a partial deficit in complexes I/III and I/III RS activity. These ETC abnormalities, along with other biomarkers of mitochondrial dysfunction, are consistent with PPA interfering with mitochondrial metabolism, potentially through the TCAC. The TCAC utilizes two electron carriers, nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) to shuttle electrons to complexes I and II of the ETC, respectively. Normally TCAC reactions produce 3 NADH and 1 FADH2, resulting in a 3:1 NADH to FADH2 ratio (Figure 6a).
Figure 6.

The tricarboxylic-acid cycle during (a) typical metabolism and (b) with high levels of propionic acid. Propionic acid is metabolized to propionyl-CoA, which inhibits the proximal portion of the tricarboxylic-acid cycle and enhances the distal portion of the tricarboxylic-acid cycle (see discussion for details). FADH2, flavin adenine dinucleotide; NADH, nicotinamide adenine dinucleotide.
PPA is metabolized to propionyl-CoA, which is further metabolized to produce methymalonic-CoA. Methymalonic-CoA enters the TCAC half way through the cycle as succinyl-CoA, thereby essentially ‘short circuiting' the TCAC (Figure 6b). Elevated succinyl-CoA enhances the distal half of the TCAC and inhibits the proximal half of the TCAC. As the distal half of the TCAC produces 1 NADH and 1 FADH2, if the proximal half of the TCAC is inhibited, the NADH to FADH2 ratio will change from 3:1 to 1:1. As NADH is metabolized by complex I a reduction in the production in NADH will result in a relative deficit in complex I, consistent with the findings from the ETC muscle studies. In addition, inhibition of the proximal portion of the TCAC will also result in a build-up of the first metabolites in the TCAC, consistent with the elevations in citrate and isocitrate in our patients. Furthermore, the end product of the fatty-acid oxidation pathway, acetyl-CoA, is the first metabolite of the proximal half of the TCAC. Thus, inhibition of the proximal half of the TCAC inhibits the fatty-acid oxidation pathway. This is consistent with the fact that examination of the fatty-acid oxidation pathway in fibroblast culture did not reveal any abnormalities to explain the elevations in acyl-carnitines.
Examination of fibroblasts, which occurs after 6 or more weeks of fibroblast growth in culture, demonstrated above average activity of complexes I/III RS and IV and a partial deficit in complex II/III. Interestingly, overactivity in complexes I and IV have been reported in children with ASD/MD.41, 42 The disparity in the ETC findings between muscle and fibroblast culture can be explained by the alternative use of citrate synthase, the first enzyme in the TCAC, for metabolizing propionyl-CoA. Normally citrate synthase produces citrate from acetyl-CoA and oxaloacetate. In the context of high levels of propionyl-CoA, citrate synthase produces methyl citrate, a dead end metabolite, from propionyl-CoA. This will also result in a competition for citrate synthase by both propionyl-CoA and acetyl-CoA, further blocking the metabolism of metabolic pathways that produce acetyl-CoA as an end product, such as the fatty-acid oxidation pathway. This overuse of citrate synthase will most likely also result in an upregulation of citrate synthase over time.
If an agent, such as PPA, that suppressed mitochondrial function was present in vivo but not in vitro, mitochondrial function in the muscle, but not the fibroblast culture, would be more compromised. If upregulation of citrate synthase occurred due to excess PPA in vivo and then PPA was removed in vitro, the high activity of citrate synthase in vitro would overproduce citrate and enhance the proximal portion of the TCAC, which preferentially produces NADH, the electron carrier metabolized by complex I. This is consistent with the observed I/III RS overactivity seen in fibroblast culture.
One common theme of the observed ETC dysfunction in both muscle and fibroblast culture was that complex dysfunction primarily occurred when evaluating the function of complex III with complex I or II, suggesting that it is the interaction between complex III and complex I or II rather than at complex I or II specifically. Interesting, the 15533A>G cytochrome b mutation identified in one of the cases has been shown to have a complicated effect on complex III function. Rather than causing a frank decrease in complex III function, this mutation appears to result in delayed assembly of the I,III,IV supercomplex,43 thus influencing the interaction of complex III with other complexes rather than specifically affecting only complex III. Interestingly, PPA has been shown to have its detrimental effect on the ETC through inhibition of complex III function.44 Furthermore, alterations in brain omega 3/6 cardiolipin profiles found in the PPA rodent model could change inner mitochondrial membrane fluidity, and, thus, could potentially affect mitochondrial ETC complex interactions.13
Mitochondrial abnormalities are consistent with acquired MD
All patients that underwent a workup for MD demonstrated probable or definite MD by standardized criteria.2, 40 However, the majority of the patients in this study did not have any identifiable genetic causes for their MD. This is not surprising as only 23% of children with ASD and MD have a known mtDNA abnormality.3 The effect of the cytochrome b mutation found in two boys is complicated.45, 46 For example, delayed supercomplex assembly associated with the 15533A>G gene mutation in a child with a neurodevelopment disorder was restored when mutant transmitochondrial cybrids were developed from the 15533A>G case.43 This suggests that this mtDNA mutation is a risk factor that requires interactions with nuclear mutations, polymorphisms or epigenetics and/or environmental triggers or modulators in order for the disease phenotype to be expressed.43 Thus, the characteristics of this series of patient are consistent with the notion that the systemic abnormalities seen in this subgroup of ASD patients may arise from environmental triggers7 in genetically sensitive subpopulations.8, 17, 47, 48
Glutathione metabolism and oxidative stress abnormalities in ASD patients
Four patients in the series underwent measurements of glutathione metabolism. Overall, there was a marked decreased in total and free-reduced glutathione with a slight increase in GSSG, suggesting a primary deficit in the production of glutathione and increase in utilization. This finding parallels the PPA rodent animal model. Indeed, intracerebroventricular infusion of PPA in rats decreases total glutathione in brain homogenates.11 Further evidence for increased oxidative stress has been demonstrated in the rodent PPA model, including increased brain protein carbonylation and lipid peroxidation, altered phospholipid profiles and increased activated microglia.11, 13, 16 Such findings are all consistent with those from ASD patients.49, 50, 51
Abnormalities in zinc and copper in ASD patients
Interestingly, a large proportion of the patients in the series demonstrated mild abnormalities in zinc and/or copper concentrations. Such abnormalities have been reported in the ASD population previously52, 53, 54 and zinc supplementation (along with B6) has been shown to decrease copper levels and improve function in ASD in an uncontrolled study.54 Some have hypothesized that abnormalities in zinc and copper metabolism could result in poor metallothionein function, leading to susceptibility to environmental toxicants through increased oxidative stress or mitochondrial dysfunction.52 Interestingly, low zinc levels have been associated with pediatric inflammatory bowel disease55 and increased inflammation in animal models of colitis56 and zinc supplementation appears to be protective of bowel inflammation in clinical57 and animal studies.58 Although speculative, this may occur via a decrease in the activity of coloprotective metallothionein59 in the intestinal mucosa, or impairment of T- and B-cell interaction60 that may contribute to gut dysbiosis favoring ASD-associated bacteria.
Potential links to unique ASD microbial populations
Enteric bacterial populations found in increased numbers in stool samples of ASD patients (Clostridia, Desulfovibrio) are known to produce PPA from fermentation of dietary carbohydrates.18, 19, 31 Impaired carbohydrate digestion and transport in children with ASD can result in a higher concentration of dietary carbohydrates for these bacterial populations to ferment.18, 19, 61 A recent study has shown that stool from ASD patients have elevations in PPA and other short-chain fatty-acids.62 In addition, Desulfovibrio is capable of producing PPA from fermentation of peptones and can produce hydrogen sulfide, a potential mitochondrial toxin, which may act synergistically with PPA to promote mitochondrial dysfunction.17, 18, 63, 64
Interestingly, administration of common antibiotics (that is, beta lactams) for routine pediatric infections alters gut flora favoring PPA-producing species. This could be significant considering the reported high incidence of antibiotic use in some ASD patients.18, 19, 65, 66 In addition, this offers a potential explanation for temporary behavioral improvements in some patients following vancomycin or metronidazole treatment, which eradicates these bacteria18, 19, 66 and profoundly reduces stool PPA.67 Furthermore, removal of refined carbohydrates from the diet, which has been suggested as an empiric treatment to improve the behavioral fluctuations, gastrointestinal symptoms and dysbiosis in ASD,8 may act by reducing substrate for these bacteria to produce PPA.11
Although low concentrations of PPA may be beneficial, humans with impairments in PPA metabolism (that is, propionic or methymalonic acidemia, holocarboxylase, biotinidase or B12 deficiency, valproate or ethanol exposure) exhibit neurodevelopmental conditions with behavioral and biochemical similarities to ASD.68, 69 PPA and related short-chain fatty-acids (that is, butyrate and acetate) have broad effects on cellular systems.13, 20, 26, 27, 48, 70, 71, 72 They are actively taken up into the brain73 and can affect diverse physiological processes such as cell signaling,71 neurotransmitter synthesis and release,70 mitochondrial function,20 lipid metabolism,13, 74 immune function,75 cell–cell interactions76 and gene expression.72 Thus, there are many potential mechanisms where metabolic end products of the enteric microbiome can alter host physiology.17
Of particular interest, PPA is a known inhibitor of mitochondrial function, through sequestration of carnitine and the production of propionyl-CoA, a potential cytotoxin.44, 77 Methymalonic acid, a metabolite of PPA, results in abnormal mitochondrial morphology,78 tissue specific ETC dysfunction,79 inhibition of the complex I and II function when interacting with complex III in the brain80 and reductions in reduced glutathione,78 similar to the patient cohort presented and the PPA animal model. As PPA is metabolized through the TCAC, we propose that excess exogenous PPA or related short-chain fatty-acid metabolites interfere with mitochondrial metabolism, thereby causing acquired mitochondrial dysfunction.
Impaired carnitine metabolism can act synergistically with PPA-producing bacteria
The Na+ dependent organic cation/carnitine transporter 2 transports carnitine across the gut-blood and blood-brain barriers.81 Antibiotics (that is, beta lactams) commonly used to treat pediatric, infections directly inhibit the organic cation/carnitine transporter 2 transporter, thus directly impairing carnitine reabsorption.81 This could be significant considering the high incidence of antibiotic use in ASD patients, which can also promote gut dysbiosis favoring ASD-associated gut bacterial populations that produce PPA.18, 19, 65, 66 Given that both carnitine deficiency and PPA can be detrimental to mitochondrial metabolism, it is possible that antibiotic overuse can cause these two effects to act synergistically to cause an acquired mitochondrial disorder, especially in genetically susceptible individuals.
Interestingly, children with ASD, as a group, have been found to have reduced blood carnitine3 and a X-linked inborn error of carnitine biosynthesis has been shown to be a risk factor for ASD.82 Furthermore, oral carnitine, and its derivative acetyl-ℒ-carnitine, have both neuroprotective83, 84 and coloprotective properties.85 Given that carnitine supplementation improves function in children with ASD,86, 87 it deserves further investigation as a therapeutic agent in ASD.3, 86, 87
Summary
This study has demonstrated that ∼17% of children with ASD manifest biomarkers of abnormal mitochondrial fatty-acid metabolism that parallel similar biomarkers in the PPA rodent model of ASD. Detailed examination of a subset of these patients indicates that these metabolic abnormalities are at least partly due to TCAC and ETC dysfunction. Genetic disorders do not appear to account for the majority of these cases and the two individuals with abnormalities in mtDNA suggest dysfunction in the interaction of complex III with complex I and/or II. For the cases in which genetic abnormalities have not been found it is very likely that MD is acquired. As this subgroup of ASD patients have several parallels with the rodent PPA model of ASD,13 this rodent model may be a useful tool to further examine the temporal relation of behavioral bouts in relation to carnitine-acyl-carnitine fluctuations, and their possible response to therapeutic compounds thought to be useful in the treatment of ASD and mitochondrial dysfunction such as carnitine supplementation.
It is important to note that PPA affects multiple systems in a complex manner and the evidence of increased PPA or other short-chain fatty-acids being involved in the pathophysiology of ASD, although compelling, is circumstantial at this stage. Thus, future studies should identify additional parallels between the PPA rodent model of ASD and individuals with ASD who manifest similar biomarkers. Further study of this model and this subgroup of ASD patients should improve our understanding of the pathophysiology and potential risk factors that lead to the metabolic, brain and behavior abnormalities associated with ASD.
Acknowledgments
This research was supported in part from generous contributions from GoodLife's Children's Foundation and Autism Research Institute to DFM. We thank Dr Raymond Thomas (postdoctoral fellow) for published data on brain lipids and Drs Stephan Kahler and Anirudh Saronwala for their insight into propionic metabolism.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Translational Psychiatry website (http://www.nature.com/tp)
Supplementary Material
References
- APA Diagnostic and statistical manual of mental disorders4th ednDC: American Psychiatric Association: Washington; 1994 [Google Scholar]
- Frye RE, Rossignol DA. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatr Res. 2011;69 (5 Pt 2:41R–47RR. doi: 10.1203/PDR.0b013e318212f16b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol D, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol Psychiatry. 2012;17:290–314. doi: 10.1038/mp.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry. 2012;17:389–401. doi: 10.1038/mp.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman JR, Kelley RI, Bauman ML, Cohen BH, Murray KF, Mitchell RL, et al. Mitochondrial disease in autism spectrum disorder patients: a cohort analysis. PLoS ONE. 2008;3:e3815. doi: 10.1371/journal.pone.0003815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulivi C, Zhang YF, Omanska-Klusek A, Ross-Inta C, Wong S, Hertz-Picciotto I, et al. Mitochondrial dysfunction in autism. JAMA. 2010;304:2389–2396. doi: 10.1001/jama.2010.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert MR, Russo JP, Yang S, Roohi J, Blaxill M, Kahler SG, et al. Autism and environmental genomics. Neurotoxicology. 2006;27:671–684. doi: 10.1016/j.neuro.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Ashwood P, Anthony A, Torrente F, Wakefield AJ. Spontaneous mucosal lymphocyte cytokine profiles in children with autism and gastrointestinal symptoms: mucosal immune activation and reduced counter regulatory interleukin-10. J Clin Immunol. 2004;24:664 73. doi: 10.1007/s10875-004-6241-6. [DOI] [PubMed] [Google Scholar]
- MacFabe DF, Rodríguez-Capote K, Hoffman JE. A novel rodent model of autism: intraventricular infusions of propionic acid increase locomotor activity and induce neuroinflammation and oxidative stress in discrete regions of adult rat brain. Am J Biochem Biotechnol. 2008;4:146–166. [Google Scholar]
- MacFabe DF, Cain DP, Rodriguez-Capote K, Franklin AE, Hoffman JE, Boon F, et al. Neurobiological effects of intraventricular propionic acid in rats: possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behav Brain Res. 2007;176:149–169. doi: 10.1016/j.bbr.2006.07.025. [DOI] [PubMed] [Google Scholar]
- Shultz SR, MacFabe DF, Ossenkopp KP, Scratch S, Whelan J, Taylor R, et al. Intracerebroventricular injection of propionic acid, an enteric bacterial metabolic end-product, impairs social behavior in the rat: implications for an animal model of autism. Neuropharmacology. 2008;54:901–911. doi: 10.1016/j.neuropharm.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Thomas RH, Foley KA, Mepham JR, Tichenoff LJ, Possmayer F, MacFabe DF. Altered brain phospholipid and acylcarnitine profiles in propionic acid infused rodents: further development of a potential model of autism spectrum disorders. J Neurochem. 2010;113:515–529. doi: 10.1111/j.1471-4159.2010.06614.x. [DOI] [PubMed] [Google Scholar]
- MacFabe DF, Cain NE, Boon F, Ossenkopp KP, Cain DP. Effects of the enteric bacterial metabolic product propionic acid on object-directed behavior, social behavior, cognition, and neuroinflammation in adolescent rats: relevance to autism spectrum disorder. Behav Brain Res. 2011;217:47–54. doi: 10.1016/j.bbr.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Shultz SR, Macfabe DF, Martin S, Jackson J, Taylor R, Boon F, et al. Intracerebroventricular injections of the enteric bacterial metabolic product propionic acid impair cognition and sensorimotor ability in the Long-Evans rat: further development of a rodent model of autism. Behav Brain Res. 2009;200:33–41. doi: 10.1016/j.bbr.2008.12.023. [DOI] [PubMed] [Google Scholar]
- Thomas RH, Meeking MM, Mepham JR, Tichenoff L, Possmayer F, Liu S, et al. The enteric bacterial metabolite propionic acid alters brain and plasma phospholipid molecular species: further development of a rodent model of autism spectrum disorders. J Neuroinflammation. 2012;9:153. doi: 10.1186/1742-2094-9-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFabe DF. Short-chain fatty acid fermentation products of the gut microbiome: implications in autism spectrum disorders. Microb Ecol Health Dis. 2012;23:19260. doi: 10.3402/mehd.v23i0.19260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegold SM, Dowd SE, Gontcharova V, Liu C, Henley KE, Wolcott RD, et al. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe. 2010;16:444–453. doi: 10.1016/j.anaerobe.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Finegold SM, Molitoris D, Song Y, Liu C, Vaisanen ML, Bolte E, et al. Gastrointestinal microflora studies in late-onset autism. Clin infect Dis. 2002;35 (Suppl 1:S6–S16. doi: 10.1086/341914. [DOI] [PubMed] [Google Scholar]
- Wajner M, Latini A, Wyse AT, Dutra-Filho CS. The role of oxidative damage in the neuropathology of organic acidurias: insights from animal studies. J Inherit Metab Dis. 2004;27:427–448. doi: 10.1023/B:BOLI.0000037353.13085.e2. [DOI] [PubMed] [Google Scholar]
- Coulter DL. Carnitine, valproate, and toxicity. J Child Neurol. 1991;6:7–14. doi: 10.1177/088307389100600102. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Rizza V. Formation of propionate after short-term ethanol treatment and its interaction with the carnitine pool in rat. Alcohol. 1999;19:169–176. doi: 10.1016/s0741-8329(99)00036-1. [DOI] [PubMed] [Google Scholar]
- Al-Owain M, Kaya N, Al-Shamrani H, Al-Bakheet A, Qari A, Al-Muaigl S, et al. Autism spectrum disorder in a child with propionic acidemia. JIMD Reports. 2012;7:63–66. doi: 10.1007/8904_2012_143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafa GA, El-Gamal HA, El-Wakkad ASE, El-Shorbagy OE, Hamza MM. Polyunsaturated fatty acids, carnitine and lactate as biological markers of brain energy in autistic children. Int J Child Neuropsychiatry. 2005;2:179–188. [Google Scholar]
- Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, et al. The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab. 2008;94:16–37. doi: 10.1016/j.ymgme.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastural E, Ritchie S, Lu Y, Jin W, Kavianpour A, Khine Su-Myat K, et al. Novel plasma phospholipid biomarkers of autism: mitochondrial dysfunction as a putative causative mechanism. Prostaglandins Leukot Essent Fatty Acids. 2009;81:253–264. doi: 10.1016/j.plefa.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Clark-Taylor T, Clark-Taylor BE. Is autism a disorder of fatty acid metabolism? possible dysfunction of mitochondrial beta-oxidation by long chain acyl-CoA dehydrogenase. Med Hypotheses. 2004;62:970–975. doi: 10.1016/j.mehy.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Frye RE. Biomarkers of abnormal energy metabolism in children with autism spectrum disorder. NAJ Med Sci. 2012;5:141–147. [Google Scholar]
- Munnich A, Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. Am J Med Genet. 2001;106:4–17. doi: 10.1002/ajmg.1391. [DOI] [PubMed] [Google Scholar]
- Mitochondrial Medicine Society's Committee on D. Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, et al. The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab. 2008;94:16–37. doi: 10.1016/j.ymgme.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LJ. Pathogenic mitochondrial DNA mutations in protein-coding genes. Muscle Nerve. 2007;36:279–293. doi: 10.1002/mus.20807. [DOI] [PubMed] [Google Scholar]
- Wong LJ, Cobb BR, Chen TJ. Molecular analysis of mitochondrial DNA point mutations by polymerase chain reaction. Methods Mol Biol. 2006;336:135–143. doi: 10.1385/1-59745-074-X:135. [DOI] [PubMed] [Google Scholar]
- Bai RK, Perng CL, Hsu CH, Wong LJ. Quantitative PCR analysis of mitochondrial DNA content in patients with mitochondrial disease. Ann N Y Acad Sci. 2004;1011:304–309. doi: 10.1007/978-3-662-41088-2_29. [DOI] [PubMed] [Google Scholar]
- Bai RK, Wong LJ. Simultaneous detection and quantification of mitochondrial DNA deletion(s), depletion, and over-replication in patients with mitochondrial disease. J Mol Diagn. 2005;7:613–622. doi: 10.1016/S1525-1578(10)60595-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby DM, Thorburn DR, Turnbull DM, Taylor RW. Biochemical assays of respiratory chain complex activity. Methods Cell Biol. 2007;80:93–119. doi: 10.1016/S0091-679X(06)80004-X. [DOI] [PubMed] [Google Scholar]
- Roe CR, Roe DS. Recent developments in the investigation of inherited metabolic disorders using cultured human cells. Mol Genet Metab. 1999;68:243–257. doi: 10.1006/mgme.1999.2911. [DOI] [PubMed] [Google Scholar]
- James SJ, Melnyk S, Fuchs G, Reid T, Jernigan S, Pavliv O, et al. Efficacy of methylcobalamin and folinic acid treatment on glutathione redox status in children with autism. Am J Clin Nutr. 2009;89:425–430. doi: 10.3945/ajcn.2008.26615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnyk S, Pogribna M, Pogribny I, Hine RJ, James SJ. A new HPLC method for the simultaneous determination of oxidized and reduced plasma aminothiols using coulometric electrochemical detection. J Nutr Biochem. 1999;10:490–497. doi: 10.1016/s0955-2863(99)00033-9. [DOI] [PubMed] [Google Scholar]
- Corydon MJ, Vockley J, Rinaldo P, Rhead WJ, Kjeldsen M, Winter V, et al. Role of common gene variations in the molecular pathogenesis of short-chain acyl-CoA dehydrogenase deficiency. Pediatr Res. 2001;49:18–23. doi: 10.1203/00006450-200101000-00008. [DOI] [PubMed] [Google Scholar]
- Morava E, van den Heuvel L, Hol F, de Vries MC, Hogeveen M, Rodenburg RJ, et al. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006;67:1823–1826. doi: 10.1212/01.wnl.0000244435.27645.54. [DOI] [PubMed] [Google Scholar]
- Graf WD, Marin-Garcia J, Gao HG, Pizzo S, Naviaux RK, Markusic D, et al. Autism associated with the mitochondrial DNA G8363A transfer RNA(Lys) mutation. J Child Neurol. 2000;15:357–361. doi: 10.1177/088307380001500601. [DOI] [PubMed] [Google Scholar]
- Frye RE, Naviaux RK. Autistic disorder with complex IV overactivity: a new mitochondrial syndrome. J Ped Neurol. 2011;9:427–434. [Google Scholar]
- Gil Borlado MC, Moreno Lastres D, Gonzalez Hoyuela M, Moran M, Blazquez A, Pello R, et al. Impact of the mitochondrial genetic background in complex III deficiency. PloS one. 2010;5:e12801. doi: 10.1371/journal.pone.0012801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, van den Heuvel LP, et al. Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J. 2006;398:107–112. doi: 10.1042/BJ20060221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Borlado MC, Moreno Lastres D, Gonzalez Hoyuela M, Moran M, Blazquez A, Pello R, et al. Impact of the mitochondrial genetic background in complex III deficiency. PLoS ONE. 2010;5:e12801. doi: 10.1371/journal.pone.0012801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye RE. Novel cytochrome b gene mutations causing mitochondrial disease in autism. J Ped Neurol. 2012;10:1–6. [Google Scholar]
- Ashwood P, Van de Water J. Is autism an autoimmune disease? autoimmunity reviews. 2004;3:557–562. doi: 10.1016/j.autrev.2004.07.036. [DOI] [PubMed] [Google Scholar]
- Ashwood P, Van de Water J. A review of autism and the immune response. Clin Dev Immunol. 2004;11:165–174. doi: 10.1080/10446670410001722096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James SJ, Melnyk S, Jernigan S, Cleves MA, Halsted CH, Wong DH, et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am J Med Genet B, Neuropsychiatr Genet. 2006;141B:947–956. doi: 10.1002/ajmg.b.30366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13:171–181. doi: 10.1016/j.pathophys.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Kuchna I, Nowicki K, Imaki H, Wegiel J, Marchi E, et al. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010;119:755–770. doi: 10.1007/s00401-010-0655-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber S, Zinn GM, Kern JC, Kingston HM. The plasma zinc/serum copper ratio as a biomarker in children with autism spectrum disorders. Biomarkers. 2009;14:171–180. doi: 10.1080/13547500902783747. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Yoshida K, Yasuda Y, Tsutsui T. Infantile zinc deficiency: association with autism spectrum disorders. Scientific reports. 2011;1:129. doi: 10.1038/srep00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo AJ, Devito R. Analysis of copper and zinc plasma concentration and the efficacy of zinc therapy in individuals with asperger's syndrome, pervasive developmental disorder not otherwise specified (PDD-NOS) and autism. Biomark Insights. 2011;6:127–133. doi: 10.4137/BMI.S7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora SK, Spady D, Prosser C, El-Matary W. Trace elements and vitamins at diagnosis in pediatric-onset inflammatory bowel disease. Clin Pediatr. 2011;50:488–492. doi: 10.1177/0009922810397041. [DOI] [PubMed] [Google Scholar]
- Iwaya H, Kashiwaya M, Shinoki A, Lee JS, Hayashi K, Hara H, et al. Marginal zinc deficiency exacerbates experimental colitis induced by dextran sulfate sodium in rats. J Nutr. 2011;141:1077–1082. doi: 10.3945/jn.111.138180. [DOI] [PubMed] [Google Scholar]
- Scrimgeour AG, Condlin ML. Zinc and micronutrient combinations to combat gastrointestinal inflammation. Curr Opin Clin Nutr Metab Care. 2009;12:653–660. doi: 10.1097/MCO.0b013e3283308dd6. [DOI] [PubMed] [Google Scholar]
- Barollo M, Medici V, D'Inca R, Banerjee A, Ingravallo G, Scarpa M, et al. Antioxidative potential of a combined therapy of anti TNFalpha and Zn acetate in experimental colitis. World J Gastroenterol. 2011;17:4099–4103. doi: 10.3748/wjg.v17.i36.4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran CD, Ball JM, Sundar S, Coyle P, Howarth GS. The role of zinc and metallothionein in the dextran sulfate sodium-induced colitis mouse model. Dig Dis Sci. 2007;52:2113–2121. doi: 10.1007/s10620-007-9765-9. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Okamura S, Kudo T, Masuo T, Mori M. Calcineurin inhibition by polaprezinc in rats with experimentally-induced colitis. Life Sci. 2011;88:432–439. doi: 10.1016/j.lfs.2010.12.018. [DOI] [PubMed] [Google Scholar]
- Williams BL, Hornig M, Buie T, Bauman ML, Cho Paik M, Wick I, et al. Impaired carbohydrate digestion and transport and mucosal dysbiosis in the intestines of children with autism and gastrointestinal disturbances. PLoS One. 2011;6:e24585. doi: 10.1371/journal.pone.0024585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Christophersen CT, Sorich MJ, Gerber JP, Angley MT, Conlon MA. Elevated fecal short chain fatty acid and ammonia concentrations in children with autism spectrum disorder. Dig Dis Sci. 2012;57:2096–2102. doi: 10.1007/s10620-012-2167-7. [DOI] [PubMed] [Google Scholar]
- Barton LL, Fauque GD. Biochemistry, physiology and biotechnology of sulfate-reducing bacteria. Adv Appl Microbiol. 2009;68:41–98. doi: 10.1016/S0065-2164(09)01202-7. [DOI] [PubMed] [Google Scholar]
- Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta. 2010;1797:1500–1511. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Fallon J. Could one of the most widely prescribed antibiotics amoxicillin/clavulanate "augmentin" be a risk factor for autism. Med Hypotheses. 2005;64:312–315. doi: 10.1016/j.mehy.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Sandler RH, Finegold SM, Bolte ER, Buchanan CP, Maxwell AP, Vaisanen ML, et al. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J Child Neurol. 2000;15:429–435. doi: 10.1177/088307380001500701. [DOI] [PubMed] [Google Scholar]
- Mellon AF, Deshpande SA, Mathers JC, Bartlett K. Effect of oral antibiotics on intestinal production of propionic acid. Arch Dis Child. 2000;82:169–172. doi: 10.1136/adc.82.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorifuji T, Kawai M, Muroi J, Mamada M, Kurokawa K, Shigematsu Y, et al. Unexpectedly high prevalence of the mild form of propionic acidemia in Japan: presence of a common mutation and possible clinical implications. Hum Genet. 2002;111:161–165. doi: 10.1007/s00439-002-0761-z. [DOI] [PubMed] [Google Scholar]
- Perez B, Desviat LR, Rodriguez-Pombo P, Clavero S, Navarrete R, Perez-Cerda C, et al. Propionic acidemia: identification of twenty-four novel mutations in Europe and North America. Mol Genet Metab. 2003;78:59–67. doi: 10.1016/s1096-7192(02)00197-x. [DOI] [PubMed] [Google Scholar]
- DeCastro M, Nankova BB, Shah P, Patel P, Mally PV, Mishra R, et al. Short chain fatty acids regulate tyrosine hydroxylase gene expression through a cAMP-dependent signaling pathway. Brain Res Mol Brain Res. 2005;142:28–38. doi: 10.1016/j.molbrainres.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Nakao S, Moriya Y, Furuyama S, Niederman R, Sugiya H. Propionic acid stimulates superoxide generation in human neutrophils. Cell Biol Int. 1998;22:331–337. doi: 10.1006/cbir.1998.0263. [DOI] [PubMed] [Google Scholar]
- Nguyen NH, Morland C, Gonzalez SV, Rise F, Storm-Mathisen J, Gundersen V, et al. Propionate increases neuronal histone acetylation, but is metabolized oxidatively by glia. Relevance for propionic acidemia. J Neurochem. 2007;101:806–814. doi: 10.1111/j.1471-4159.2006.04397.x. [DOI] [PubMed] [Google Scholar]
- Conn AR, Fell DI, Steele RD. Characterization of alpha-keto acid transport across blood-brain barrier in rats. Am J Physiol. 1983;245:E253–E260. doi: 10.1152/ajpendo.1983.245.3.E253. [DOI] [PubMed] [Google Scholar]
- Hara H, Haga S, Aoyama Y, Kiriyama S. Short-chain fatty acids suppress cholesterol synthesis in rat liver and intestine. J Nutr. 1999;129:942–948. doi: 10.1093/jn/129.5.942. [DOI] [PubMed] [Google Scholar]
- Le Poul E, Loison C, Struyf S. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. 2003;278:25481–25489. doi: 10.1074/jbc.M301403200. [DOI] [PubMed] [Google Scholar]
- Rorig B, Klausa G, Sutor B. Intracellular acidification reduced gap junction coupling between immature rat neocortical pyramidal neurones. J Physiol. 1996;490 (Pt 1:31–49. doi: 10.1113/jphysiol.1996.sp021125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass EP. Interaction of carnitine and propionate with pyruvate oxidation by hepatocytes from clofibrate-treated rats: importance of coenzyme A availability. J Nutr. 1992;122:234–240. doi: 10.1093/jn/122.2.234. [DOI] [PubMed] [Google Scholar]
- Chandler RJ, Zerfas PM, Shanske S, Sloan J, Hoffmann V, DiMauro S, et al. Mitochondrial dysfunction in mut methylmalonic acidemia. The FASEB Journal. 2009;23:1252–1261. doi: 10.1096/fj.08-121848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettenuzzo LF, Ferreira Gda C, Schmidt AL, Dutra-Filho CS, Wyse AT, Wajner M. Differential inhibitory effects of methylmalonic acid on respiratory chain complex activities in rat tissues. International J Dev Neurosci. 2006;24:45–52. doi: 10.1016/j.ijdevneu.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Brusque AM, Borba Rosa R, Schuck PF, Dalcin KB, Ribeiro CA, Silva CG, et al. Inhibition of the mitochondrial respiratory chain complex activities in rat cerebral cortex by methylmalonic acid. Neurochem Int. 2002;40:593–601. doi: 10.1016/s0197-0186(01)00130-9. [DOI] [PubMed] [Google Scholar]
- Miecz D, Januszewicz E, Czeredys M, Hinton BT, Berezowski V, Cecchelli R, et al. Localization of organic cation/carnitine transporter (OCTN2) in cells forming the blood-brain barrier. J Neurochem. 2008;104:113–123. doi: 10.1111/j.1471-4159.2007.05024.x. [DOI] [PubMed] [Google Scholar]
- Celestino-Soper PB, Violante S, Crawford EL, Luo R, Lionel AC, Delaby E, et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc Nat Acad Sci USA. 2012;109:7974–7981. doi: 10.1073/pnas.1120210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scafidi S, Racz J, Hazelton J, McKenna MC, Fiskum G. Neuroprotection by acetyl-L-carnitine after traumatic injury to the immature rat brain. Dev Neurosci. 2010;32:480–487. doi: 10.1159/000323178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Lyttle TS, Rabchevsky AG. Acetyl-ℒ-carnitine ameliorates mitochondrial dysfunction following contusion spinal cord injury. J Neurochem. 2010;114:291–301. doi: 10.1111/j.1471-4159.2010.06764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin G, Yurchenko K, Collette C, Rubio M, Villani AC, Bitton A, et al. L-carnitine, a diet component and organic cation transporter OCTN ligand, displays immunosuppressive properties and abrogates intestinal inflammation. Clin Exp Immunol. 2009;156:161–171. doi: 10.1111/j.1365-2249.2009.03879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geier DA, Kern JK, Davis G, King PG, Adams JB, Young JL, et al. A prospective double-blind, randomized clinical trial of levocarnitine to treat autism spectrum disorders. Med Sci Monit. 2011;17:PI15–PI23. doi: 10.12659/MSM.881792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol DA. Novel and emerging treatments for autism spectrum disorders: a systematic review. Ann Clin Psychiatr. 2009;21:213–236. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.