

Computational enzyme design holds promise for the production of renewable fuels, drugs, and chemicals. De novo enzyme design has generated catalysts for several reactions, but with lower catalytic efficiencies than naturally occurring enzymes 1–4. Here we report the use of crowdsourcing to enhance the activity of a computationally designed enzyme through the functional remodeling of its structure. Players of the online game Foldit 5, 6 were challenged to remodel the backbone of a computationally designed bimolecular Diels-Alderase 3 to enable additional interactions with substrates. Several iterations of design and characterization generated a 24 residue helix-turn-helix motif, including a 13 residue insertion, that increased enzyme activity over 18-fold. X-ray crystallography showed that the large insertion adopts a helix-turn-helix structure positioned as in the Foldit model. These results demonstrate that human creativity with design problems can extend beyond the macroscopic problems of everyday life to less familiar molecular scale protein design problems.
Previous computational enzyme design methods have kept the backbone fixed, but it is clear from natural evolution that the optimization of new functions generally involves some backbone remodeling 7. De novo design methods have been used to create new loops and protein structures 8, and motif directed design methods can introduce new functional loops when specific interactions are used to direct the modeling 9, 10. However, undirected remodeling of a protein backbone structure to improve function has not yet been achieved. A primary challenge is that the number of possibilities for undirected remodeling, once large insertions and sequence variability are allowed, is too large to be systematically searched by automated methods.
Recent work has demonstrated that crowdsourcing protein modeling problems to an online community through the game Foldit is an effective way to solve difficult protein structure modeling problems 5, 6. However, it was unclear whether players’ modeling expertise, which relies on human creativity and spatial intuition to direct search through alternative protein structures, could be extended to protein design, which involves a much more open ended search through protein sequence and structure space. To explore if human creativity could help guide the search in this significantly larger space, new tools allowing insertions, deletions and sequence substitutions were incorporated into Foldit, to supplement the existing tools available for manipulating protein conformation. To integrate players into the experimental design process, we presented them with a series of puzzles. To connect Foldit player iterative exploration with experimental testing, we established an advanced Foldit player as an intermediary between the Foldit community and the experimental lab who presented players with puzzles at each stage of the design process. Using Foldit, the advanced player analyzed the top ranking community designs and built sequence libraries around the structures in order to stabilize favorable interactions. The designs were then experimentally tested, and the best were used as input for the next puzzle posted to the online community (Supplementary Fig. 1).
We challenged Foldit players to remodel the active site loops of a computationally designed enzyme that catalyzes the Diels-Alder reaction, DA_20_10 3. The Diels-Alder reaction, a cornerstone of organic synthesis, creates two new carbon-carbon bonds and up to four stereocenters in one step. DA_20_10 catalyzes the well studied reaction between 4-carboxybenzyl trans-1,3-butadiene-1-carbamate (diene) and N,N-dimethylacrylamide (dienophile, Supplementary Fig. 2). Despite significant catalytic activity, the DA_20_10 active site is open on one side leaving the substrates quite solvent exposed (Fig. 1a). We reasoned that redesigning active site loops to make additional contacts with the substrates could improve catalytic activity and specificity. However, the previously developed mass spectrometry based assay for detecting Diels-Alderase activity only allows screening of ~200 variants at one time, and hence screening large libraries11 is not feasible. Instead, we chose to enlist Foldit players to guide the search for remodeled loops producing higher activities.
Figure 1.
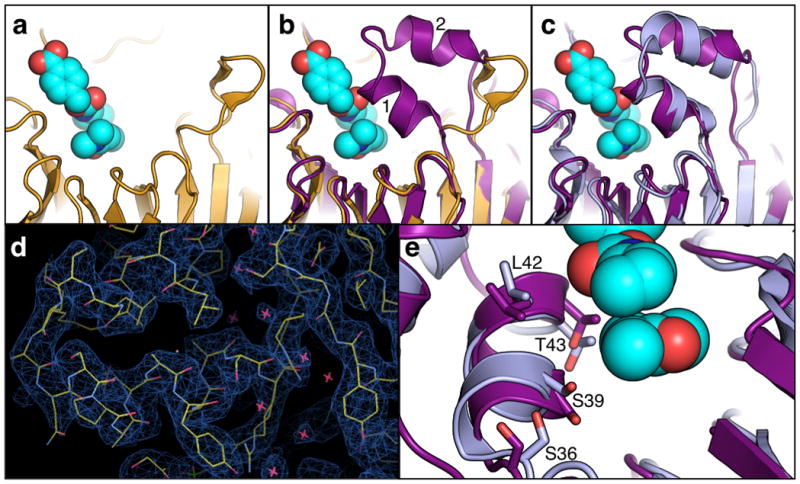
Crystal structure of Foldit design is similar to player model. (a) Crystal structure of starting designed Diels Alderase. The structure of DA_20_10 has not been solved, shown in the Figure is the structure of a precursor (3I1C, yellow) which differs by only three amino acid substitutions. The modeled transition state of the Diels-Alder reaction is depicted in cyan. (b) Structure of starting Diels Alderase (yellow) overlaid on CE6 player predicted structure (purple). CE6 has a 24 amino acid loop remodel, including a 13-residue insertion, converting an unstructured loop into a helix-turn-helix motif. (c) Crystal structure of CE6 (3U0S, light blue) overlaid with player predicted structure of CE6 (purple). The helix turn helix motif is clearly present and has a position close to that in the predicted structure. (d) Correlation between CE6 remodeled helices and corresponding experimental electron density. The electron density map is contoured to 1.0 σ. Sidechain alanine 45 is near the center of the picture on the left, with Helix 1 above and Helix 2 below. (e) The player designed side chains in CE6 that form the interface between Helix 1 (purple) and the transition state model (cyan) are found in the same rotameric conformation as in the crystal structure of CE6 (3U0S, light blue). Figure 1d generated in Coot 13. All other Figures generated in PyMol 14.
As it was not clear which loop to engineer, the first Foldit puzzle, “Cover the Ligand” asked players to remodel any of four active site loops in DA_20_10 to make additional molecular contacts to the ligand. Players were allowed to add or delete up to 5 amino acids in addition to mutating residues in the active site (Supplementary Fig. 1a). After a week of game play, the 69,773 designs made by the players were ranked by energy, the lowest 50 were visually assessed, and four designs that made particularly favorable interactions with the ligands were chosen to undergo additional rounds of refinement (Supplementary Fig. 1b and 3). Starting with these four loops, the advanced player designed a library of 36 sequences predicted to interact favorably with the substrates and/or stabilize the designed structure (Supplementary Library 1 and Supplementary Fig. 1c). While most variants exhibited no significant levels of activity, one (CE0) had a catalytic efficiency of 0.5 s−1M−1M−1, roughly a 10-fold decrease relative to DA_20_10, which has a catalytic efficiency of 4.7 s−1M−1M−1 (Table 1). We hypothesized that the designed insertion may have the desired structure, but the current amino acids interacting with the substrates or transition state were suboptimal. We explored this design further by making and testing an additional 500 sequence variants predicted to make favorable interactions with the modeled ligands (Supplementary Library 2). The most active of these designs, CE4, consisted of a helix buttressing the ligands, followed by an unstructured loop, and is 9-fold more active than DA_20_10 with a catalytic efficiency of 42.4 s−1M−1M−1 (Table 1).
Table 1.
Kinetic constants derived from the non-linear regression fits in Figure 2. Errors for kcat/KM-diene/KM-dienophile were calculated using: δkcat/KM-diene/KM-dienophile = |kcat/KM/KM|*√{(δkcat/kcat)2 + (δKM-diene/KM-diene)2 + (δKM- dienophile/KM- dienophile)2}. The kuncat for the Diels-Alder reaction at 298 K was found to be 2.2 × 10−2 M−1 hour−1. n.d. not detectable.
| Protein | KM-Diene (mM) | KM-Dienophile (mM) | kcat (hr−1) | kcat/KM-Diene/KM-Dienophile (s−1M−1M−1) |
|---|---|---|---|---|
| DA2010 | 1.2±0.2 | 101±21 | 2.1±0.3 | 4.7±1.5 |
| CE0 | n.d | n.d | n.d | 0.5±0.05 |
| CE4 | 0.5±0.03 | 31±3.0 | 2.4±0.1 | 42.4±5.7 |
| CE6 | 0.2±0.03 | 35±1.4 | 2.2±0.1 | 87.3±13.9 |
A second puzzle, “Back Me Up”, was then posted to the Foldit community asking players to stabilize the initially designed helix by transforming an unstructured loop into an additional neighboring structured helix. They were allowed to change the structure, sequence and length as before, but only for the unstructured loop. After another week and 109,421 designs, the top designs had converged on a helix-turn-helix motif, as requested (Supplementary Fig. 1d). Again, the advanced player constructed two libraries based on the community designed helix-turn-helix motif, each consisting of roughly 200 sequences (Supplementary Library 3). The most active design from these libraries was identified as CE6, with a catalytic efficiency of 87.3 s−1M−1M−1 (Table 1). This corresponds to over a 150-fold increase in activity relative to the initial player designed model (CE0) and over a 18-fold improvement relative to the original enzyme, DA_20_10. The third and final puzzle challenged players to predict the structure of the large insertion in CE6 starting from the crystal structure of the original design (Supplementary Fig. 1e). After a week players generated 335,697 solutions, and the lowest energy of these was selected as the player predicted structure of CE6 (Fig. 1b and Supplementary Fig. 1f).
To validate the accuracy of the top scoring CE6 model, the structure of CE6 was then determined by x-ray crystallography (Supplementary Table 1). The designed helices are well resolved in the electron density (Fig. 1d), and the player-designed helix-turn-helix model is remarkably close to the actual structure (Fig. 1c and Supplementary Fig. 1g). Helix 1 has the correct secondary structure, register, placement, and orientation resulting in a Cα-RMSD of 1.21 Å across the length of the designed helical element (spanning residues 36 to 44 in the design). All three designed residues in the interface between Helix 1 and the modeled transition state (Ser 39, Leu 42, and Thr 43) are in the same rotameric conformation as predicted in the final designed model (Fig. 1e). In addition Serine 36, which was designed to cap the N-terminus of Helix 1, is also modeled correctly (Fig. 1e).
Helix 2 (which was designed to interact with Helix 1, and corresponds to residues 48 to 56 in the designed enzyme) is well ordered in the crystal structure and has the same overall placement as in the Foldit model, but its packing angle and orientation relative to Helix 1 differ somewhat from the model. The design of Helix 2 was predicted to have a packing angle of approximately 30 degrees relative to Helix 1, whereas in the crystal structure the two helices are parallel. The difference in the position of the designed helix versus the crystal structure results from a small rotation around the center of Helix 2 (near alanine 51).
The backbone RMSD over the full 24 residue designed helix-turn-helix motif is 3.13Å, but the majority of this increased RMSD is a result of the shifted orientation of Helix 2. Some of the differences between the final design and the experimentally determined crystal structure are located near crystal contacts with the C-terminus of a second molecule of CE6 in the asymmetric unit (Supplementary Fig. 4). To evaluate if the observed crystal contacts occur in solution we mutated the most buried residues in the crystal interface (F324R, I326G and F327K). The activity of this mutant was indistinguishable from CE6, suggesting that the interface does not form in solution (Supplementary Fig. 5).
The Michaelis constants, KM-diene and KM-dienophile (Table 1), of CE6 are improved 6 fold and 3-fold respectively compared to the starting design, but the turnover number (kcat) is unchanged. The improvement in KM but lack of change in kcat are consistent with the design model and crystal structure. The designed loop interacts with and likely increases affinity for both substrates consistent with the decrease in KM. The catalytic residues that stabilize the transition state are on the opposite side of the active site, and these residues have almost identical locations in CE6 and the original design (Supplemental Fig. 6) despite the large scale remodeling of the loop. Given this similarity, and the fact that the conformation of the substrates and transition state are very similar in the region of the designed loop, it is not surprising that, as suggested by the lack of change in kcat, the loop does not selectively stabilize the transition state.
In addition to increases in activity, we hypothesized that the increased buried surface area and hydrophobicity of the binding pocket would increase dienophile specificity for other hydrophobic substrates. We tested this hypothesis by assaying enzyme activity of CE6 with a series of modified dienophiles previously described 3. Relative to DA_20_10, the player-designed CE6 exhibited a 3-fold increase in binding specificity for the hydrophobic dienophile 2A over the hydrophilic dienophile 2E, (Supplementary Fig. 7) consistent with our prediction. However, CE6 shows no significant preference for 2A when compared to the similar-sized hydrophobic 2B and 2C substrates. The increase in specificity for hydrophilic substrates, but loss of specificity for hydrophobic substrates suggests that while the desired hydrophobic pocket is formed, further improvements to the shape complementarily between the substrate and the engineered enzyme remain possible. Given the new backbone structure of the active site, future studies will explore additional backbone remodeling to modulate substrate specificity.
Insertion of helix-turn-helix motifs may be broadly useful in computational protein design. An advantage of helical hairpins is that they are to a large extent self-stabilizing and do not require additional tertiary interactions to form 12. This allows most of the experimental sampling to be focused on introducing new functional interactions with ligands. The highly ordered and predictable helical register enable sampling to be focused on a small subset of positions predicted to be pointing directly towards the ligand of interest.
We have demonstrated that crowdsourcing complex computational protein design problems can be an effective way of creatively sampling the potential sequence space for the design of active site loops that modulate enzyme activity. To our knowledge, this is the most extensive remodeling by design of a functional protein structure to date, and was accomplished by screening fewer than 1,000 sequences. The ability of an online community to successfully guide large-scale protein design problems suggests that human creativity can extend down to molecular scale when given the appropriate tools.
Supplementary Material
Figure 2.
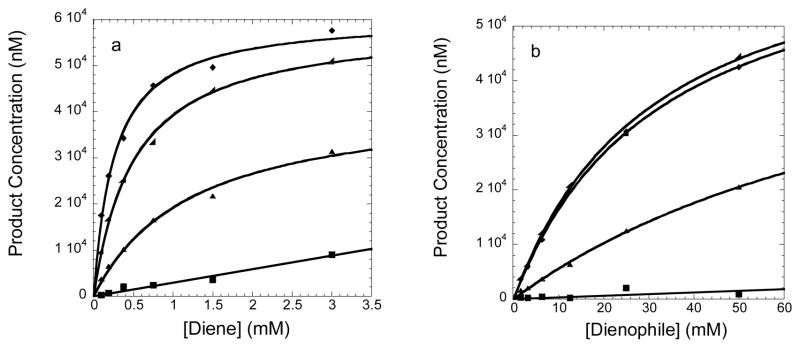
Kinetic characterization for DA_20_10 (▲), CE0 (■), CE4 (◢), and CE6 (◆). (a) Product observed after three hours with the dieneophile held constant at 50mM and the diene at 3mM, 1.5mM, 0.8mM, 0.4mM, 0.2mM, and 0.1mM. (b) Product observed after three hours with the diene held constant at 3mM and the dienophile at 50mM, 25mM, 12mM, 6mM, 3mM, and 1.5mM. The kinetic constants were determined using 10μM purified protein (Supplementary Fig. 8) and a standard Michaelis–Menten assay as described in the supplemental material.
Acknowledgments
We would like to acknowledge the members of the Foldit team for their help designing and developing the game and all the Foldit players who volunteered to make this work possible. We would also like to thank James Thompson for help in building computational scripts to analyze protein RMSD, as well as B. Siegel and M. Eiben for helpful comments on the manuscript. The x-ray crystallographic coordinates have been deposited in the Protein Data Bank with accession ID 3U0S. This work was supported by the Center for Game Science at the University of Washington, US Defense Advanced Research Projects Agency (DARPA) grant N00173-08-1-G025, the DARPA PDP program, the Howard Hughes Medical Institute (D.B.), a National Science Foundation graduate research fellowship to J.B.B. (grant NO DGE-0718124), and a National Science Foundation Grant for F.K. (grant NO 0906026).
Footnotes
AUTHOR CONTRIBUTIONS
C.B.E. Analyzed community models, in addition to designing, building, and testing the enzyme libraries.
J.B.S. Designed the experimental and computational methods, built the initial computational models.
F.K. Set up the Foldit puzzles and curated the player results for analysis by C.B.E;
S.C. Led design and development of Foldit;
B.L.S., J.B.B., and B.W.S grew the crystals and collected X-ray diffraction data; processed X-ray data and analyzed the structure;
Z.P. and D.B. contributed to the writing of the manuscript.
References
- 1.Rothlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 2.Jiang L, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel JB, et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science. 2010;329:309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bar-Even A, et al. The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry-Us. 2011;50:4402–4410. doi: 10.1021/bi2002289. [DOI] [PubMed] [Google Scholar]
- 5.Khatib F, et al. Crystal structure of a monomeric retroviral proteasesolved by protein folding game players. Nat Struct Mol Biol. 2011;18:1175–1177. doi: 10.1038/nsmb.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper S, et al. Predicting protein structures with a multiplayer online game. Nature. 2010;466:756–760. doi: 10.1038/nature09304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sterner R, Hocker B. Catalytic versatility, stability, and evolution of the (βα)8-barrel enzyme fold. Chemical Reviews. 2005;105:4038–4055. doi: 10.1021/cr030191z. [DOI] [PubMed] [Google Scholar]
- 8.Hu X, Wang H, Ke H, Kuhlman B. High-resolution design of a protein loop. P Natl Acad Sci USA. 2007;104:17668–17673. doi: 10.1073/pnas.0707977104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy PM, Bolduc JM, Gallaher JL, Stoddard BL, Baker D. Alteration of enzyme specificity by computational loop remodeling and design. P Natl Acad Sci USA. 2009;106:9215–9220. doi: 10.1073/pnas.0811070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Havranek JJ, Baker D. Motif-directed flexible backbone design of functional interactions. Protein science: a publication of the Protein Society. 2009;18:1293–1305. doi: 10.1002/pro.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park HS, et al. Design and evolution of new catalytic activity with an existing protein scaffold. Science. 2006;311:535–538. doi: 10.1126/science.1118953. [DOI] [PubMed] [Google Scholar]
- 12.Religa TL, et al. The helix-turn-helix motif as an ultrafast independently folding domain: the pathway of folding of Engrailed homeodomain. P Natl Acad Sci USA. 2007;104:9272–9277. doi: 10.1073/pnas.0703434104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 14.The PyMOL Molecular Graphics System, Version 1.3. Schrödinger, LLC; [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.