

Abstract
Mutations in Cu,Zn superoxide dismutase (SOD1) cause familial amyotrophic lateral sclerosis (FALS), a rapidly fatal motor neuron disease. Mutant SOD1 has pleiotropic toxic effects on motor neurons, among which mitochondrial dysfunction has been proposed as one of the contributing factors in motor neuron demise. Mitochondria are highly dynamic in neurons; they are constantly reshaped by fusion and move along neurites to localize at sites of high-energy utilization, such as synapses. The finding of abnormal mitochondria accumulation in neuromuscular junctions, where the SOD1-FALS degenerative process is though to initiate, suggests that impaired mitochondrial dynamics in motor neurons may be involved in pathogenesis. We addressed this hypothesis by live imaging microscopy of photo-switchable fluorescent mitoDendra in transgenic rat motor neurons expressing mutant or wild-type human SOD1. We demonstrate that mutant SOD1 motor neurons have impaired mitochondrial fusion in axons and cell bodies. Mitochondria also display selective impairment of retrograde axonal transport, with reduced frequency and velocity of movements. Fusion and transport defects are associated with smaller mitochondrial size, decreased mitochondrial density, and defective mitochondrial membrane potential. Furthermore, mislocalization of mitochondria at synapses among motor neurons, in vitro, correlates with abnormal synaptic number, structure, and function. Dynamics abnormalities are specific to mutant SOD1 motor neuron mitochondria, since they are absent in wild-type SOD1 motor neurons, they do not involve other organelles, and they are not found in cortical neurons. Together, these results suggest that impaired mitochondrial dynamics may contribute to the selective degeneration of motor neurons in SOD1-FALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease affecting motor neurons. The disease begins in mid-life and is rapidly progressive, leading to paralysis and death within a few years of onset (Rowland and Shneider, 2001). While the majority of the cases of ALS are sporadic with unknown etiology, ∼10% are inherited (familial ALS, FALS), of which ∼20% have mutations in the Cu,Zn superoxide dismutase gene (SOD1) (Rosen et al., 1994).
There are several, interconnected, pathogenic pathways leading to SOD1-FALS, since mutant SOD1 has a multitude of pathogenic effects in neurons and in surrounding cells, such as astrocytes and microglia (Ilieva et al., 2009). Among the several proposed mechanisms of toxicity, which include protein aggregation, neuroinflammation, excitotoxicity, oxidative stress, growth factor deficiency, glial pathology, cytoskeletal alteration, and axonal transport abnormalities (Rothstein, 2009), mitochondrial defects are thought to play a contributing role (Magrané and Manfredi, 2009; Shi et al., 2010). The toxic effects of mutant SOD1 on mitochondria occur at multiple levels, affecting their bioenergetics (Mattiazzi et al., 2002; Damiano et al., 2006), their channel properties (Israelson et al., 2010), protein import (Li et al., 2010), and the balance of anti- and proapoptotic proteins (Pedrini et al., 2010). In neurons, mitochondria form a highly dynamic and interconnected network that undergoes transport and remodeling by fusion and fission (Chan et al., 2006). Therefore, in mutant SOD1 neurons mitochondrial impairment may also be linked to defects in their dynamics (Magrané and Manfredi, 2009).
A potential consequence of dynamics impairment in neurons is the failure to provide healthy mitochondria to synaptic terminals and other sites of high-energy consumption. SOD1-FALS originates as a distal axonopathy, where neuromuscular junctions (NMJs) are the first regions of the motor neurons to degenerate (Frey et al., 2000; Fischer et al., 2004; Schaefer et al., 2005). Abnormal mitochondria at the NMJs of mutant SOD1 mice appear early on in the course of the disease, and correlate with the beginning of denervation (Vande Velde et al., 2004; Gould et al., 2006), suggesting that a defect in the maintenance of functional mitochondria may be participating in the pathogenic process.
To address the effects of mutant SOD1 on mitochondrial dynamics we investigated motor neurons derived from animal models of SOD1-FALS (G93A mutant and wild-type SOD1) using time-lapse confocal microscopy and the photo-switchable fluorescent protein, Dendra (Gurskaya et al., 2006), targeted to mitochondria (mitoDendra). Dendra naturally fluoresces green, but has the distinctive property of changing to red when photoactivated with 488 nm light. This photoconversion is irreversible, nontoxic, and stable, which makes Dendra an ideal tool for real-time and long-term imaging.
We demonstrated in this study that mitochondrial fusion was decreased and axonal transport impaired in G93A mutant SOD1 motor neurons. These abnormalities correlated with reduced mitochondrial size and density along axons, and with impaired mitochondrial bioenergetics. Changes in mitochondrial dynamics and morphology affected motor neurons, but not cortical neurons. Furthermore, we found that axonal transport defects were specific to mitochondria, since transport of other organelles was intact. Finally, we showed that mitochondrial defects were associated with functional and morphological abnormalities in the synaptic structures that are formed in vitro among motor neurons, suggesting that these defects may play a role in motor neuron synaptic degeneration.
Materials and Methods
Materials.
Live imaging experiments were performed in Lab-Tek 4-well chambered glass slides (Nalge Nunc International). The following antibodies were used: cytochrome c (BD Pharmingen), MnSOD (Stressgen), SOD1 (Calbiochem), β-actin (Sigma), SV2 (Developmental Studies Hybridoma Bank), synapsin I (Sigma), and secondary Cy2-, Cy3-, Cy5- and HRP-conjugated IgG (Jackson Immunochemicals). Fluorescent dyes MitoTracker Red CMXROS and tetramethyl rhodamine methyl ester (TMRM) were from Invitrogen.
In the pTurbo-mitoDendra expression vector, Dendra coding region substituted the GFP at the AgeI and NotI sites of the pTurboGFP-mito (Evrogen). In the amyloid precursor protein (APP695)-Dendra expression vector, APP695 cDNA was cloned into pTurbo-DendraV-N using HindIII and AgeI sites. All constructs were verified by sequencing.
Animal models.
Transgenic rats expressing human mutant G93A SOD1 or wild-type (WT) SOD1 were on a Sprague Dawley genetic background (Chan et al., 1998; Howland et al., 2002). All experiments were performed on neurons derived from at least three independent isolations. Each isolation consisted of motor neurons prepared by mixing all littermate embryos with the same genotype (transgenic or non-transgenic) from one pregnant rat. Genotyping was performed by PCR using the following pair of primers for human SOD1 transgene: IMR 113 (5′-catcagccctaatccatctga) and IMR 114 (5′-cgcgactaacaatcaaagtga). All experiments were approved by the Weill Cornell Medical College Institutional Animal Care and Use Committee.
Primary motor neuron cultures.
Purified motor neurons were prepared from E14.5 rat embryo spinal cords, and cultured as previously described (Estévez et al., 1998). Embryos were kept at 4°C in Hybernate E medium (BrainBits) supplemented with B27 (Invitrogen), while genotype analysis was performed. Motor neurons (7500–10,000 per chamber; 5000 per coverslip) were plated onto polyornithine- and laminin-coated surfaces (3 μg/ml each). Transgenic and non-transgenic control embryos were from the same litters. Motor neurons were transfected after 5–7 days in vitro (div) with 1 μg of cDNA and 1 μl of Lipofectamine 2000 (Invitrogen); medium was changed after 4–6 h. Imaging was performed 24–36 h later.
Primary cortical neuron cultures.
Primary rat cortical neuron cultures were prepared as described previously (Magrané et al., 2004), and plated onto polyornithine- and laminin-coated surfaces (3 and 5 μg/ml, respectively). Cortical neurons were transfected with mitoDendra after 7 div, and imaged 24–36 h later. For live imaging studies, axons were identified according to morphological criteria (long neurites, constant thin diameter, no branching, perpendicular emergence from the cell body).
Live-cell imaging.
The confocal microscope and live imaging station used were previously described (Magrané et al., 2009). The 488 nm laser intensity was kept <0.5 mW to prevent spontaneous photoactivation of Dendra. A 63× oil-immersion lens, and a pinhole of 2.89 AU (optical slice <2 μm) were applied for mitochondrial imaging; pinhole opening was set to 5.78 AU (optical slice < 4 μm) for membrane-bound organelle (MBO) imaging. Images were collected every 5 s for 5–7 min. Imaged axonal segments were defined as proximal (starting within 20 μm from the cell body) or distal (ending within 20 μm from the axon terminal).
Photoactivation of mitoDendra and analysis of mitochondrial fate.
Photoactivation of mitochondria was achieved in half of the motor neuron soma by exposing mitoDendra to 30–50 iterations using the 488 nm laser (7.5 mW). The entire thickness of the half soma was photoconverted by moving the laser to the first, mid, and last z-positions (1.6 μm intervals).
To study fusion and motility of mitochondria in the motor neuron somas both intact and photoconverted mitochondria pools were simultaneously imaged using 488 (0.25 mW) and 543 nm (0.5 mW) excitation, respectively. Frames were acquired prior (−1 min) and after photoactivation (0, 10, 40, 70, and 100 min). Single optical z-sections were used for the analysis, and the same thresholds were applied over time for each channel. Regions of interest (ROIs) were drawn for both photoactivated and non-photoactivated areas. Colocalization and Integrated Morphometric Analysis applications were from MetaMorph software (Universal Imaging Co.).
Colocalization (fusion rate) was measured within the photoactivated area, and expressed as percentage of the area of red overlapping green fluorescent mitochondria. Analysis of motility was performed on the green and red channels independently. First, the transport of non-photoactivated (green fluorescent) mitochondria over the photoactivated area (no green mitochondria present) was measured by quantifying the number of pixels appearing over time relative to the total amount of mitochondria present before photoactivation (t = −1). Similarly, the transport of photoactivated (red fluorescent) mitochondria over the non-photoactivated area (no red mitochondria present) was analyzed, and the appearance of pixels relative to the total amount of pixels (green and red) present in each time point was measured.
Analyses of organellar transport dynamics.
A mitochondrion was defined as mobile if it changed its position in at least three consecutive frames (taken at 5 s intervals). Transport of mitochondria and MBOs along neuronal processes was analyzed using MetaMorph software, as described previously (Magrané et al., 2009). Briefly, we generated kymographs (a graph of position vs time) and studied several transport parameters for anterograde or retrograde moving mitochondria, in proximal and distal regions of the motor axons. Distance was defined as the distance (in micrometers) traveled by a mitochondrion in either direction from origin to end of the moving event (i.e., distance traveled between two pauses); only distances longer than 3 μm were analyzed. Persistence of movement was the duration (in seconds) of a moving event. Velocity of mitochondria and MBOs was measured during sustained movement along the axon. Pauses were defined as absence of movement for more than three consecutive frames in mitochondria that were previously moving during the recording. The frequency of pauses was expressed as the ratio number of pauses/number of moving events; fusion events were not considered as pauses. Duration of pauses (in seconds) was also measured. Fusion events were identified when two mitochondria merged and became a single one for >3 consecutive frames; data were plotted as the ratio frequency of fusion/moving mitochondria.
Analyses of mitochondrial morphology in neurites and mitochondrial density in somas.
Proximal and distal segments of axons (see above for definition) were imaged. Criteria for the analysis of mitochondrial length and density were as previously described (Magrané et al., 2009). Mitochondrial distribution in neurites was expressed as the number of mitochondria per 50 μm of neurite segments. Mitochondrial distribution in distal axons was also measured by binning data into 25 μm segments. To study density of mitochondria in the motor neuron somas, ROIs in single optical z-sections were defined; threshold was set to twice the background fluorescence. Mitochondrial density was determined by measuring the percentage of thresholded area in the defined ROI using MetaMorph.
Analyses of synaptic structure and function.
Neuron-to-neuron synapses were identified by fixing 9 div motor neurons in 3% PFA and immunolabeling them with antibodies against the presynaptic proteins SV2 and synapsin I. A 63× oil-immersion lens, standard pinhole of 1 and optical intervals of 0.5 μm were used for confocal imaging; 16-bit, nonsaturated, images were acquired. Threshold was set to twice the fluorescence intensity of regions of the neuritic shaft devoid of synapses (background). The number and size of the thresholded puncta were determined by using the integrated morphometric analysis feature in MetaMorph. To study the relative distribution of mitochondria around synaptic puncta, cells were labeled with 250 nm MitoTracker Red CMXROS for 10 min at 37°C, followed by several washes with medium, fixation, and immunolabeling of synaptic proteins as described above. Circular ROIs with a radius of 1 μm from the synaptic puncta were placed around each identified puncta; mitochondria occupying at least half of the radius of an ROI were considered to be localized in that ROI.
Synaptic function was assed in living motor neurons at 9 div by following the endocytosis of the fixable, activity-dependent fluorescent nerve terminal probe AM4-65 (Biotium). Briefly, cells were incubated for 90 s at room temperature with 4 μm AM4-65, which was prepared in high potassium Ringer's solution (containing, in mm: 119 NaCl, 70 KCl, 2 CaCl2, 2 MgCl2, HEPES, pH 7.4 5, and 30 glucose) to depolarize the neurons. Motor neurons were washed twice for 4 min in low potassium (2.5 mm) Ringer's solution containing 1 μm tetrodotoxin (TTX), and then fixed as described above.
Analyses of mitochondrial membrane potential (ΔΨm).
The potentiometric fluorescent dye TMRM was used to assess changes in ΔΨm. Briefly, motor neurons (8 div) were loaded with 5 nm TMRM at 37°C. Confocal imaging of TMRM fluorescence was performed using low 543 nm excitation (0.02 mW intensity) to keep fluorescent values within a nonsaturated range (0–4095 units). A 63× oil-immersion lens and a pinhole of 2.89 AU were used. Images (12-bit) were taken. Average fluorescence in each mitochondrion was determined after thresholding of the images. The threshold was set to twice the fluorescence from the neuritic shaft in areas devoid of mitochondria to provide adequate background subtraction.
Western blot analysis.
Cortices were dissected and cortical neurons were pelleted from individual rat embryos. The ventral part of spinal cords was dissected away, cords from embryos with the same genotype (non-transgenic or transgenic) were pooled after genotyping, and an enriched motor neuron fraction (purity >80%) was obtained by density centrifugation (bovine serum albumin gradient followed by OptiPrep gradient) as previously described (Bishop et al., 2009). Cells pellets were solubilized, electrophoresed in 12% SDS polyacrylamide gels, and immunoblotted for the indicated proteins, as described previously (Magrané et al., 2004). Band intensities were quantified by densitometry using ImageJ software.
Electron microscopy.
Primary motor neurons growing on glass coverslips were fixed at 9 div with a modified Karmovsky's solution of 2.5% glutaraldehyde, 4% paraformaldehyde and 0.02% picric acid in 0.1 m sodium cacodylate buffer at pH 7.2. After a secondary fixation in 1% osmium tetroxide and 1.5% potassium ferricyanide, samples were dehydrated through a graded ethanol series, and embedded in an Epon analog resin. After polymerization, the coverslip glass was etched away with hydrofluoric acid. Ultrathin sections were cut using a Diatome diamond knife on a Leica Ultracut S ultramicrotome. Sections were collected on copper grids and further contrasted with lead citrate, and viewed on a JSM 100 CX-II electron microscope (JEOL USA) operated at 80 kV. Images were recorded on Kodak 4489 Electron Image film then digitized on an Epson Expression1600 Pro scanner at 900 dpi for publication.
Statistical analyses.
Data were expressed as the mean ± SE. Differences between two groups were measured by the Student's two-tailed, unpaired t test, with significance set at p < 0.05. When these two groups were followed over time, an ANOVA with repeated measurements followed by Fisher post hoc test was used, and significance set at p < 0.05.
Results
MitoDendra, a fluorescent protein for live imaging of mitochondrial dynamics
To develop appropriate imaging tools for live imaging of mitochondrial fusion and transport in motor neurons, the photo-switchable fluorescent protein Dendra (Gurskaya et al., 2006) was targeted to the mitochondrial matrix (mitoDendra) by adding a cleavable N-terminal presequence derived from subunit VIII of cytochrome c oxidase (COX VIII). A linker peptide was introduced between the presequence and Dendra to improve fusion protein cleavage (Fig. 1A). MitoDendra was transfected into primary embryonic rat neurons, and the mitochondrial localization of mitoDendra was confirmed by immunostaining and colocalization with the mitochondrial proteins MnSOD and cytochrome c (Fig. 1B).
Figure 1.

Study of mitochondrial dynamics in motor neurons using the photoactivatable fluorescent protein Dendra. A, Schematic representation of the mitoDendra construct used to label mitochondria. Restrictions sites are indicated. B, Costaining with MnSOD and cytochrome c confirms the mitochondrial localization pattern of mitoDendra in a transfected neuron. Scale bar, 2 μm. C, There was no photoconversion of Dendra during live imaging at low laser intensity (see absence of red fluorescence before photoactivation). Upon photoactivation of mitochondria in a ROI, green and red fluorescence were imaged simultaneously. Scale bar, 10 μm. D, Time-lapse microscopy of two-colored mitoDendra-labeled mitochondria in the soma of a living neuron after photoactivation of a subpopulation of mitochondria (t = 0 min). Frames were acquired every 30 s for a total recording time of 105 min (the panels show selected frames at 15 min intervals). Fusion events of mitochondria were identified by the appearance of yellow color, which resulted from merging green and red mitochondria. Examples of fusion are indicated by arrows at 15 min after photoactivation of mitoDendra. Images from single z-sections are shown. Numbers refer to time in minutes. Scale bar, 5 μm.
First, we determined the optimal confocal microscopy imaging conditions to visualize and photoactivate mitoDendra in living neurons. Low-power 488 nm laser excitation (<0.5 mW) did not cause changes in the morphology of the transfected neurons or their mitochondria (data not shown). Furthermore, there was no photoconversion to red (Fig. 1C, left), even after repetitive imaging for extended periods of time (250 times over 2 h, data not shown). However, exposure to 10–30 iterations at higher 488 nm laser intensity (2.5 mW) was sufficient to completely and irreversibly photoconvert mitoDendra from green to red fluorescence at a defined ROI (Fig. 1C, right).
Then, we demonstrated the effectiveness of mitoDendra in studying the dynamics of mitochondria in primary neurons by time-lapse in vivo imaging (Fig. 1D). The ability to generate two differentially labeled (green or red) populations of mitochondria within the same neuron by photoconversion of mitoDendra, allowed for detection of mitochondrial fusion events. In the example shown in Figure 1D, the neuronal mitochondrial network underwent extensive fusion, as demonstrated by the mixing of red and green fluorescence (Fig. 1D). We also determined that repetitive imaging did not cause photobleaching of mitoDendra fluorescence (Fig. 1D).
Impaired mitochondria fusion in G93A SOD1 motor neurons
Mitochondrial fusion is thought to be essential for neuronal survival, since genetic defects of components of the fusion machinery, such as Opa1 and Mfn2, result in neurodegeneration (Chen and Chan, 2006; Chen et al., 2007). To determine whether mitochondrial fusion is affected by mutant SOD1 we analyzed the reshaping of mitochondrial network in the soma and axons of motor neurons.
Primary motor neurons from the spinal cords of transgenic rat E14.5 embryos expressing either WT or G93A mutant SOD1, and respective non-transgenic embryos, were isolated and transfected with mitoDendra after 5–7 days in vitro (div) for mitochondrial imaging.
In the soma, it was difficult to follow the fate of individual mitochondria, because of the high density of the mitochondrial network, which does not allow for single mitochondria resolution. Therefore, taking advantage of the biophysical properties of mitoDendra, we photoconverted subpopulations of mitochondria in motor neuron cell bodies from green to red fluorescence, and followed their fate by time-lapse confocal microscopy. As a result of the mixing of green and red fluorescence by fusion, yellow mitochondria were detectable in the merged time-lapse images of non-transgenic neurons 40 min after photoconversion and increased over time (Fig. 2A, top). However, in G93A SOD1 motor neuron somas, yellow fluorescence was not apparent until 100 min after photoactivation (Fig. 2A, bottom). Quantification of the fusion rates (colocalization of red over green fluorescence) demonstrated a linear increase (correlation coefficient, r ≥ 0.99) in the amount of colocalization over time, in both control and mutant SOD1 somas, but the rate of fusion was slower (p = 0.013 by ANOVA with repeated measurements; n = 7–8) in G93A motor neuron somas (Fig. 2B).
Figure 2.
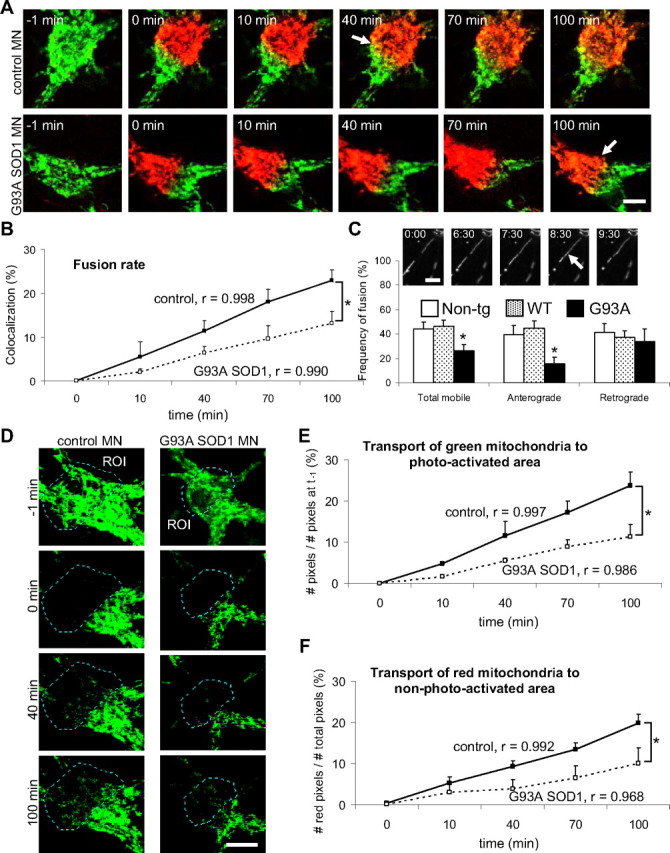
Impaired mitochondrial fusion in the soma and motor axons of G93A SOD1 motor neurons. A, Cell bodies from non-transgenic control and G93A SOD1 motor neurons containing mitoDendra-labeled mitochondria before (−1 min) and after (0 min) photoactivation. Subsets of mitochondria (green and red fluorescence) were followed over time (10, 40, 70, and 100 min) by live imaging microscopy. Note that the appearance of yellow fluorescence, as a result of the mixing between green and red mitochondria (i.e., fusion, indicated by arrows), was delayed in mutant SOD1 motor neurons. Scale bar, 10 μm. B, Fusion rates were obtained in single optical z-sections by measuring the colocalization (in percentage) of red over green fluorescent mitochondria at the indicated time points. The correlation coefficient (r) for each group is indicated. n (somas) = 8 non-transgenic, and 7 G93A. *p < 0.05 by ANOVA with repeated measurements. C, In the inset, example of a time-lapse recording of axonal mitochondria (numbers indicate time in minutes) showing a fusion event (arrow). Scale bar, 5 μm. The graph shows the analysis of fusion (% of fusion events of total moving mitochondria) in control and G93A SOD1 motor axons. n (axons) = 24 non-transgenic, 28 WT and 16 G93A axonal segments. *p < 0.05 versus non-transgenic. D, Time-lapse microscopy of mitochondrial transport in the soma of non-transgenic and G93A SOD1 motor neurons. All mitochondria in a ROI of the soma were photoconverted, while only non-photoconverted (green fluorescent) mitochondria were followed over time. Note a decrease of mobile mitochondria toward the ROI in mutant SOD1 motor neurons compared with controls. Scale bar, 10 μm. E, Analysis of the transport of green mitochondria over the photoactivated area (no green mitochondria present). The correlation coefficient (r) for each group is indicated. n (somas) = 8 non-transgenic, and 7 G93A. *p < 0.05 by ANOVA with repeated measurements. F, Analysis of the transport of red mitochondria over the non-photoactivated area (no red mitochondria present). The correlation coefficient (r) for each group is indicated. n (somas) = 8 non-transgenic, and 7 G93A. *p < 0.05 by ANOVA with repeated measurements. All data obtained from 3–5 independent experiments. The error bars represent ± SE. Non-tg, Non-transgenic.
In the motor axons, identified by morphological criteria, fusion events involving individual mitochondria could be visualized during time-lapse recordings without applying photoactivation (Fig. 2C, insets). Quantification of fusion events involving mobile mitochondria revealed that G93A SOD1, but not WT SOD1, axons had less mitochondrial fusion than non-transgenic controls (p = 0.03 by Student's t test; n = 16–28). The fusion impairment affected anterograde (p = 0.02 by Student's t test; n = 12–26), but not retrograde, moving axonal mitochondria (Fig. 2C).
Together these results indicate that in G93A SOD1 motor neurons there is a defect of mitochondrial dynamics, which involves fusion and affects both axons and cell bodies.
Defective transport of mitochondria in G93A SOD1 motor neurons
Mitochondrial fusion defects in neurons can be accompanied by impaired mitochondrial motility, although whether the two defects are mechanistically linked or if they can develop independently is still unclear (Misko et al., 2010). Therefore, we investigated mitochondrial transport dynamics in mutant SOD1 motor neurons. The ability to discriminate between different populations of mitochondria within the same cell using mitoDendra photoactivation was used to study mitochondrial transport in the motor neuron somas. We followed the transport of green mitochondria into the photoconverted (red) areas (Fig. 2D) and, vice versa, the transport of red mitochondria into the non-photoconverted (green) areas (data not shown) over time. We found that the rate of transport of both green (Fig. 2E) and red (Fig. 2F) mitochondria within the cell soma was reduced in G93A motor neurons, compared with non-transgenic controls (p = 0.011 and p = 0.039, respectively, by Student's t test at 100 min; p = 0.025 and p = 0.032, respectively, by ANOVA with repeated measurements; n = 7–8).
To quantify fast axonal transport of individual mitochondria in the axons of motor neurons we generated kymographs from time-lapse recordings. Kymographs provide graphical representations of spatial position over time, where the y-axis represents time and the x-axis, distance. A decrease in the number of diagonal traces indicated a reduction in the number of moving mitochondria in G93A axons (Fig. 3A). Mitochondrial motility, defined as the proportion of mitochondria moving in any direction during the time of the recording (5 min), was decreased in G93A SOD1 motor neurons (17.23 ± 2.29%) compared with non-transgenic controls (29.01 ± 2.36%; P = 0.0008 by Student's t test; n = 18–29), while the number of stationary mitochondria in G93A SOD1 motor neurons was increased (82.77 ± 2.29% mutant SOD1 vs 70.99 ± 2.36% non-transgenic controls; P = 0.0008 by Student's t test), consistently with the decreased motility. Motility in WT SOD1 motor neurons was not significantly different from non-transgenic controls.
Figure 3.
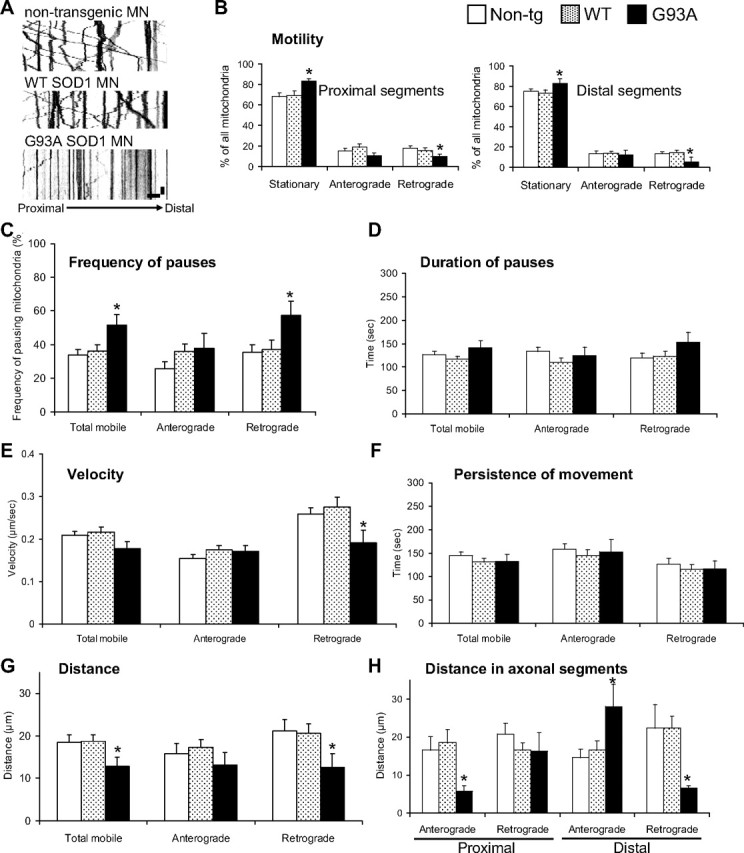
Mitochondrial dynamics abnormalities in mutant SOD1 motor neuron axons. A, Representative kymographs of mitoDendra-labeled mitochondria recorded in distal axons from non-transgenic, WT SOD1, and G93A SOD1 motor neurons (MN) at 5 s intervals for 5 min showed reduced motility in mutant motor neurons. Color in the image was inverted (negative image) for clarity. Scale bars, 10 μm (horizontal), and 1 min (vertical). B, Analysis of mitochondrial transport by direction of movement (anterograde and retrograde) and axonal regions (proximal and distal). Fewer mitochondria moved retrogradely in both proximal and distal axonal segments. n (axons) = 29 non-transgenic, 26 WT and 18 G93A. C, Analysis of the frequency at which mitochondria pause (expressed as percentage of all moving mitochondria). n (axons) = 29 non-transgenic, 28 WT and 20 G93A axonal segments. D, Duration of pauses was unchanged in G93A SOD1 motor neurons. n (pauses) = 74 non-transgenic, 85 WT and 23 G93A. E, Analysis of the velocities of mobile mitochondria in each direction. n (moving events) = 355 non-transgenic, 261 WT and 123 G93A. F, Analysis of the persistence (duration) of movement showed no significant differences in mutant SOD1 motor neurons. n (moving events) = 128 non-transgenic, 78 WT and 56 G93A. G, Analysis of the distance traveled (during movement without pause) achieved by mobile mitochondria. n (moving events) = 118 non-transgenic, 105 WT and 59 G93A. H, Analysis of the distance traveled by mitochondria in axonal regions showed that mitochondrial movement in distal parts was unbalanced, with increased anterograde and decreased retrograde movements. n (moving events) = 118 non-transgenic, 105 WT and 59 G93A. Data were obtained from 4–5 independent motor neuron isolations. The error bars represent ± SE in all graphs. *p < 0.05 versus non-transgenic. Non-tg, Non-transgenic.
To study whether mitochondrial transport abnormalities were widespread along the motor axons or limited to specific axonal regions, we defined two regions: proximal (starting within 20 μm from the cell body and spanning 152 ± 34 μm; n = 45 axons), and distal (ending within 20 μm from the axon terminal and spanning 156 ± 41 μm; n = 36). By focusing the analysis on proximal or distal segments of the motor neuron axons and on the direction of the movement, we identified a selective decrease of retrograde transport, in both proximal and distal regions of G93A SOD1 (P = 0.013 and 0.017, respectively, by Student's t test; n = 6–17), but not in WT SOD1 motor neurons (Fig. 3B), compared with non-transgenic controls.
To further characterize the effects of mutant SOD1 on mitochondrial transport, we analyzed the frequency and duration of pauses, the velocity of transport, the distance traveled between pauses, and the persistence (duration) of movement. We observed that mitochondria paused more often in the retrograde direction in mutant SOD1 motor neurons (Fig. 3C; p = 0.016 by Student's t test; n = 18–27 axons), while there were no significant differences in the duration of the pauses (Fig. 3D). Moreover, the velocities in the retrograde direction were reduced in mutant motor neurons (Fig. 3E; p = 0.04 by Student's t test; n = 123–355 moving events). Since there were no significant differences in the persistence of movement of mitochondria (Fig. 3F), the distance traveled by mobile mitochondria was shorter in G93A motor neurons (Fig. 3G; p = 0.048 by Student's t test; n = 56–128 moving events). Finally, we observed an imbalance in the distance traveled by mitochondria in distal segments of G93A SOD1 axons: retrograde distances were shorter (p = 0.033 by Student's t test; n = 59–118 moving events), whereas anterograde distances were longer (Fig. 3H; p = 0.013 by Student's t test; n = 59–118 moving events).
To determine whether fast axonal transport defects affected other cargos, we labeled MBOs with amyloid precursor protein (APP)-Dendra (Fig. 4A) (Koo et al., 1990; Kaether et al., 2000). Kymographs of APPDendra-labeled MBOs demonstrated higher motility of these vesicles (Fig. 4B) compared with mitochondria (70 ± 4.0% of MBOs in control motor neurons moved within the time-frame of the recording, compared with ∼30% of mitochondria). Unlike mitochondria, the anterograde and retrograde motility of MBOs was unchanged in proximal and distant segments of G93A SOD1 motor neurons (Fig. 4C). Velocity of MBO transport was also unaffected (Fig. 4D), in both directions and axonal segments. These results suggest that in motor neurons the fast axonal transport of mitochondria, but not that of other fast moving organelles, such as MBOs, is affected by mutant SOD1.
Figure 4.

No abnormalities in the transport of MBOs. A, Schematic representation of the APPDendra construct used to label MBOs. B, Representative kymographs of APPDendra-labeled MBOs recorded from control and G93A SOD1 motor neurons at 5 s intervals for 5 min. Color in the image was inverted (negative image) for clarity. Scale bars, 10 μm (horizontal), and 1 min (vertical). C, Percentage of stationary and mobile (anterograde or retrograde) MBOs in proximal and distal segments of control and G93A SOD1 motor axons. No significant differences were observed. n (axonal segments) = 24 control and 21 G93A. D, Velocities in proximal and distal segments of control and G93A SOD1 motor axons. No significant differences were found. n (MBOs) = 228 control and 272 G93A. The error bars represent ± SE. Data obtained from 4 independent motor neuron isolations.
Reduced mitochondrial length and density in G93A SOD1 motor neurons
A logical consequence of impaired mitochondrial transport and fusion could be the disruption of normal mitochondrial morphology and distribution along axonal processes. Therefore, to first assess mitochondrial morphology we measured the size of mitochondria in proximal and distal regions of motor neuron axons. Mitochondrial average length was reduced in the distal segments of G93A SOD1 motor neuron axons, but not in WT SOD1 (Fig. 5A,B), compared with non-transgenic controls (p = 0.05−10 by Student's t test; n = 192–331 mitochondria). Furthermore, while at 5 div mitochondria in the proximal segments of G93A SOD1 motor axons were of normal size (Fig. 5B), at 10 div their length had significantly decreased compared with controls (3.33 ± 0.24 μm mutant SOD1 vs 4.03 ± 0.24 μm non-transgenic controls; P = 0.04 by Student's t test; n = 113–138), suggesting that these abnormalities progress from distal to proximal segments as the neurons age in culture. No significant decrease in mitochondrial length was detected in dendrites of mutant SOD1 motor neurons (Fig. 5C), indicating that mitochondrial morphological abnormalities were limited to motor axons.
Figure 5.

Mitochondrial morphological abnormalities in axons, but not dendrites, of mutant SOD1 motor neurons. A, Images of mitoDendra-labeled mitochondria in distal axonal segments of non-transgenic, WT SOD1, and G93A SOD1 motor neurons (MN). Scale bar, 5 μm. B, Quantification of mitochondrial length from proximal and distal axonal segments in non-transgenic, WT SOD1, and mutant motor neurons. Mitochondria were significantly smaller in distal portions of G93A axons. n (mitochondria) = 533 non-transgenic, 566 WT and 381 G93A. *p < 0.0005 versus non-transgenic. C, Quantification of mitochondrial length in dendrites. No significant differences were observed. n (mitochondria) = 309 non-transgenic, and 367 G93A. D, Analysis of mitochondria distribution in controls and mutant motor neurons revealed no differences among groups. n (axons) = 29 non-transgenic, 29 WT and 23 G93A. E, Representative fluorescence (top) and electron microscopy (bottom) images of the distal tip of axons of non-transgenic and G93A SOD1 motor neurons. Dashed lines delineate the axon terminals. Note the similar mitochondrial distribution pattern in control and mutant motor neurons. Scale bars, 5 μm for fluorescence, and 1 μm for electron microscopy images. F, Analysis of mitochondria distribution in non-transgenic, WT SOD1, and mutant motor neuron distal axonal segments. Distribution measurements were binned into 25 μm segments. No significant differences were observed among groups. n (axons) = 15 non-transgenic, 16 WT and 14 G93A. G, Analysis of mitochondrial density in controls and mutant motor neurons. Mitochondrial density was significantly reduced distally in G93A SOD1 motor neurons. n (axons) = 29 non-transgenic, 29 WT and 23 G93A. *p < 0.05 versus non-transgenic. Data obtained from 3–5 independent motor neuron isolations. The error bars represent ± SE. Non-tg, Non-transgenic.
Second, to assess mitochondrial distribution in motor neuron axons we measured the average number of mitochondria contained in proximal and distal axonal segments. Despite the alterations in retrograde transport, there were no significant differences, either in proximal or distal segments, in the number of mitochondria among mutant SOD1, WT, and non-transgenic control axons (Fig. 5D). We confirmed this lack of changes in mitochondrial distribution in G93A SOD1 motor neurons by labeling mitochondria with MitoTracker and cytochrome c antibodies (data not shown). Moreover, the distribution of mitochondria at the axon terminals, assessed by fluorescence (Fig. 5E, top) and electron microscopy (Fig. 5E, bottom) did not appear altered in G93A SOD1 motor neurons. Further quantification of mitochondrial distribution in defined 25 μm segments did not reveal significant differences among groups at the axonal terminals (Fig. 5F).
We then analyzed mitochondrial “density,” expressed as the sum of all the mitochondrial lengths in 50 μm axonal segments. Mitochondrial density was reduced in the distal segments of G93A SOD1 axons, but not WT SOD1, compared with non-transgenic controls (Fig. 5G, p = 0.027 by Student's t test; n = 23–29 axons). However, no differences were observed between control and mutant SOD1 when measuring the density of mitochondria in the motor neuron cell bodies (data not shown).
These results suggest that mutant SOD1 may cause an imbalance in the distribution of mitochondria along the axons, which appears first at the periphery of the motor neurons. Although the overall number of mitochondria was unchanged in mutant SOD1 and control axons, there was a decrease of mitochondrial density, as measured by mass, in the mutants due to smaller average mitochondrial size.
Mitochondrial dynamics and morphology are not altered in G93A SOD1 cortical neurons
To investigate whether abnormalities in mitochondrial dynamics and morphology are specific to motor neurons or they affect other neuronal types, we studied cortical neurons from G93A SOD1 and control rat embryos at 8 div. Kymographs revealed a similar motility pattern for mitochondria in both non-transgenic controls and G93A SOD1 cortical neurons (Fig. 6A). After analysis of time-lapse recordings, no significant differences in the frequency of mitochondrial movement were found in either proximal or distal segments of the axons (Fig. 6B), and in either anterograde or retrograde directions (data not shown).
Figure 6.

No abnormalities in transport or morphology of mitochondria in mutant SOD1 cortical neurons. A, Representative kymographs from proximal segments of non-transgenic and G93A SOD1 cortical neurons at 8 div. Color in the image was inverted (negative image) for clarity. Scale bars, 10 μm (horizontal), and 1 min (vertical). B, Analysis of the frequency of movement from time-lapse recordings (5 s intervals for 5 min) of mitoDendra-labeled mitochondria in proximal and distal axonal segments of non-transgenic controls and G93A SOD1 cortical neurons. No significant differences were found in mobile or stationary mitochondria in mutant SOD1 cortical neurons, compared with controls. n (axons) = 16 control and 31 G93A SOD1.Data from 4 independent cortical neuron isolations. The error bars represent ± SE. C, Images of mitoDendra-labeled mitochondria in distal axonal segments of non-transgenic and G93A SOD1 cortical neurons at 8 div. Scale bar, 10 μm. D, Analysis of mitochondrial length in axons of cortical neurons revealed no significant abnormalities in G93A SOD1 neurons. n (mitochondria) = 298 control and 324 G93A. Data from 4 independent cortical neuron isolations. The error bars represent ± SE. E, Analysis of human SOD1 protein levels in cortical neuron and motor neuron lysates isolated from non-transgenic (1) and G93A SOD1 (2) rat embryos. Note that both human SOD1 (hSOD1) and endogenous rat SOD1 (rSOD1) were detected. β-actin was used as loading control. Each lane contains 20 μg of protein. F, Densitometric analysis of relative human SOD1 expression protein levels in purified cortical neurons and motor neurons. Data obtained from 3 independent neuron isolations. The error bars represent ± SE.
The average mitochondrial length in both proximal and distal axonal segments was also unchanged in G93A SOD1 cortical neurons at 8 div (Fig. 6C,D), and no differences in the number of mitochondria and mitochondrial density were found (data not shown).
To exclude that abnormalities developed in cortical neurons at later time points, we analyzed mitochondrial frequency of transport and morphological parameters in proximal and distal segments of neurons at 15 div, and confirmed that there were no changes in G93A SOD1, compared with non-transgenic controls (data not shown).
We then compared SOD1 expression levels in purified cortical neurons and spinal motor neurons by Western blot (Fig. 6E). The relative amount of G93A SOD1 in purified cortical neurons was higher than in motor neurons (Fig. 6F), excluding that the lack of mitochondrial abnormalities in cortical neurons was related to lower mutant protein load.
Accumulation of dysfunctional mitochondria in distal regions of mutant SOD1 motor neurons
Abnormalities of mitochondrial fusion and transport in mutant SOD1 motor neurons could be linked, either as a cause or as a consequence, to mitochondrial functional impairment. Thus, to explore whether there is a functional correlate to the abnormalities in mitochondrial dynamics and morphology in G93A SOD1 motor axons we investigated mitochondrial membrane potential (ΔΨ). Motor neurons were loaded with the potentiometric fluorescent dye TMRM (5 nm) and the intensity of fluorescence was used as a relative measure of ΔΨ (Vives-Bauza et al., 2008). We observed reduced average TMRM intensity in the mitochondria in distal segments of mutant SOD1 motor neurons (Fig. 7A, p = 0.001 by Student's t test; n = 97–145), suggesting that bioenergetic dysfunction in distal motor axon segments may be correlated with reduced size and impaired dynamics of mitochondria. There was a slight but statistically significant increase of TMRM intensity of mutant SOD1 mitochondria in proximal segments (Fig. 7A, p = 0.022 by Student's t test; n = 110–140). The reasons for this increase are not clear, but it could be due to the increase of the ratio of stationary over retrograde moving mitochondria, which are thought to have lower ΔΨ than stationary ones, in neurons (Miller and Sheetz, 2004).
Figure 7.
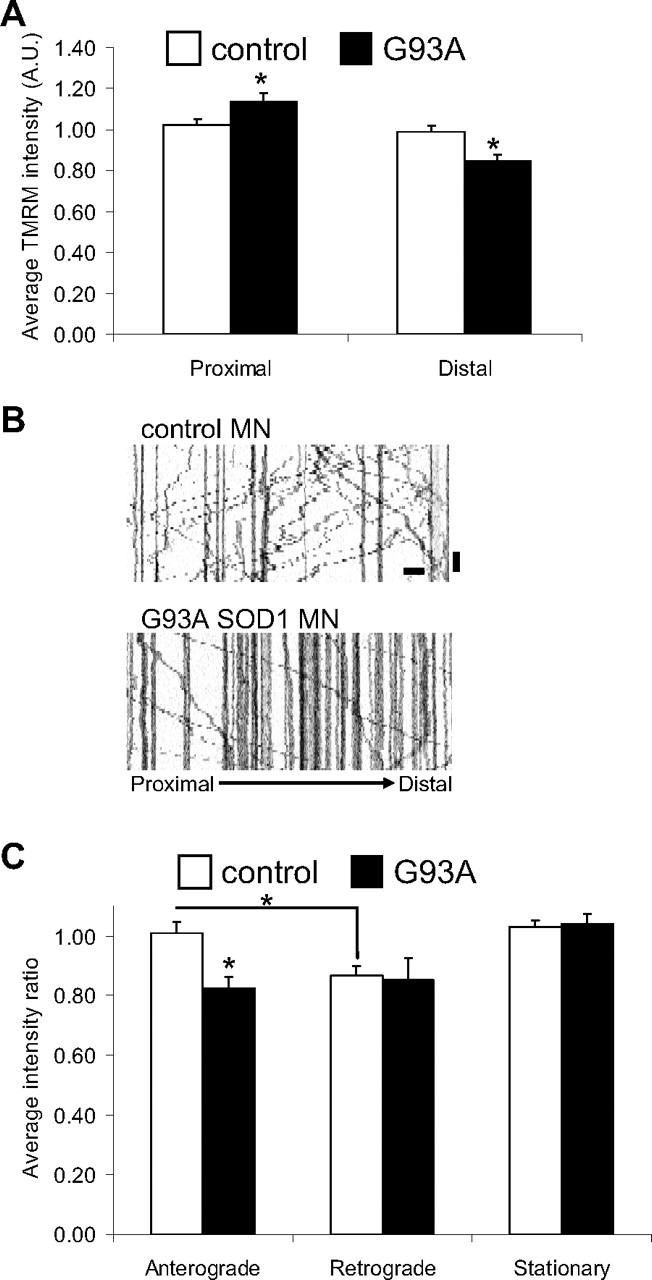
Impairment of mitochondrial bioenergetics in mutant SOD1 motor neuron axons. A, Average TMRM fluorescence intensity (mitochondrial membrane potential) of all mitochondria in proximal and distal segments of non-transgenic and G93A SOD1 motor neurons (8 div). n (mitochondria) = 285 control and 207 G93A. B, Representative kymographs of TMRM-labeled mitochondria from distal axonal segments of control and mutant SOD1 motor neurons, used to correlate membrane potential and mitochondrial transport. Scale bars, 10 μm (horizontal), and 1 min (vertical). C, TMRM intensity measured as the integrated fluorescence in each mitochondrion/area in pixels and expressed as a proportion of average potential in all mitochondria. n (axons) = 12 control and 10 G93A. Data from 3 independent motor neuron isolations. The error bars represent ± SE. *p < 0.05.
Next, TMRM-labeled mitochondria were imaged by time-lapse microscopy, and kymographs were obtained to simultaneously assess mitochondrial transport and ΔΨ. For each genotype, the average TMRM intensities measured for mobile or stationary mitochondria were expressed relative to the average TMRM intensity of all mitochondria analyzed (set at 1). The kymographs confirmed the reduced motility of mitochondria in distal regions of mutant SOD1 motor neurons (Fig. 7B). Anterograde-moving mitochondria in mutant SOD1 motor neurons had lower ΔΨ than anterograde-moving mitochondria in controls (p = 0.0017 by Student's t test; n = 30–37), and were similar to retrograde-moving mitochondria in both mutant and control neurons (Fig. 7C). This finding has potential pathogenic significance, because it suggests that the pool of upcoming mitochondria destined to replace or fuse with distal aging mitochondria in mutant SOD1 motor axons may be functionally impaired.
Fewer and smaller synaptic puncta in G93A SOD1 motor neurons
Since neuronal mitochondria are often localized in proximity of synapses to provide the necessary energy and calcium buffering, we hypothesized that impaired mitochondrial transport in mutant SOD1 motor neurons could be associated with synaptic abnormalities. To investigate mitochondrial localization at synaptic sites we looked at the synaptic contacts that are spontaneously formed among motor neurons in vitro, and used these structures as reference sites, where to study mitochondrial localization. We labeled the neuron-to-neuron synapses with antibodies against the presynaptic proteins SV2 and synapsin I, and measured both the number and the size of synaptic puncta. G93A SOD1 motor neurons presented a markedly decreased number of SV2- and synapsin I-positive puncta (p = 0.05−11 and p = 0.000004 by Student's t test for SV2 and synapsin I labeling, respectively; n = 21–27 axons) and smaller puncta sizes (p = 0.0006 and p = 0.00011 by Student's t test for SV2 and synapsin I labeling, respectively; n = 190–385 puncta) compared with controls (Fig. 8A–C).
Figure 8.
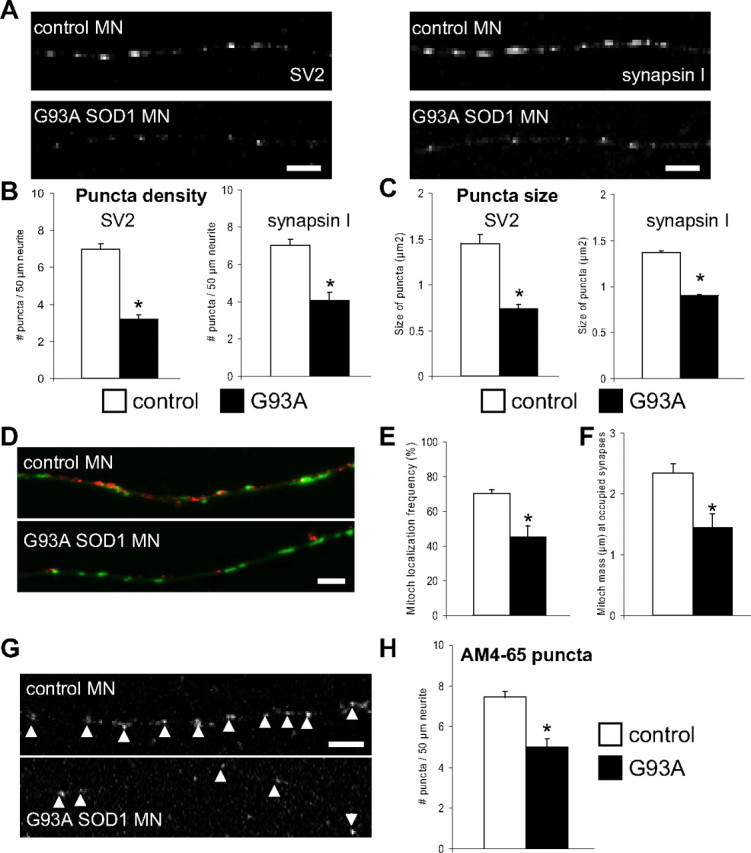
Synaptic abnormalities in G93A SOD1 motor neurons. A, Representative images of synaptic structures labeled with SV2 and synapsin I in control and G93A SOD1 motor neurons. Scale bar, 5 μm. B, Analysis of puncta density (number of puncta per 50 μm of axon) indicated fewer puncta in G93A SOD1 motor neurons compared with non-transgenic controls. For SV2 labeling, n (axons) = 27 control and 26 mutant SOD1; for synapsin I labeling, n (axons) = 24 control and 21 mutant SOD1. *p > 0.0005. C, Quantification of size of puncta demonstrated a reduction in size in G93A SOD1 motor neurons. For SV2 labeling, n (puncta) = 385 control and 190 mutant SOD1. *p > 0.005; for synapsin I labeling, n (puncta) = 331 control and 302 mutant SOD1. *p > 0.0005. D, Representative images of mitochondria (in green) and SV2-labeled puncta (in red) in control and mutant SOD1 motor neuron axons. Scale bar, 5 μm. E, Analysis of the number of synaptic puncta having mitochondria within a 1 μm distance (frequency of mitochondrial localization) revealed that fewer puncta in G93A motor neurons contained mitochondria, compared with non-transgenic motor neurons. n (axons) = 20 control and 20 mutant SOD1. *p > 0.005. F, Quantification of synaptic mitochondrial density identified a decrease in the mass (μm) of mitochondria at occupied synapses from mutant SOD1 motor neurons. n (axons) = 22 control and 22 mutant SOD1. *p > 0.005. G, Representative images of functional synaptic clusters labeled with AM4-65 in control and mutant SOD1 motor axons. Arrowheads indicate active synapses. Scale bar, 5 μm. H, Analysis of puncta density indicated fewer functional synapses in G93A SOD1 motor neurons. n (axons) = 29 control and 20 mutant SOD1. *p > 0.005. Data from 3 independent motor neuron isolations. The error bars represent ± SE.
To determine whether abnormal mitochondrial distribution was associated with the observed synaptic abnormalities, we measured the colocalization of mitochondria and SV2-labeled puncta (Fig. 8D). Two parameters were used for the measurements: the first was the probability of finding a mitochondrion localized within 1 μm from the puncta; the second was the mitochondrial density at the puncta, defined as the sum of all the mitochondrial lengths within 5 μm from each puncta. Both the probability of mitochondria-puncta colocalization (Fig. 8E) and the puncta mitochondrial density (Fig. 8F) were reduced in the axons of G93A SOD1 motor neurons (p = 0.018 and p = 0.010 by Student's t test, respectively; n = 20–22 axons).
To focus on functional synapses and to investigate differences in synaptic function, we used the dye AM4-65. AM4-65 is loaded into synaptic vesicles at functional synapses under synaptic activation (Cochilla et al., 1999). We analyzed axonal segments from mutant SOD1 and control motor neurons after incubation with AM4-65 under depolarizing conditions. Quantification of the density of AM4-65 puncta revealed a reduction in the number of synaptic puncta in G93A SOD1 motor neurons (p = 0.000007 by Student's t test; n = 20–29 axons; Fig. 8G,H), which indicates that the synaptic morphological changes correlated with synaptic dysfunction.
These findings suggest the possibility that a lack of appropriate supply of functional mitochondria is involved in causing synaptic abnormalities in mutant SOD1 motor neuron axons.
Discussion
We used time-lapse confocal microscopy and mitoDendra labeling to investigate mitochondrial dynamics in motor neurons from transgenic rats expressing ALS-linked G93A mutant or WT SOD1. We identified abnormalities involved in fundamental aspects of mitochondrial biology, such as fusion and transport, in mutant, but not in WT, SOD1 motor neurons. In association with these defects, mitochondria in axons had decreased size and density, and impaired bioenergetics. Moreover, abnormal mitochondrial distribution in mutant SOD1 motor neurons correlated with neuron-to-neuron synaptic dysfunction.
Mitochondrial fusion is a process that appears to be necessary for the maintenance of healthy mitochondria and to ensure neuronal viability (Chen and Chan, 2006; Chen et al., 2007). By time-lapse microscopy of individual motor neurons, we showed decreased mitochondrial fusion in cell bodies and axons of mutant SOD1 motor neurons. Defective fusion may derive from impaired transport, if mitochondria are not brought in close proximity then fusion cannot be initiated. On the other hand, it is possible that defective fusion is a primary pathogenic event, leading to defective mitochondrial morphology and function, which then results in transport defects. In favor of this interpretation, ablation of Mfn2 gene in neurons has been shown to cause mitochondrial hyper-fragmentation and impaired transport (Chen et al., 2007).
The apparent transport defects affected mitochondria but not MBOs, which were normally trafficked along motor neuron axons. This indicates that mutant SOD1 does not impair fast axonal transport globally, but selectively targets specific organelles. Furthermore, mitochondrial abnormalities appeared to spare dendritic processes of motor neurons as well as cortical neuron axons, suggesting that motor neuron axons are highly susceptible to mitochondrial damage induced by mutant SOD1. The selective impairment of mitochondrial transport in spinal motor neurons may relate to a preferential binding of mutant SOD1 to mitochondria of spinal cord (Liu et al., 2004; Pasinelli et al., 2004), although the molecular determinants of these interactions involved in transport defects remain to be further investigated.
Earlier studies of mitochondrial transport in mutant SOD1 models had shown defects in anterograde axonal transport of mitochondria (De Vos et al., 2007). In those studies, non-transgenic rat cortical neurons were investigated after transient transfection with exogenous human mutant SOD1, which resulted in impaired mitochondrial transport. In our study, we did not detect mitochondrial transport or morphological abnormalities in primary cortical neurons derived from the same strain of mutant SOD1 transgenic rats used for the spinal motor neuron studies. The difference between our results and those by De Vos et al. (2007) may be due to our use of a homologous model system, where expression levels of the mutant protein were driven by the same transgenic construct in all tissues. Also, the main direction of the motility defect was different, since we identified a defect predominantly in retrograde transport. Our results are consistent with the finding of Zhang et al. that an aberrant interaction exists between mutant SOD1 and components of the dynein complex that are involved in retrograde mitochondrial transport (Zhang et al., 2007). Furthermore, vesicular axonal retrograde transport defects have been described in detail in motor neurons expressing mutant SOD1 by Perlson et al. (2009). MBO axonal transport defect was also reported in both anterograde and retrograde directions in cortical neurons transiently transfected with mutant SOD1 (De Vos et al., 2007). We investigated MBOs in spinal cord motor neurons and did not detect transport abnormalities. Again, these differences underline the importance of investigating homogeneous model systems.
There is evidence that argues in favor of a cause-effect relationship between mitochondrial dynamics impairment and distal motor neuron degeneration. For instance, such a relationship has been directly demonstrated in a Drosophila model, where inactivation of the cargo adaptor for fast mitochondrial anterograde transport, dMiro, resulted in mitochondrial accumulation in the cell body, NMJ degeneration, and muscle denervation and atrophy (Guo et al., 2005). Our findings seem consistent with the hypothesis that mitochondrial transport may be involved in distal motor neuron degeneration, since in mutant SOD1 motor axons mitochondrial abnormalities were prominent in the distal regions. This could be an indication that distal axonal mitochondria suffer from accelerated degeneration, as a consequence of impaired transport and fusion, possibly because of inadequate fusion with new mitochondria originated in the soma.
Impaired mitochondrial fusion and transport, and abnormal morphology correlated with reduced mitochondrial ΔΨ in mutant SOD1 motor neurons. The finding that only mitochondria moving in an anterograde direction had a reduction of ΔΨ may suggest that mitochondria moving toward the distal portions of the axon were damaged by mutant SOD1, and incapable of providing for the bioenergetic needs of the axonal terminals and to reactivate aging mitochondria by fusing with them. These findings agree with the observations that mutant SOD1 axons start degenerating at their terminals, where damaged mitochondria accumulate early on in the course of degeneration (Vande Velde et al., 2004; Gould et al., 2006).
Mitochondrial bioenergetic impairment has been extensively demonstrated in the mutant SOD1 mouse (for review, see Magrané and Manfredi, 2009), but the causal relationship between impaired bioenergetics and defective transport needs to be further clarified. While it is possible that the two defects originate independently and proceed in parallel, it might also be that transport defects contribute to the bioenergetic problem. It could be hypothesized that mitochondria are not effectively regenerated and fail to maintain sufficient metabolic activity, and eventually succumb when challenged with heavy workloads, such as buffering of intracellular Ca2+ spikes. Dysfunctional mitochondria that are not efficiently recycled by fusion with healthy mitochondria may become fragmented and targeted for mitophagy (Twig et al., 2008).
The molecular determinants of impaired mitochondrial dynamics in mutant SOD1 motor axons have not yet been characterized. We observed that mutant SOD1 caused a predominant effect on retrograde transport of mitochondria, with reduced motility, increased frequency of pauses, decreased velocity and decreased distance traveled. The observation that some of these abnormalities were present predominantly in either proximal or distal regions of the axons suggests that different players may be involved in regulating transport of mitochondria in these regions. Furthermore, the balance between competing anterograde and retrograde forces driving organelle transport (Ally et al., 2009; Hendricks et al., 2010; Schuster et al., 2011) could be altered, if mutant SOD1 interferes preferentially with the molecular machinery for anterograde or retrograde transport. Normal MBO transport seems to exclude abnormalities in microtubule tracks, which would likely impair the transport of all cargoes and not mitochondria specifically. In addition to a direct interaction with the transport machinery, it is possible that SOD1 interferes with the complex signaling mechanisms regulating mitochondrial motility. For example, inactivation of the cargo adaptor Miro1 by increasing local Ca2+ concentration has been recently identified (Liu and Hajnóczky, 2009), suggesting that impaired mitochondrial Ca2+ handling in mutant SOD1 motor neurons (Kawamata and Manfredi, 2010) could contribute to transport defects. Furthermore, mutant SOD1 may affect mitochondrial motility by impairing the regulation of molecular motors by cytosolic kinases (De Vos et al., 2000; Morfini et al., 2006; Bosco et al., 2010).
What are the consequences of mitochondrial mislocalization and dysfunction in motor neuron axons? Published reports suggest that mitochondrial Ca2+ uptake is impaired in brain, spinal cord and motor nerve terminals in G93A SOD1 mice (Vila et al., 2003; Damiano et al., 2006). Furthermore, in presymptomatic mutant mice, an altered response to Ca2+ loads in mitochondria within motor neurons involving altered mitochondrial membrane potential has been described (Nguyen et al., 2009). Based on these findings and on our observations of mitochondrial dynamics abnormalities, it is possible that mitochondrial Ca2+ handling is defective at the NMJs of mutant SOD1 mice, resulting in impaired local Ca2+ homeostasis. Damaged mitochondria may also be incapable of providing adequate ATP supplies for synaptic transmission, and may become the source of toxic free radicals. Although the motor neuron-to-motor neuron synaptic changes observed in vitro have unknown pathological significance for ALS, the correlation between abnormal mitochondrial distribution at presynaptic sites and reduction of size, number, and function of synapses supports the view that mitochondrial dynamics defects may contribute to synaptic abnormalities.
Together, our observations suggest that perturbation of axonal mitochondrial dynamics may contribute to motor neuron degeneration in familial ALS with SOD1 mutations. Future studies will have to clarify the molecular mechanisms underlying mitochondrial dynamics impairment in mutant SOD1 motor neurons. They will also need to determine whether impaired mitochondrial dynamics is a common factor in other motor neuron diseases, including other familial forms and sporadic ALS.
Footnotes
This work was supported by grants from the Muscular Dystrophy Association (to J.M., S.P., G.M.), the Robert Packard Center for ALS Research “The New York Community Trust” (to G.M.), the National Institutes of Health (Grants NS051419, NS062055 to G.M.; Grants NS042269, NS064191, NS38370, NS070276, and NS072182 to S.P.; Grant NS36761 to A.G.E), the Department of Defense (Grants W81XWH-08-1-0522, W81XWH-08-1-0465, W81XWH-09-1-0245 to S.P.), and Project A.L.S. (to S.P.). We thank Dr. Konstantin A. Lukyanov (Russian Academy of Sciences, Moscow, Russia) for Dendra cDNA, and Leona Cohen-Gould (Cornell University, New York, NY) for preparation and imaging of samples for transmission electron microscopy. We also thank Dr. Geri E. Kreitzer (Cornell University, New York, NY) and Dr. Roy C. Smith (Tufts University, Boston, MA) for discussion and critical reading of the manuscript.
The authors declare no competing financial interests.
References
- Ally S, Larson AG, Barlan K, Rice SE, Gelfand VI. Opposite-polarity motors activate one another to trigger cargo transport in live cells. J Cell Biol. 2009;187:1071–1082. doi: 10.1083/jcb.200908075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop A, Hobbs KG, Eguchi A, Jeffrey S, Smallwood L, Pennie C, Anderson J, Estévez AG. Differential sensitivity of oligodendrocytes and motor neurons to reactive nitrogen species: implications for multiple sclerosis. J Neurochem. 2009;109:93–104. doi: 10.1111/j.1471-4159.2009.05891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien JP, Brady ST, Brown RH., Jr Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D, Frank S, Rojo M. Mitochondrial dynamics in cell life and death. Cell Death Differ. 2006;13:680–684. doi: 10.1038/sj.cdd.4401857. [DOI] [PubMed] [Google Scholar]
- Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, Reola L, Carlson E, Epstein CJ. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC. Critical dependence of neurons on mitochondrial dynamics. Curr Opin Cell Biol. 2006;18:453–459. doi: 10.1016/j.ceb.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Cochilla AJ, Angleson JK, Betz WJ. Monitoring secretory membrane with FM1-43 fluorescence. Annu Rev Neurosci. 1999;22:1–10. doi: 10.1146/annurev.neuro.22.1.1. [DOI] [PubMed] [Google Scholar]
- Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, Flint Beal M, Manfredi G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem. 2006;96:1349–1361. doi: 10.1111/j.1471-4159.2006.03619.x. [DOI] [PubMed] [Google Scholar]
- De Vos K, Severin F, Van Herreweghe F, Vancompernolle K, Goossens V, Hyman A, Grooten J. Tumor necrosis factor induces hyperphosphorylation of kinesin light chain and inhibits kinesin-mediated transport of mitochondria. J Cell Biol. 2000;149:1207–1214. doi: 10.1083/jcb.149.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CC, Grierson AJ. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007;16:2720–2728. doi: 10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estévez AG, Spear N, Thompson JA, Cornwell TL, Radi R, Barbeito L, Beckman JS. Nitric oxide-dependent production of cGMP supports the survival of rat embryonic motor neurons cultured with brain-derived neurotrophic factor. J Neurosci. 1998;18:3708–3714. doi: 10.1523/JNEUROSCI.18-10-03708.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Gurskaya NG, Verkhusha VV, Shcheglov AS, Staroverov DB, Chepurnykh TV, Fradkov AF, Lukyanov S, Lukyanov KA. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 2006;24:461–465. doi: 10.1038/nbt1191. [DOI] [PubMed] [Google Scholar]
- Hendricks AG, Perlson E, Ross JL, Schroeder HW, 3rd, Tokito M, Holzbaur EL. Motor coordination via a tug-of-war mechanism drives bidirectional vesicle transport. Curr Biol. 2010;20:697–702. doi: 10.1016/j.cub.2010.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V, Cleveland DW. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67:575–587. doi: 10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaether C, Skehel P, Dotti CG. Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol Biol Cell. 2000;11:1213–1224. doi: 10.1091/mbc.11.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata H, Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech Ageing Dev. 2010;131:517–526. doi: 10.1016/j.mad.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Vande Velde C, Israelson A, Xie J, Bailey AO, Dong MQ, Chun SJ, Roy T, Winer L, Yates JR, Capaldi RA, Cleveland DW, Miller TM. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci U S A. 2010;107:21146–21151. doi: 10.1073/pnas.1014862107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brännström T, Gredal O, Wong PC, Williams DS, Cleveland DW. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Liu X, Hajnóczky G. Ca2+-dependent regulation of mitochondrial dynamics by the Miro-Milton complex. Int J Biochem Cell Biol. 2009;41:1972–1976. doi: 10.1016/j.biocel.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrané J, Manfredi G. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009;11:1615–1626. doi: 10.1089/ars.2009.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrané J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci. 2004;24:1700–1706. doi: 10.1523/JNEUROSCI.4330-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrané J, Hervias I, Henning MS, Damiano M, Kawamata H, Manfredi G. Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum Mol Genet. 2009;18:4552–4564. doi: 10.1093/hmg/ddp421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010;30:4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci. 2006;9:907–916. doi: 10.1038/nn1717. [DOI] [PubMed] [Google Scholar]
- Nguyen KT, García-Chacón LE, Barrett JN, Barrett EF, David G. The Psi(m) depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals. Proc Natl Acad Sci U S A. 2009;106:2007–2011. doi: 10.1073/pnas.0810934106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Pedrini S, Sau D, Guareschi S, Bogush M, Brown RH, Jr, Naniche N, Kia A, Trotti D, Pasinelli P. ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum Mol Genet. 2010;19:2974–2986. doi: 10.1093/hmg/ddq202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, Jeong GB, Ross JL, Dixit R, Wallace KE, Kalb RG, Holzbaur EL. A switch in retrograde signaling from survival to stress in rapid-onset neurodegeneration. J Neurosci. 2009;29:9903–9917. doi: 10.1523/JNEUROSCI.0813-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Sapp P, O'Regan J, McKenna-Yasek D, Schlumpf KS, Haines JL, Gusella JF, Horvitz HR, Brown RH., Jr Genetic linkage analysis of familial amyotrophic lateral sclerosis using human chromosome 21 microsatellite DNA markers. Am J Med Genet. 1994;51:61–69. doi: 10.1002/ajmg.1320510114. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65(Suppl 1):S3–S9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Sanes JR, Lichtman JW. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J Comp Neurol. 2005;490:209–219. doi: 10.1002/cne.20620. [DOI] [PubMed] [Google Scholar]
- Schuster M, Lipowsky R, Assmann MA, Lenz P, Steinberg G. Transient binding of dynein controls bidirectional long-range motility of early endosomes. Proc Natl Acad Sci U S A. 2011;108:3618–3623. doi: 10.1073/pnas.1015839108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Wei Y, Zhang J, Gal J, Zhu H. Mitochondrial dysfunction is a converging point of multiple pathological pathways in amyotrophic lateral sclerosis. J Alzheimers Dis. 2010;20(Suppl 2):S311–S324. doi: 10.3233/JAD-2010-100366. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde C, Garcia ML, Yin X, Trapp BD, Cleveland DW. The neuroprotective factor Wlds does not attenuate mutant SOD1-mediated motor neuron disease. Neuromolecular Med. 2004;5:193–203. doi: 10.1385/NMM:5:3:193. [DOI] [PubMed] [Google Scholar]
- Vila L, Barrett EF, Barrett JN. Stimulation-induced mitochondrial [Ca2+] elevations in mouse motor terminals: comparison of wild-type with SOD1–G93A. J Physiol. 2003;549:719–728. doi: 10.1113/jphysiol.2003.041905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Anand M, Shirazi AK, Magrane J, Gao J, Vollmer-Snarr HR, Manfredi G, Finnemann SC. The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J Biol Chem. 2008;283:24770–24780. doi: 10.1074/jbc.M800706200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Ström AL, Fukada K, Lee S, Hayward LJ, Zhu H. Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex: implications of retrograde axonal transport in ALS. J Biol Chem. 2007;282:16691–16699. doi: 10.1074/jbc.M609743200. [DOI] [PubMed] [Google Scholar]