

Abstract
Aiming at restricting the conformational freedom of tryptophan-containing peptide ligands, we designed a THBC (tetrahydro-β-carboline)-DKP (diketopiperazine)-based peptidomimetic scaffold capable of arranging in an unusual α-turn conformation. The synthesis is based on a diastereoselective Pictet–Spengler condensation to give the THBC core, followed by an intramolecular lactamization to complete the tetracyclic THBC-DKP fused ring system. The presence of conformers bearing the intramolecular thirteen-membered hydrogen bond that characterizes the α-turn structure is confirmed by 1H NMR conformational studies. To the best of our knowledge, this scaffold represents one of the rare examples of a designed constrained α-turn mimic.
Keywords: α-turn, conformational analysis, diketopiperazine, peptidomimetics, tetrahydro-β-carboline
Introduction
From a long time, the alkaloids containing the 1,2,3,4-tetrahydro-β-carboline (THBC) skeleton have represented important lead structures in view of their wide range of biological activities [1], mainly due to their interaction with the central nervous system [2–7]. Moreover, recently some tetracyclic β-carbolines have been described to act as selective inhibitors in the anticancer field [8–9], or to be endowed with antimalarial properties [10]. 2,5-Diketopiperazine (DKP)-based compounds are heterocyclic scaffolds structurally similar to peptides. They have attracted attention in recent years because of their broad biological activities [11–12] and therapeutic applications, ranging from antibiotics [13] to anticancer agents [14]. Moreover, the DKP moiety has been exploited as a peptidomimetic scaffold [15–17]. Structural unification of THBC and DKP pharmacophores has led to new classes of biologically active tetracyclic compounds, both naturally occurring and synthetically made [18]. Highly complex natural products displaying the fused, tetracyclic THBC-DKP ring system were recently isolated from the fungus Aspergillus fumigatus [19] and have been shown to exhibit significant cell growth inhibitory activities against various cell lines [20]. Specifically designed THBC-DKP-based compounds have received considerable attention over the past few years for their valuable biological activities, ranging from the inhibition of the cyclic guanosine monophosphate type 5 specific phosphodiesterase (PDE 5) for the treatment of erectile dysfunction (Tadalafil) [21], and the inhibition of plasmodial PDE activity for antimalarial drugs [22–23], to topoisomerase II inhibition [24], and oral antithrombotic properties [25] (Figure 1).
Figure 1.

THBC-DKP-based natural and synthetically made compounds.
In our ongoing program of design of pharmacophore-based combinatorial libraries [26] and identification of new peptidomimetic scaffolds of potential interest in drug discovery [27–30], we evaluated the THBC-DKP-based scaffold as a potential suitable motif for the creation of unusual reverse-turn nucleators.
Reverse turns are structural motifs commonly found in bioactive peptides, which, besides being fundamental in protein folding, play a central role as molecular-recognition elements [31–34]. In addition to the most frequently occurring β and γ-turns, reversal of the polypeptide chain direction in globular proteins can also occur thanks to less common substructures, for example involving five amino acids residues, such as the case of the α-turns.
Even if a very large majority of α-turn segments form a part of regular α-helices, isolated α-turns have been reported [35], which are stabilized by a 5→1 hydrogen bond between the carbonyl oxygen at position i and the amide at position i+4.
Quite recently the presence of α-turns in constrained peptides has been associated with various relevant biological activities, highlighting potential applications in the field of bacteriolytic [36], antiviral [37] and anti-HIV compounds [38]. Other examples of α-turn conformations are described in synthetic peptidomimetics [36,39–42].
Despite the growing interest in this kind of reverse turn and the need for all kinds of conformationally constrained mimics as tools for medicinal chemistry, the development of constrained α-turn mimetics has received little attention until now. We report here the synthesis and conformational evaluation of the THBC-DKP-based peptidomimetic 1a (Figure 2), for which molecular modeling allowed us to envisage a 13-membered hydrogen-bond-stabilized α-turn conformation.
Figure 2.
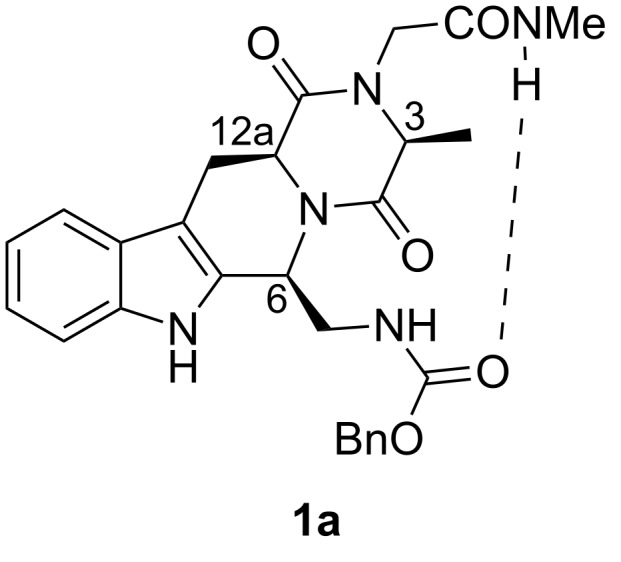
THBC-DKP-based peptidomimetic 1a.
Results and Discussion
In order to investigate the presence of a preferred conformation able to mimic an ordered protein secondary structure, a computer-aided conformational analysis was performed on 1a (6S,12aS-configuration, IUPAC atom numbering as in Figure 2) and diastereoisomeric 1b (6R, 12aS). Compounds 1a and 1b were submitted to an unconstrained Monte Carlo (MC) conformational search combined with Molecular Mechanics (MM) minimization (see Table 1 for results). As a main indication of a stable secondary structure, the presence of intramolecular hydrogen bonds was first evaluated. Two H-bonds have been identified, i.e., a 7-membered-ring H-bond (H-bond A, Figure 3) between the N5H hydrogen and the C3 carbonyl and a 13-membered-ring H-bond (H-bond B) between the N5H hydrogen and the C1 carbonyl, here represented by the Cbz carbonyl. To assess the presence of turn conformations we also measured the interatomic distance dα between the terminal Cα5 atom and the benzyl oxygen of the Cbz (which emulates the Cα1 atom), assuming a value dα < 7 Å as a probe of a generic reverse turn. Results are reported as the number of conformers that meet the geometric requirements.
Table 1.
MC/EM conformational analysis for peptidomimetics 1a and 1b. The + symbol indicates the presence in the global minimum.
| Conf. within 6 kcal/mol | H-bond A | H-bond B | dα < 7 Å | |
| 1a | 35 | 17 | 4 + | 4 + |
| 1b | 33 | 22 + | 1 | 1 |
Figure 3.
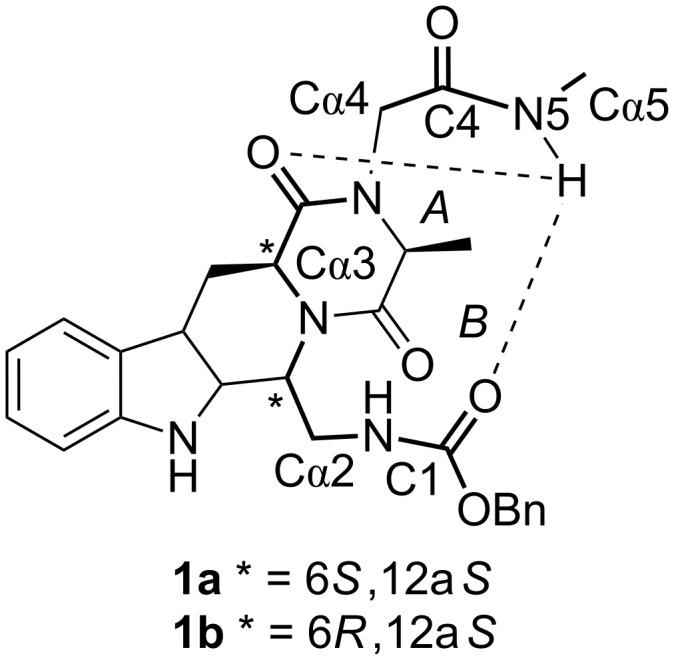
Geometric parameters for mimics 1a,b.
The most frequently observed is the 5→3 H-bond (H-bond A), which is related to a classical γ-turn around these residues, and a 5→1 H-bond (H-bond B), which can be identified as an α-turn (this conformation is present in four conformers for compound 1a). Inspection of the global minima showed the α-turn conformation only for compound 1a. For this diastereoisomer the first four low-energy conformers all adopt the α-turn conformation (presence of the 5→1 H-bond and average dα = 5.1 Å): according to a Boltzmann distribution analysis, these conformers take into account 94.5% of all the conformers obtained by the MC/MM analysis. On the other hand, for compound 1b, the lowest energy conformers all have the 5→3 H-bond (classical γ-turn), with the only α-turn conformation lying 5.9 kcal/mol above the global minimum. In Figure 4 the low-energy conformers of 1a and 1b are represented. These pictures highlight well the crucial role of the C6-configuration in favoring either an α-turn conformation (1a) or a γ-turn conformation (1b).
Figure 4.

Perspective view of the low-energy conformers of 1a,b. Hydrogen atoms are omitted for clarity.
Finally, having ascertained by means of further calculations the irrelevance of the C3 configuration on the expected secondary structure, we fixed it as 3S. Being interested in unusual reverse turns, we then pursued the synthesis of peptidomimetic 1a (Scheme 1). Starting from L-tryptophan methyl ester and N-Cbz-aminoacetaldehyde dimethyl acetal [43], tetrahydro-β-carboline 2 was obtained in good yield and high diastereoselectivity (dr 70% from 1H NMR) by means of Pictet–Spengler reaction [44] and subsequent chromatographic separation.
Scheme 1.

Synthesis of the THBC-DKP-based peptidomimetic 1a.
The 6S,12aS-configuration of the prevailing diastereoisomer was easily ascertained by application of the protocol of Ungemach et al. [45] on the 13C NMR spectrum of compound 2 and, conclusively, by the observation of an intense NOE contact between H-6 and H-12a in the 2D NOESY spectrum. Alkylation of L-alanine benzyl ester with ethyl bromoacetate afforded amine 3 [46], on which N-Fmoc protection and subsequent carefully conducted hydrogenolysis of the benzyl ester were performed [47], to give acid 4 in acceptable overall yield.
Condensation of acid 4 with secondary amine 2 proved to be troublesome under a wide range of conditions, probably due to the severe steric hindrance of both the amine and acid coupling partners. At the end, acceptable yields could be obtained via formation of the chloride intermediate [48], by reaction of acid 4 with thionyl chloride, and subsequent coupling with 2 in CH2Cl2 and 2,6-lutidine to give 5. The formation of the fused, tetracyclic THBC-DKP ring system was then easily achieved by removal of the N-Fmoc protecting group [49] and spontaneous lactamization to give the diketopiperazine ring of 6. The obtained compound 6 represents a valuable peptidomimetic, whose potential is also related to the possibility of further derivatization with desired pharmacophoric groups, on both the terminal acid and amine functional groups, for the development of conformationally constrained tryptophan-containing peptide ligands.
To investigate the actual secondary structure of the THBC-DKP scaffold also in solution, compound 6 was converted into the N-methyl carboxyamide derivative 1a, by a two-step procedure (0.5 M LiOH, 0 °C, then MeNH2, TBTU, DIPEA), which was carefully conducted in order to avoid the easy epimerization of the C3 and C12a stereocenters. Spin-system identification and assignment of individual resonances for peptidomimetic model 1a was straightforward with 1H COSY. The study of the conformational behavior was conducted in CDCl3, to identify possible intramolecular hydrogen bonding.
The involvement of the NH amide protons in such bonding was first estimated from evaluation of their chemical shift value (δ) and of the temperature coefficients Δδ/ΔT (between 263 and 328 K). All data were measured at 2.0 mM concentration, that is, in the absence of any noticeable intermolecular aggregation. While both NHMe and NHCbz chemical-shift values are low (6.78 ppm for NHMe and 5.53 ppm for NHCbz), a significant difference could be appreciated between their temperature coefficients, ranging from 7.0 ppb K−1 (in absolute value) for the NHMe signal to 2.3 ppb K−1 (in absolute value) for the NHCbz signal. According to the literature [50], these data could be attributed to a situation of equilibrium between hydrogen-bonded and non-hydrogen-bonded states for the NHMe amide proton and a completely non-hydrogen-bonded state for the NHCbz amide proton. In addition, a supplementary indication of the different hydrogen-bonding state for the two NH protons was obtained from DMSO titration studies in CDCl3 [51], indicating that the chemical shift of the NHMe has a minor variation (0.24 ppm) upon addition of up to 30% of the competitive solvent DMSO, with respect to the chemical shift of NHCbz, i.e., varying by 0.82 ppm.
Taking into account the suggestions from the molecular modeling and these experimental results, for 1a we can presume the presence in solution of conformers bearing a 13-membered intramolecular hydrogen bond involving the NHMe proton and the Cbz carbonyl group, as visualized in the perspective view of the low energy conformer of 1a.
Conclusion
In conclusion, we realized the synthesis of a new constrained THBC-DKP-based scaffold able to mimic an α-turn. It was designed with the aid of computational tools, which highlight the relative cis arrangement of the substituents on the THBC piperidine ring as being a crucial requirement in order to obtain the correct geometry for mimicry. Following these studies, the desired isomer 1a of the THBC-DKP based peptidomimetic was synthesized. 1H NMR conformational studies confirmed the presence of the intramolecular thirteen-membered hydrogen bond that characterizes the α-turn conformation, even if a situation of equilibrium between hydrogen-bonded and non-hydrogen-bonded states can be observed. Nevertheless, this scaffold represents one of the rare examples of a designed constrained α-turn mimic. Its application to the synthesis of biologically valuable peptides embodying an α-turn core is currently under way.
Experimental
General information: All commercial materials (Aldrich, Fluka) were used without further purification. All solvents were reagent grade or HPLC grade. All reactions were carried out under a nitrogen atmosphere unless otherwise noted. All reactions were monitored by thin-layer chromatography (TLC) on precoated silica gel 60 F254; spots were visualized with UV light or by treatment with 1% aqueous KMnO4 solution. Products were purified by flash chromatography on silica gel 60 (230–400 mesh). 1H NMR spectra and 13C NMR spectra were recorded on 300, 400 and 500 MHz spectrometers. Chemical shifts are reported in parts per million relative to the residual proton resonance of the solvent. 13C NMR spectra were recorded by using the APT pulse sequence. Multiplicities in 1H NMR are reported as follows: s = singlet, d = doublet, t = triplet, m = multiplet, br s = broad singlet. High-resolution MS spectra were recorded with a FT-ICR (Fourier Transform Ion Cyclotron Resonance) instrument, equipped with an ESI source, or a standard MS instrument, equipped with an EI source. Yields refer to chromatographically and spectroscopically pure compounds.
(1S,3S)-Methyl 1-(benzyloxycarbonylaminomethyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (2): Under a nitrogen atmosphere, L-tryptophan methyl ester (755 mg, 3.46 mmol, 1.0 equiv) was dissolved in CH2Cl2 (30 mL, 0.12 M), then N-Cbz-aminoacetaldehyde dimethyl acetal (912 mg, 3.81 mmol, 1.1 equiv) was added. The solution was cooled to −30 °C, then TFA was added (1.97 mg, 1.3 mL, 17.3 mmol, 5 equiv). The mixture was kept for 2 h at this temperature then reacted overnight at room temperature. The solution was diluted with AcOEt and washed three times with a saturated solution of NaHCO3 (3 × 15 mL), dried over Na2SO4, filtered and evaporated under reduced pressure. The crude mixture was purified by chromatographic column (n-hexane/AcOEt 6:4), and 808 mg of (1S,3S)-2 and 143 mg of the (1R,3S)-diastereoisomer were obtained (70% overall yield, 70% dr).
Characterization of 2. Oil; [α]D −44.4 (c 1, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 8.96 (1H, br s), 7.55–7.10 (9H, m), 6.02 (1H, br, dd, NH-Cbz), 5.03 (2H, s), 4.40 (1H, br s), 3.90–3.60 (6H, m), 3.18 (1H, dd, J = 15.0, 1.6 Hz), 2.88 (1H, dd, J = 15.0, 11.2 Hz), 2.77 (1H, br s); 13C NMR (CDCl3, 100 MHz) δ 173.5, 157.3, 136.5, 136.3, 132.3, 128.5–127.8 (5C), 127.0, 122.0, 119.5, 118.0, 111.3, 109.2, 66.9, 56.3, 53.6, 52.3, 44.3, 25.5; IR (cm−1): 3018, 1709, 1514, 1362, 1267, 1228; HRMS–EI (m/z): [M+] calcd for C22H23N3O4, 393.1689; found, 393;1702.
(S)-Benzyl 2-((2-ethoxy-2-oxoethyl)amino)propanoate (3): Under a nitrogen atmosphere L-alanine-benzyl ester (500 mg, 2.79 mmol, 1 equiv) was dissolved in CH3CN (6 mL, 0.47 M), then K2CO3 (3.85 g, 27.90 mmol, 10 equiv) was added. Ethyl bromoacetate (466 mg, 2.79 mmol, 310 µL, 1 equiv) was added to the suspension, and the mixture was reacted overnight at room temperature. The solvent was evaporated under reduced pressure, and the crude material was dissolved in AcOEt (20 mL). The solution was extracted with an aqueous solution of H3PO4 5% (3 × 25 mL), and the reunited aqueous layers were basified with Na2CO3 until pH 9 and extracted repeatedly with AcOEt. The reunited organic phases were dried with Na2SO4, filtered and evaporated under reduced pressure, to give compound 3 (503 mg, 67% yield), as an oil. [α]D −17.11 (c 1, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 7.4–7.28 (5H, m), 5.2–5.1 (2H, m), 4.15 (2H, q, J = 8.6 Hz), 3.53 (1H, q, J = 9.6 Hz), 3.49 (1H, d, J = 17.3 Hz), 3.39 (1H, d, J = 17.3 Hz), 2.37 (1H, br s), 1.38 (3H, d, J = 9.6 Hz), 1.25 (3H, t, J = 8.6 Hz); 13C NMR (CDCl3, 75.4 MHz) δ 174.3, 171.4, 135.6, 128.6–128.2 (5C), 66.7, 61.0, 55.9, 48.7, 18.6, 14.2; HRMS–EI (m/z): [M+] calcd for C14H19NO4, 265.1314; found, 265.1328.
(S)-2-((9H-Fluoren-9-ylmethoxycarbonyl)(2-ethoxy-2-oxoethyl)amino)propanoic acid (4): Under a nitrogen atmosphere, compound 3 (1.49 g, 5.62 mmol, 1 equiv) and DIPEA (871 mg, 6.74 mmol, 1.17 mL, 1.2 equiv) were dissolved in CH2Cl2 (40 mL, 0.14 M), then FmocCl (1.48 g, 5.73 mmol, 1.02 equiv) was added. The mixture was stirred at room temperature overnight. Then CH2Cl2 (10 mL) was added, the solution was washed with HCl 0.5 M (3 × 30 mL), and the organic layer was dried with Na2SO4, filtered and evaporated under reduced pressure. The crude was dissolved in MeOH (100 mL, 0.045 M), and then, under a nitrogen atmosphere, Pd/C 5% w/w (150 mg) was added. The resulting suspension was stirred for 3 h under a hydrogen atmosphere at room temperature. The reaction was accurately monitored by TLC in order to prevent hydrogenolysis of the Fmoc group. The suspension was filtered on a layer of celite, the solution was evaporated under reduced pressure, and the crude was dissolved in AcOEt and then extracted three times with a saturated solution of NaHCO3. The reunited aqueous phases were acidified with HCl (0.5 M) and extracted with AcOEt (3 × 35 mL), then dried with Na2SO4, filtered and evaporated under reduced pressure, to give pure 4 (1.76 g, 79% overall yield) as an oil. [α]D −9.42 (c 1, MeOH); 1H NMR (DMSO, 300 MHz, 95 °C) δ 7.88 (2H, d, J = 7.5 Hz), 7.64 (2H, d, J = 7.4 Hz), 7.43 (2H, t, J = 7.3 Hz), 7.34 (2H, t, J = 7.3 Hz), 4.58 (1H, q, J = 7.3 Hz), 4.38 (2H, d, J = 6.3 Hz), 4.26 (1H, t, J = 6.3 Hz), 4.12 (2H, q, J = 7.1 Hz), 3.98 (2H, AB system, J = 18.0 Hz), 1.37 (3H, d, J = 7.3 Hz), 1.21 (3H, t, J = 7.1 Hz); 13C NMR (CDCl3, 75.4 MHz, mixture of two rotamers) δ 175.4 and 174.5, 171.8 and 170.7, 155.7 and 155.6, 143.7, 143.6, 141.2 (2C), 127.7 (2C), 127.1 (2C), 124.9 (2C), 120.0 (2C), 68.6 and 68.2, 62.2 and 61.8, 55.7 and 55.5, 47.6 and 47.1, 47.0, 15.5 and 14.9, 14.1; IR (cm−1): 2954, 1750, 1709, 1467, 1451, 1325, 1204; HRMS–EI (m/z): [M+] calcd for C22H23NO6, 397.1525; found, 397.1541.
(1S,3S)-Methyl 2-((S)-2-((9H-fluoren-9-ylmethoxycarbonyl)(2-ethoxy-2-oxoethyl)amino)propanoyl)-1-(benzyloxycarbonylaminomethyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (5): Under a nitrogen atmosphere, compound 4 (302 mg, 0.76 mmol, 1 equiv) was dissolved in CH2Cl2 (4 mL, 0.19 M), the solution was cooled to 0 °C, and then SOCl2 (904 mg, 7.60 mmol, 550 µL, 10 equiv) was added. The mixture was stirred at room temperature for 5 h then evaporated under reduced pressure. The obtained crude material was dissolved in CH2Cl2 (3 mL) and slowly added to a solution of 2 (299 mg, 0.76 mmol, 1 equiv) and 2,6-lutidine (110 mg, 1.03 mmol, 120 μL, 1.35 equiv) in CH2Cl2 (5 mL), cooled at 0 °C under nitrogen atmosphere. The mixture was stirred at room temperature overnight, then CH2Cl2 (10 mL) was added, the organic layer was washed with HCl (0.5 M, 5 mL), saturated aq NaHCO3 (5 mL) and brine (5 mL). The organic phase was dried with Na2SO4, filtered and evaporated under reduced pressure. The crude material was purified by column chromatography (n-hexane/AcOEt 6:4), affording 5 (321 mg, 55% yield) as an oil. [α]D +17.87 (c 1 CHCl3), 1H NMR (CDCl3, 300 MHz, major rotamer) δ 9.87 (1H, br s), 7.88–7.00 (17H, m), 6.07 (1H, br s), 5.45 (1H, br q), 5.32 (1H, br s), 5.23 (2H, br s), 4.71–4.32 (2H, m), 4.20–4.10 (5H, m), 4.08–3.74 (3H, m), 3.74–3.08 (5H, m), 1.48 (3H, d, J = 7.5 Hz), 1.25 (3H, d, J = 7.3 Hz); 13C NMR (CDCl3, 100 MHz, major rotamer) δ 171.1, 169.3, 169.1, 155.9, 154.2, 143.9, 141.2, 136.4, 130.6, 128.5–111.5 (22C), 68.8, 67.1, 61.4, 56.3, 54.8, 52.1, 51.0, 47.1, 47.0, 46.6, 24.4, 15.3, 14.2; HRMS–EI (m/z): [M+] calcd for C44H44N4O9, 772.3108; found, 772.3095.
Ethyl 2-((3S,6S,12aS)-6-(benzyloxycarbonylaminomethyl)-3-methyl-1,4-dioxo-3,4,12,12a-tetrahydropyrazino[1',2':1,6]pyrido[3,4-b]indol-2(1H,6H,7H)-yl)acetate (6): Compound 5 (300 mg, 0.39 mmol) was dissolved in THF (13 mL, 0.03 M), and then piperidine was added (2.6 mL). The solution was reacted overnight, then HCl (0.5 M) was added, and the mixture was extracted with AcOEt (3 × 30 mL). The reunited organic layers were dried with Na2SO4, filtered and evaporated under reduced pressure. The crude material was purified by column chromatography (AcOEt/n-hexane, 4:6 to 6:4), to give 6 (165 mg, 81% yield) as an oil. [α]D −59.5 (c 1, CHCl3); 1H NMR (CDCl3, 400 MHz, major rotamer) δ 9.27 (1H, br s), 7.55–7.10 (9H, m), 5.91 (1H, br s), 5.15–5.09 (3H, m), 4.50 (1H, d, J = 17.2 Hz), 4.26 (4H, m), 4.05 (1H, m), 3.97 (1H, d, J = 17.5 Hz), 3.68 (1H, m), 3.57 (1H, br d, J = 15.6 Hz), 3.07 (1H, dd, J = 15.6, 11.8 Hz), 1.58 (3H, d, J = 12.8 Hz), 1.31 (3H, t, J = 7.2 Hz); 13C NMR (CDCl3, 75.4 MHz) δ 168.9, 168.5, 167.3, 157.6, 136.5, 136.3, 130.7, 128.6, 128.2, 128.0, 126.2, 122.5, 119.9, 119.8, 118.3, 111.7, 107.7, 67.1, 61.8, 57.6, 57.2, 56.3, 45.0, 44.7, 31.6, 25.2, 17.9, 14.2; IR (cm−1): 3016, 1742, 1667, 1506, 1455, 1326, 1222. HRMS–EI (m/z): [M+] calcd for C28H30N4O6, 518.2165; found, 518.2178.
Benzyl (((3S,6S,12aS)-3-methyl-2-(2-(methylamino)-2-oxoethyl)-1,4-dioxo-1,2,3,4,6,7,12,12a-octahydropyrazino[1',2':1,6]pyrido[3,4-b]indol-6-yl)methyl)carbamate (1a): Compound 6 (165 mg, 0.32 mmol, 518 g/mol) was dissolved in EtOH (3 mL). Aqueous LiOH (0.5 M, 2.54 mmol, 4.6 mL) was added and the reaction was stirred at 0 °C for 1 h. After then, the solution was acidified with HCl (1 N, 8 mL), extracted with AcOEt (3 × 20 mL), dried, filtered and concentrated. The crude material was suspended in CH2Cl2 (10 mL) under N2, DIPEA (208 µL, 1.20 mmol, 129 g/mol) and TBTU (384 mg, 1.20 mmol, 321 g/mol) were added. After stirring for 30 min, MeNH2 (2.0 M THF solution, 1.20 mmol, 601 µL) was added. The reaction was stirred for 24 h under N2, and then it was poured into water (20 mL) and extracted with AcOEt (3 × 20 mL), dried, filtered and concentrated. Purification by flash chromatography (AcOEt) afforded product 1a (110 mg, 68% overall yield) as an oil. [α]D −98.3 (c 0.75, MeOH); 1H NMR (500 MHz, CD3CN, major rotamer) δ 9.41 (1H, br s), 7.55 (1H, d, J = 7.6 Hz), 7.38 (1H, d, J = 7.8 Hz), 7.34–7.23 (5H, m), 7.13 (1H, ddd, J = 7.8, 7.6 and 0.9 Hz), 7.06 (1H, dt, J = 7.6 and 0.9 Hz), 6.95 (1H, br q, J = 4.7 Hz), 5.83 (1H, br q, J = 8.2 and 5.1 Hz), 5.43 (1H, br t, J = 3.4 Hz), 4.90 (1H, d, J = 12.3 Hz), 4.87 (1H, d, J = 12.3 Hz), 4.26 (1H, d, J = 17.0 Hz), 4.16 (1H, q, J = 7.0 Hz), 4.14 (1H, dd, J = 11.4 and 5.5 Hz), 3.98 (1H, ddd, J = 14.2, 8.2 and 3.8 Hz), 3.94 (1H, d, J = 17.0 Hz), 3.43 (1H, dd, J = 15.6 and 5.5 Hz), 3.26 (1H, ddd, J = 14.2, 5.1 and 3.2 Hz), 3.00 (1H, dd, J = 15.6 and 11.4 Hz), 2.69 (3H, d, J = 4.7 Hz), 1.48 (3H, d, J = 7.0 Hz); 13C NMR (125.8 MHz, CD3CN) δ 170.4, 170.0, 169.8, 157.8, 137.5, 135.2, 132.0, 129.0 (2C), 128.5, 128.3 (2C), 126.9, 122.2, 119.8, 118.7, 112.0, 108.1, 66.7, 55.4, 55.3, 53.5, 46.7, 44.7, 22.7, 25.9, 14.5; HRMS–EI (m/z): [M+] calcd for C27H29N5O5, 503.2169; found, 503.2186.
Computational details
An unconstrained Monte Carlo (MC) conformational search combined with Molecular Mechanics (MM) minimization was performed by using the software Spartan’08 [52]. Images were elaborated with the software PyMol [53]. For each compound 972 conformers were generated, and after removal of duplicated minima, only conformations within 6 kcal/mol of the global minimum were considered. Thirty-five and thirty-three conformers were kept for compounds 1a and 1b, respectively. Inspection of the virtual torsion angles of the amide backbone revealed the preference for an inverse γ-turn of the low-energy conformers of 1b (φ = −80.1° and ψ = 79.6° on average, based on the conformers accounting 93% of all the conformers from a Boltzmann distribution analysis). For compound 1a the averages of the torsion angles were φ (Cα2) = −81.5°, ψ (Cα2) = 64.5°, φ (Cα3) = 146.3°, ψ (Cα3) = 36.9°, φ (Cα4) = 120.5°, and ψ (Cα4) = −29.3°. According to the classification of α-turns in protein structures [32], compound 1a is an α-turn mimetic with a B-αL-X designation.
Contributor Information
Alessandro Sacchetti, Email: alessandro.sacchetti@polimi.it.
Alessandra Silvani, Email: alessandra.silvani@unimi.it.
References
- 1.Cordell G A, editor. The Alkaloids: Chemistry and Pharmacology. Vol. 43. New York: Academic Press; 1993. [Google Scholar]
- 2.Oakley N, Jones B. Eur J Pharmacol. 1980;68:381. doi: 10.1016/0014-2999(80)90538-5. [DOI] [PubMed] [Google Scholar]
- 3.Khorana N, Smith C, Herrick-Davis K, Purohit A, Teitler M, Grella B, Dukat M, Glennon R A. J Med Chem. 2003;46:3930–3937. doi: 10.1021/jm030080s. [DOI] [PubMed] [Google Scholar]
- 4.Audia J E, Evrard D A, Murdoch G R, Droste J J, Nissen J S, Schenk K W, Fludzinski P, Lucaites V L, Nelson D L, Cohen M L. J Med Chem. 1996;39:2773–2780. doi: 10.1021/jm960062t. [DOI] [PubMed] [Google Scholar]
- 5.Abou-Gharbia M, Patel R U, Webb M B, Moyer J A, Andree T H, Muth T A. J Med Chem. 1987;30:1818. doi: 10.1021/jm00393a023. [DOI] [PubMed] [Google Scholar]
- 6.Callaway J J, Gynther J, Poso A, Vepsäläinen J, Airksinen M M. J Heterocycl Chem. 1994;31:431. doi: 10.1002/jhet.5570310231. [DOI] [Google Scholar]
- 7.Ho B T. J Pharm Sci. 1972;61:821. doi: 10.1002/jps.2600610602. [DOI] [PubMed] [Google Scholar]
- 8.Jenkins P R, Wilson J, Emmerson D, Garcia M D, Smith M R, Gray S J, Britton R G, Mahale S, Chaudhuri B. Bioorg Med Chem. 2008;16:7728–7739. doi: 10.1016/j.bmc.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Sunder-Plassmann N, Sarli V, Gartner M, Utz M, Seiler J, Huemmer S, Mayer T U, Surrey T, Giannis A. Bioorg Med Chem. 2005;13:6094–6111. doi: 10.1016/j.bmc.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 10.Gupta L, Srivastava K, Singh S, Puri S K, Chauhan P M S. Bioorg Med Chem Lett. 2008;18:3306–3309. doi: 10.1016/j.bmcl.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 11.Wang S, Golec J, Miller W, Milutinovic S, Folkes A, Williams S, Brooks T, Hardman K, Charlton P, Wren S, et al. Bioorg Med Chem Lett. 2002;12:2367–2370. doi: 10.1016/S0960-894X(02)00389-X. [DOI] [PubMed] [Google Scholar]
- 12.Houston D R, Synstad B, Eijsink V G H, Stark M J R, Eggleston I M, Van Aalten D M F. J Med Chem. 2004;47:5713–5720. doi: 10.1021/jm049940a. [DOI] [PubMed] [Google Scholar]
- 13.Fdhila F, Vazquez V, Sanchez J L, Riguera R. J Nat Prod. 2003;66:1299–1301. doi: 10.1021/np030233e. [DOI] [PubMed] [Google Scholar]
- 14.Nicholson B, Lloyd G K, Miller B R, Palladino M A, Kiso Y, Hayashi Y, Neuteboom S T C. Anti-Cancer Drugs. 2006;17:25–31. doi: 10.1097/01.cad.0000182745.01612.8a. [DOI] [PubMed] [Google Scholar]
- 15.Chanda K, Chou C-T, Lai J-J, Lin S-F, Yellol G S, Sun C-M. Mol Diversity. 2011;15:569–581. doi: 10.1007/s11030-010-9284-z. [DOI] [PubMed] [Google Scholar]
- 16.Ressurreicao A S M, Bordessa A, Civera M, Belvisi L, Gennari C, Piarulli U. J Org Chem. 2008;73:652–660. doi: 10.1021/jo702072z. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Brahimi F, Saragovi H U, Burgess K. J Med Chem. 2010;53:5044–5048. doi: 10.1021/jm100148d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maity P, König B. Org Lett. 2008;10:1473–1476. doi: 10.1021/ol8002749. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Li Z-L, Bai J, Zhang L-M, Wu X, Zhang L, Pei Y-H, Jing Y-K, Hua H-M. Chem Biodiversity. 2012;9:385–393. doi: 10.1002/cbdv.201100061. [DOI] [PubMed] [Google Scholar]
- 20.Deveau A M, Costa N E, Joshi E M, Macdonald T L. Bioorg Med Chem Lett. 2008;18:3522–3525. doi: 10.1016/j.bmcl.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 21.Daugan A, Grondin P, Ruault C, Gouville A C, Le M, Coste H, Linget J M, Kirilovsky J, Hyafil F, Labaudiniere R. J Med Chem. 2003;46:4533–4542. doi: 10.1021/jm0300577. [DOI] [PubMed] [Google Scholar]
- 22.Beghyn T B, Charton J, Leroux F, Henninot A, Reboule I, Cos P, Maes L, Deprez B. J Med Chem. 2012;55:1274–1286. doi: 10.1021/jm201422e. [DOI] [PubMed] [Google Scholar]
- 23.Beghyn T B, Charton J, Leroux F, Laconde G, Bourin A, Cos P, Maes L, Deprez B. J Med Chem. 2011;54:3222–3240. doi: 10.1021/jm1014617. [DOI] [PubMed] [Google Scholar]
- 24.Deveau A M, Labroli M A, Dieckhaus C M, Barthen M T, Smith K S, Macdonald T L. Bioorg Med Chem Lett. 2001;11:1251–1255. doi: 10.1016/S0960-894X(01)00136-6. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Wu G, Cui G, Wang W X, Zhao M, Wang C, Zhang Z, Peng S. Bioorg Med Chem. 2007;15:5672–5693. doi: 10.1016/j.bmc.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 26.Danieli B, Giovanelli P, Lesma G, Passarella D, Sacchetti A, Silvani A. J Comb Chem. 2005;7:458–462. doi: 10.1021/cc049881m. [DOI] [PubMed] [Google Scholar]
- 27.Lesma G, Landoni N, Sacchetti A, Silvani A. Tetrahedron. 2010;66:4474–4478. doi: 10.1016/j.tet.2010.04.077. [DOI] [Google Scholar]
- 28.Lesma G, Landoni N, Pilati T, Sacchetti A, Silvani A. J Org Chem. 2009;74:8098–8105. doi: 10.1021/jo901480d. [DOI] [PubMed] [Google Scholar]
- 29.Lesma G, Landoni N, Sacchetti A, Silvani A. J Org Chem. 2007;72:9765–9768. doi: 10.1021/jo701581j. [DOI] [PubMed] [Google Scholar]
- 30.Lesma G, Meschini E, Recca T, Sacchetti A, Silvani A. Tetrahedron. 2007;63:5567–5578. doi: 10.1016/j.tet.2007.04.024. [DOI] [Google Scholar]
- 31.Ball J B, Hughes R A, Alewood P F, Andrews P R. Tetrahedron. 1993;49:3467–3478. doi: 10.1016/S0040-4020(01)90207-8. [DOI] [Google Scholar]
- 32.Grison C, Coutrot P, Genève S, Didierjean C, Marraud M. J Org Chem. 2005;70:10753–10764. doi: 10.1021/jo051483y. [DOI] [PubMed] [Google Scholar]
- 33.Trabocchi A, Occhiato E G, Potenza D, Guarna A. J Org Chem. 2002;67:7483–7492. doi: 10.1021/jo0202132. [DOI] [PubMed] [Google Scholar]
- 34.Chakraborty T K, Srinivasu P, Vengal Rao R, Kiran Kumar S, Kunwar A C. J Org Chem. 2004;69:7399–7402. doi: 10.1021/jo048893r. [DOI] [PubMed] [Google Scholar]
- 35.Nataraj D V, Srinivasan N, Sowdhamini R, Ramakrishnan C. Curr Sci. 1995;69:434–446. [Google Scholar]
- 36.Chakraborty T K, Koley D, Ravi R, Krishnakumari V, Nagaraj R, Chand Kunwar A. J Org Chem. 2008;73:8731–8744. doi: 10.1021/jo801123q. [DOI] [PubMed] [Google Scholar]
- 37.Shepherd N E, Hoang H N, Desai V S, Letouze E, Young P R, Fairlie D P. J Am Chem Soc. 2006;128:13284–13289. doi: 10.1021/ja064058a. [DOI] [PubMed] [Google Scholar]
- 38.Bell C H, Pantophlet R, Schiefner A, Cavacini L A, Stanfield R L, Burton D R, Wilson I A. J Mol Biol. 2008;375:969–978. doi: 10.1016/j.jmb.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saha I, Raghothama S, Aravinda S, Rai R, Shamala N, Balaram P. Chem–Eur J. 2008;14:6192–6204. doi: 10.1002/chem.200702029. [DOI] [PubMed] [Google Scholar]
- 40.Srinivas D, Vijayadas K N, Gonnade R, Phalgune U D, Rajamohanan P R, Sanjayan G J. Org Biomol Chem. 2011;9:5762. doi: 10.1039/c1ob05553d. [DOI] [PubMed] [Google Scholar]
- 41.Hoang H N, Driver R W, Beyer R L, Malde A K, Le G T, Abbenante G, Mark A E, Fairlie D P. Angew Chem, Int Ed. 2011;50:11107–11111. doi: 10.1002/anie.201105119. [DOI] [PubMed] [Google Scholar]
- 42.Kelso M J, Beyer R L, Hoang H N, Lakdawala A S, Snyder J P, Oliver W V, Robertson T A, Appleton T G, Fairlie D P. J Am Chem Soc. 2004;126:4828–4842. doi: 10.1021/ja037980i. [DOI] [PubMed] [Google Scholar]
- 43.Boisbrun M, Jeannin L, Toupet L, Laronze J-Y. Eur J Org Chem. 2000:3051–3057. doi: 10.1002/1099-0690(200009)2000:17<3051::AID-EJOC3051>3.0.CO;2-3. [DOI] [Google Scholar]
- 44.Pulka K, Tymecka D, Frankiewicz L, Wilczek M, Kozminski W, Misicka A. Tetrahedron. 2008;64:1506–1514. doi: 10.1016/j.tet.2007.11.037. [DOI] [Google Scholar]
- 45.Ungemach F, Soerens D, Weber R, Di Pierro M, Campos O, Mokry P, Cook J M, Silverton J V. J Am Chem Soc. 1980;102:6976–6984. doi: 10.1021/ja00543a012. [DOI] [Google Scholar]
- 46.Coppola G M, Iwaki Y, Karki R G, Kawanami T, Ksander G M, Mogi M, inventors. Substituted aminopropionic derivatives as neprilysin inhibitors. WO2010136493 A1. WO Patent Application. 2010 Dec 2;
- 47.Shoulders M D, Kotch F W, Choudhary A, Guzei I A, Raines R T. J Am Chem Soc. 2010;132:10857–10865. doi: 10.1021/ja103082y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tantry S T, Venkataramanarao R, Chennakrishnareddy G, Sureshbabu V V. J Org Chem. 2007;72:9360–9363. doi: 10.1021/jo701329w. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Ganesan A. Tetrahedron Lett. 1997;38:4327–4328. doi: 10.1016/S0040-4039(97)00892-7. [DOI] [Google Scholar]
- 50.Kessler H. Angew Chem, Int Ed Engl. 1982;21:512–523. doi: 10.1002/anie.198205121. [DOI] [Google Scholar]
- 51.For DMSO-d6/CDCl3 solutions, chemical shifts were standardized by reference to residual proton resonance for CHD2CD3SO (2.49 ppm).
- 52.Spartan 08. Irvine, CA: Wavefunction, Inc.; [Google Scholar]
- 53.The PyMOL Molecular Graphics System. Portland, OR: Schrödinger, LLC; [Google Scholar]