

Abstract
Mammalian DNA replication initiates at multiple sites along chromosomes at different times during S phase, following a temporal replication program. The specification of replication timing is thought to be a dynamic process regulated by tissue-specific and developmental cues that are responsive to epigenetic modifications. However, the mechanisms regulating where and when DNA replication initiates along chromosomes remains poorly understood. Homologous chromosomes usually replicate synchronously, however there are notable exceptions to this rule. For example, in female mammalian cells one of the two X chromosomes becomes late replicating through a process known as X inactivation1. Along with this delay in replication timing, estimated to be 2-3 hr, the majority of genes become transcriptionally silenced on one X chromosome. In addition, a discrete cis-acting locus, known as the X inactivation center, regulates this X inactivation process, including the induction of delayed replication timing on the entire inactive X chromosome. In addition, certain chromosome rearrangements found in cancer cells and in cells exposed to ionizing radiation display a significant delay in replication timing of >3 hours that affects the entire chromosome2,3. Recent work from our lab indicates that disruption of discrete cis-acting autosomal loci result in an extremely late replicating phenotype that affects the entire chromosome4. Additional 'chromosome engineering' studies indicate that certain chromosome rearrangements affecting many different chromosomes result in this abnormal replication-timing phenotype, suggesting that all mammalian chromosomes contain discrete cis-acting loci that control proper replication timing of individual chromosomes5.
Here, we present a method for the quantitative analysis of chromosome replication timing combined with fluorescent in situ hybridization. This method allows for a direct comparison of replication timing between homologous chromosomes within the same cell, and was adapted from6. In addition, this method allows for the unambiguous identification of chromosomal rearrangements that correlate with changes in replication timing that affect the entire chromosome. This method has advantages over recently developed high throughput micro-array or sequencing protocols that cannot distinguish between homologous alleles present on rearranged and un-rearranged chromosomes. In addition, because the method described here evaluates single cells, it can detect changes in chromosome replication timing on chromosomal rearrangements that are present in only a fraction of the cells in a population.
Keywords: Genetics, Issue 70, Biochemistry, Molecular Biology, Cellular Biology, Chromosome replication timing, fluorescent in situ hybridization, FISH, BrdU, cytogenetics, chromosome rearrangements, fluorescence microscopy
Protocol
1. BrdU Incorporation (Terminal Labeling)
Plate cells to about 70% confluence in a 150 mm tissue culture dish 24 hr prior to the addition of BrdU.
Replace media with fresh complete media containing 20 μg/ml BrdU (Sigma) at appropriate time points prior to harvest. The length of time cells are cultured in media with BrdU will vary with cell type and species, typically the G2 phase lasts between 2 and 5 hr (Figure 1).
2. Chromosome Harvest of Monolayer Cells Cultures
Remove culture media from the plate and collect 10 ml in a 15 ml conical centrifuge tube. Discard remaining media.
Rinse cells once with 10 ml Versine [or HBSS (Sigma)].
Add 5 ml 0.25% trypsin in Versine (or HBSS) to the plate. Incubate at room temperature until cells are detached from the plate.
Add trypsinized cell suspension to the tube containing the 10 ml of media from step 2.1.
Centrifuge at 400 x g for 10 min to pellet the cells. Aspirate supernatant, leaving 0.5 ml of media plus the cell pellet.
Resuspend cells thoroughly by pipetting with a Pasteur pipette.
Add 3 drops of hypotonic solution (0.075 M KCl (Sigma) warmed to 37 °C); mix the cell suspension with a Pasteur pipette. Add 0.5 ml hypotonic solution and mix again with a Pasteur pipette. Bring volume of hypotonic solution to 5 ml and mix once again. Incubate at 37 °C for 20-45 min, depending on the cell type.
Centrifuge at 400 x g for 10 min. Aspirate supernatant, leaving 0.5 ml of media plus the cell pellet.
Resuspend the cell pellet by gently flicking the tube. The osmotically swollen cells will be fragile at this point, so care should be taken not to disrupt the cytoplasmic membranes.
Add 3 drops of Carnoy's fixative (1:3 Glacial Acetic Acid: Methanol); mix by gently pipetting with a Pasteur pipette. Add 0.5 ml fixative and mix by gently pipetting with a Pasteur pipette. Bring volume of fixative to 5 ml and mix again by gently pipetting with a Pasteur pipette. Fixed cells can be stored in the dark at -20 °C for several months.
Centrifuge and aspirate as in step 2.8.
Resuspend the cell pellet in fresh fixative; estimate the volume of the pellet and add ~10x the volume of Carnoy's fixative.
Add the cell suspension drop-wise on wet, ice-cold microscope slides; hold the slide at an ~45° angle and allow the cell suspension to flow over the surface of the slide. Lay the slide flat on paper towels and flood the slide with fixative and allow to air dry. Check slides for the presence of mitotic spreads using an inverted microscope.
3. RNase Treatment and Ethanol Dehydration
Place 200 μl of 10 μg/ml RNase A (Sigma) in 2x SSC to each slide; incubate at 37 °C for 1 hr.
Wash slides 3 times with 2x SSC, pH 7.0 at room temperature for 3 min each.
Dehydrate slides through an EtOH (Sigma) series (70%, 90% and 100%) at room temperature for 3 min each.
Air dry the slides at room temperature.
4. Preparation of Probe Cocktails for BAC Plus Chromosome-specific Centromere Enumeration Probe (CEP) or for Whole Chromosome Paints In situ Hybridization
For the BAC/CEP simultaneous hybridization, prepare two separate probe cocktails for pre-hybridization. We have found that pre-hybridizing the BAC and CEP probes separately under different stringencies results in less background and greater signal intensities. We have also found that individual Fosmids can also be used in place of BAC clones. The cocktail formulation described below represents the volumes for a single slide. If processing multiple slides using the same probes simply increase the volumes appropriately. *BAC DNA probe cocktail: 11.5 μl hybridization buffer (50% Formamide (Sigma) /2x SSC /Dextran Sulfate (Sigma)) 2 μl dH2O 2.5 μl BAC/Cot1 DNA in dH2O (see below for labeling protocol) 15 μl total volume *CEP probe cocktail: 7 μl CEP hybridization buffer (65% Formamide/2x SSC/Dextran Sulfate) 2 μl dH2O 1 μl CEP/Cot1 DNA (purchased prelabeled with Spectrum Orange/Cy3/Texas Red) 10 μl total volume *After denaturing and pre-hybridization (step 5.1) mix BAC probe with CEP probe at a 3:2 ratio 25 μl/slide and mix probe cocktails. OR
Aliquot 20 μl/slide of Whole Chromosome Paint probe (Cy3/Texas Red) into a tube. Proceed to denaturation and pre-hybridization step.
5. Slide and Probe Denaturation and In situ Hybridization
Denature probe cocktail(s) at 75 °C for 10 min. Pre-hybridize at 37 °C for 30 min to allow the Cot1 DNA to hybridize to repetitive sequences and to prevent subsequent hybridization to metaphase chromosomes.
Denature slides in Coplin jars in 70% Formamide/2x SSC, pH 7.0 at 72 °C for 3 min.
Immediately dehydrate slides through an EtOH series (70%, 90% and 100%) in Coplin jars at 4 °C for 3 min each.
Allow the slides to air dry at room temperature.
Pre-warm slides to 45 °C on slide warmer for the last 10 min of the probe pre-hybridzation (step 5.1). Mix pre-hybridized probe cocktails if needed and add 25 ul to each slide; gently place a coverslip on each slide and seal along all edges with rubber cement. Incubate overnight at 37 °C in a humidified chamber to prevent evaporation.
6. Post Hybridization Washes
Wash slides in Coplin jars 3 times in 50% formamide/2x SSC, pH 7.0 at 38-40 °C for 3 min each. The temperature of the washes may vary between probes. If there is high background hybridization you can increase the temperature of the washes. Conversely, if the signal is faint and there is no background then you can decrease the temperature of these washes.
Wash slides 1 time in PN buffer at room temperature for 3 min, and proceed to the BrdU detection step.
7. BrdU Detection
Block slides with PNM buffer for 10 min (200 μl/slide) at room temperature in the dark.
Drain slides by turning on edge on a paper towel. Add 100 μl of anti-BrdU-FITC (50 μg/ml Roche or Millipore) in PNM buffer for 30 min at 37 °C.
Wash slides 3 times in PN buffer at room temperature for 3 min each.
Drain excess PN buffer on slides one at a time by turning on edge on a paper towel. Add 20 μl DAPI/antifade mounting solution (Invitrogen). Cover the slide with a paper towel and press down on the coverslip to force out air bubbles and excess mounting solution. Proceed to image analysis.
8. Capturing Images and Quantifying BrdU Incorporation
Images are captured with an Olympus BX61 Fluorescent Microscope and Olympus CCD Camera, 100x objective, automatic filter-wheel and Cytovision software (Applied Imaging). BrdU is captured using an FITC filter; chromosome paints (Texas Red, Metasystems or Cytocell), BAC's (Cy3) and CEP (Vysis Spectrum Orange) probes are captured using a Texas Red or Cy3 filter; DAPI is captured with a DAPI Filter (see Figures 2 and 3).
Individual chromosomes of interest are identified with BAC/CEP or chromosome-specific paint probes (Figures 2B and 3B). Utilizing the Cytovision software, each chromosome of interest is "cut-out" from the metaphase spread as a whole, a line is drawn through the along the length of the chromosome from short arm to long arm (Figures 2C and 3C). The Cytovision software is used to quantify the pixel intensity profile along the length of each chromosome. The Cytovision software calculates the area occupied by the pixels and quantifies the intensity of the pixels represented by the BrdU or DAPI signals on each isolated chromosome. The average pixel intensity represented by each chromosome is then multiplied by the area occupied by those pixels to obtain the total number of pixels (Table 1 and Figure 4). In addition, this protocol allows for the visualization of the latest replicating regions of chromosomes. Accordingly, the banded pattern of BrdU incorporation allows for the detection of actively replicating regions of chromosomes, and differences in replication timing between chromosome pairs are seen as differences in this banding pattern (Figures 2 and 3). Finally, these differences in binding patterns can be compared to known replication timing maps for each chromosome6,7, which allows for an estimate of the replication timing difference between homologous chromosomes or chromosome arms4. For example, the two chromosome 6's in Figure 3A display a difference in banding pattern of >2 hr when compared to the normal replication timing of chromosome 64. In addition, a similar procedure to the one described here has been successfully used for the study of the asynchronous replication timing between the inactive and active X chromosomes8,9. The active and inactive X chromosomes were identified using X chromosome paints. Images were acquired using Quips mFISH software (Vysis) and the number of pixels occupied by the X chromosomes and the number of pixels occupied by fluorescently labeled BrdU were then calculated using NIH Image (http://rsb.info.nih.gov/nih-image/index.html).
9. Nick Translation of BAC DNA for Fluorescent Labeling (Vysis)
Add the following probe mix to chilled a microfuge tube: 70 μl (4 μg DNA) in dH2O 10 μl .2 mM dUTP-Orange or Green 20 μl .1 mM dTTP 20 μl 10x Nick Translation Buffer 40 μl NT dNTP's (minus dTTP) 40 μl nick Translation enzyme 200 μl total volume Incubate @ 16 °C O/N. Stop reaction by heating at 70 °C for 10 min. Chill on ice for 5 min.
Precipitate the DNA with ethanol by adding the following to the probe mix: 200 μl probe solution 48 μl 3 M NaOAc 160 μl Cot1DNA (.25 μg/ul) 1,200 μl 100% EtOH
Store at -80 °C for 10 min to O/N.
Spin in cold at 12,000 x g for 20 min. Decant off EtOH. Wash 1 time with 70% EtOH. Spin as before. Decant off EtOH. Air dry just until EtOH evaporates. Resuspend in 40 μl dH2O for final concentration of 100 ng/μl of BAC DNA.
Representative Results
An example of the replication timing analysis for human chromosome 6 is shown in Figure 2. Cells containing a deletion of the ASAR6 gene4, located at 6q16.1, were exposed to BrdU for 5 hr, harvested for mitotic cells and processed for FISH with a chromosome 6 paint probe (Vysis) and for BrdU incorporation. Note that there is a significant difference in the BrdU banding pattern between the two 6's, which is consistent with a delay in replication timing of >2 hr for one of the chromosome 6's [see4 for the replication timing banding pattern of chromosome 6 prior to the deletion]. In addition, there is a significant difference in the total amount of BrdU incorporation (pixels) when compared to a similar analysis of the DAPI staining (Figure 2D).
Another example of the replication timing analysis for human chromosome 6 is shown in Figure 3. Cells containing the same deletion of the ASAR6 gene4 shown if Figure 2 were exposed to BrdU for 5 hr, harvested for mitotic cells and processed for FISH with a chromosome 6 CEP probe plus a BAC containing the ASAR6 gene and for BrdU incorporation. Note that the deleted chromosome 6 (Δ6) displays more BrdU incorporation and a more extended banding pattern of BrdU incorporation than the non-deleted chromosome 6. Mitotic spreads from 7 different cells were processed as in Figure 3 and the pixel profiles for both DAPI and BrdU are shown in Table 1.
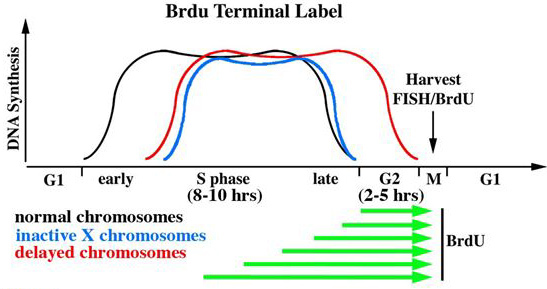 Figure 1. BrdU terminal label scheme. A typical S phase in mammalian cells last for 8 to 10 hr, and G2 is typically 2 to 5 hr. BrdU is added for increasing periods of time (green arrows) to label the last portions of the chromosomes to replicate. The chromosomes in mammalian cells replicate according to a temporal program, with early and late replication occurring at the beginning and end of S phase respectively (black line). Inactive X chromosomes are delayed in replication timing with the majority of DNA synthesis occurring during the second half of S phase (blue line). Chromosomes with delayed replication timing are delayed in both initiation and completion of DNA synthesis, with active replication continuing through G2 (red line). Asynchronous cultures are harvested for mitotic cells and processed for BrdU incorporation and FISH.
Figure 1. BrdU terminal label scheme. A typical S phase in mammalian cells last for 8 to 10 hr, and G2 is typically 2 to 5 hr. BrdU is added for increasing periods of time (green arrows) to label the last portions of the chromosomes to replicate. The chromosomes in mammalian cells replicate according to a temporal program, with early and late replication occurring at the beginning and end of S phase respectively (black line). Inactive X chromosomes are delayed in replication timing with the majority of DNA synthesis occurring during the second half of S phase (blue line). Chromosomes with delayed replication timing are delayed in both initiation and completion of DNA synthesis, with active replication continuing through G2 (red line). Asynchronous cultures are harvested for mitotic cells and processed for BrdU incorporation and FISH.
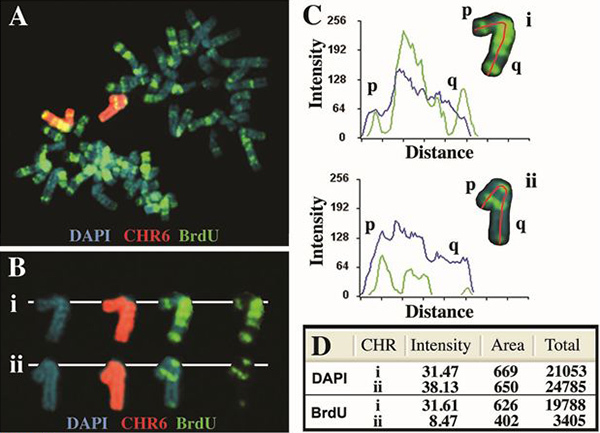 Figure 2. Asynchronous replication of human chromosome 6. Cells containing an engineered deletion of the ASAR6 gene4 were treated with BrdU for 5 hr, harvested for mitotic cells and processed for BrdU incorporation and FISH using a chromosome 6 paint as probe. The DNA was stained with DAPI (blue). A) A mitotic spread containing a typical "banded" pattern of BrdU incorporation (green) is shown. The chromosome 6 paint probe hybridized to two chromosome 6's (red) in this cell. B) The two chromosome 6's (i and ii) were "cut out" and displayed with the three fluorescent labels separated into distinct images. C) The chromosome 6's were analyzed using the Cytovision software and the signal intensity profiles for both DAPI (blue) and BrdU (green) are shown. The red line indicates the path used, from short arm (p) to long arm (q) for the quantification of both BrdU and DAPI. The Distance refers to the length of each chromosome from short arm (p) to long arm (q) in pixels. D) Quantification of the total signal for both DAPI and BrdU fluorescence. The total values represent the average pixel intensity multiplied by the area represented by those pixels.
Figure 2. Asynchronous replication of human chromosome 6. Cells containing an engineered deletion of the ASAR6 gene4 were treated with BrdU for 5 hr, harvested for mitotic cells and processed for BrdU incorporation and FISH using a chromosome 6 paint as probe. The DNA was stained with DAPI (blue). A) A mitotic spread containing a typical "banded" pattern of BrdU incorporation (green) is shown. The chromosome 6 paint probe hybridized to two chromosome 6's (red) in this cell. B) The two chromosome 6's (i and ii) were "cut out" and displayed with the three fluorescent labels separated into distinct images. C) The chromosome 6's were analyzed using the Cytovision software and the signal intensity profiles for both DAPI (blue) and BrdU (green) are shown. The red line indicates the path used, from short arm (p) to long arm (q) for the quantification of both BrdU and DAPI. The Distance refers to the length of each chromosome from short arm (p) to long arm (q) in pixels. D) Quantification of the total signal for both DAPI and BrdU fluorescence. The total values represent the average pixel intensity multiplied by the area represented by those pixels.
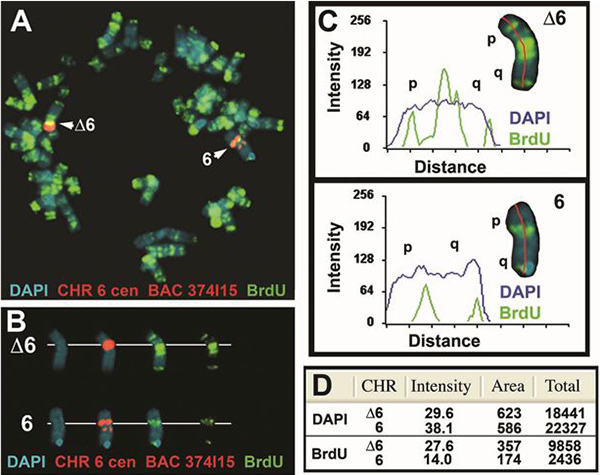 Figure 3. Delayed replication of human chromosome 6 containing a deletion of ASAR6. Cells containing an engineered deletion of the ASAR6 gene4 were treated with BrdU for 5 hr, harvested for mitotic cells and processed for BrdU incorporation and FISH using a chromosome 6 CEP plus a BAC containing the ASAR6 gene as probes. The DNA was stained with DAPI (blue). A) A mitotic spread containing a typical "banded" pattern of BrdU incorporation (green) is shown. The chromosome 6 CEP probe hybridized to two chromosome 6's (large red centromeric signal), and the BAC hybridized to a single chromosome 6 (small red signal on the long arm) in this cell. Note the difference in the BrdU banding pattern between the two 6's. B) The deleted chromosome 6 is represented by Δ6 and the non-deleted 6 by 6. The two 6's were "cut out" and displayed with the three fluorescent labels separated into distinct images. C) The chromosome 6's were analyzed using the Cytovision software and the signal intensity profiles for both DAPI (blue) and BrdU (green) are shown. The red line indicates the path used, from short arm (p) to long arm (q) for the quantification of both BrdU and DAPI. The Distance refers to the length of each chromosome from short arm (p) to long arm (q) in pixels. D) Quantification of the total signal for both DAPI and BrdU fluorescence. The total values represent the average pixel intensity multiplied by the area represented by those pixels.
Figure 3. Delayed replication of human chromosome 6 containing a deletion of ASAR6. Cells containing an engineered deletion of the ASAR6 gene4 were treated with BrdU for 5 hr, harvested for mitotic cells and processed for BrdU incorporation and FISH using a chromosome 6 CEP plus a BAC containing the ASAR6 gene as probes. The DNA was stained with DAPI (blue). A) A mitotic spread containing a typical "banded" pattern of BrdU incorporation (green) is shown. The chromosome 6 CEP probe hybridized to two chromosome 6's (large red centromeric signal), and the BAC hybridized to a single chromosome 6 (small red signal on the long arm) in this cell. Note the difference in the BrdU banding pattern between the two 6's. B) The deleted chromosome 6 is represented by Δ6 and the non-deleted 6 by 6. The two 6's were "cut out" and displayed with the three fluorescent labels separated into distinct images. C) The chromosome 6's were analyzed using the Cytovision software and the signal intensity profiles for both DAPI (blue) and BrdU (green) are shown. The red line indicates the path used, from short arm (p) to long arm (q) for the quantification of both BrdU and DAPI. The Distance refers to the length of each chromosome from short arm (p) to long arm (q) in pixels. D) Quantification of the total signal for both DAPI and BrdU fluorescence. The total values represent the average pixel intensity multiplied by the area represented by those pixels.
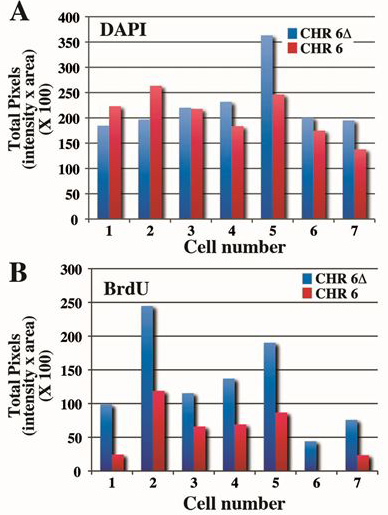 Figure 4. Quantification of the replication timing difference between chromosome 6's. Cells containing an engineered deletion of the ASAR6 gene4 were treated with BrdU for 5 hr, harvested for mitotic cells and processed for BrdU incorporation and FISH using a chromosome 6 CEP probe plus a BAC containing the ASAR6 gene as probe. Mitotic spreads were processed as in Figure 3 and the values for 7 different cells are shown (see Table 1). A) The DAPI staining was quantified and the total number of pixels is displayed for each cell. Note that there is only ~10-20% difference between chromosomes within the same cell. B) The BrdU incorporation was quantified from the same chromosomes as panel B above. Note that there is >2 fold difference between the total pixel values for the deleted and non-deleted chromosome 6's.
Figure 4. Quantification of the replication timing difference between chromosome 6's. Cells containing an engineered deletion of the ASAR6 gene4 were treated with BrdU for 5 hr, harvested for mitotic cells and processed for BrdU incorporation and FISH using a chromosome 6 CEP probe plus a BAC containing the ASAR6 gene as probe. Mitotic spreads were processed as in Figure 3 and the values for 7 different cells are shown (see Table 1). A) The DAPI staining was quantified and the total number of pixels is displayed for each cell. Note that there is only ~10-20% difference between chromosomes within the same cell. B) The BrdU incorporation was quantified from the same chromosomes as panel B above. Note that there is >2 fold difference between the total pixel values for the deleted and non-deleted chromosome 6's.
Discussion
The preparation of chromosome spreads is a critical step for the successful replication-timing assay described here. The inclusion of a colcemid pretreatment step prior to the hypotonic treatment may aid in the frequency and spreading of mitotic cells. We typically expose cells to colcemdi for 1-3 hr prior to harvest, and use colcemid at a final concentration of 10 ug/ml. However, the inclusion of a colcemid pretreatment step may alter the length of G2 and consequently may alter the apparent replication timing and state of condensation of the chromosomes3.
This procedure can be applied to many different cell types and species by varying the length of BrdU incubation, which depends on the cell cycle duration. For most human and mouse cell lines, the G2 phase is typically 3-6 hr; therefore, BrdU treatments are typically in this range. An alternative to BrdU is 5-ethynyl-2'deoxyuridine (EdU) and its subsequent detection using a fluorescent azide and "click chemistry" reaction10. The EdU detection scheme has several advantages over the BrdU detection scheme. For example, detection of EdU does not require sample fixation or DNA denaturation. Thus, the use of EdU to asses replication timing can be combined with simple G-banding techniques instead of FISH to identify the chromosomes of interest.
The replication timing protocol described here is specifically designed to assay late S phase plus any DNA synthesis extending into G2. In addition, DNA replication can be monitored throughout S phase using this procedure by using short (15-30 min) pulses of BrdU followed by relatively long chase periods of 6-10 hr. This allows for the visualization of BrdU incorporation in both early and middle S-phase. For example, certain tumor derived chromosome rearrangements are delayed in both the initiation and completion of DNA synthesis along the entire length of the chromosomes3.
One advantage of this replication timing procedure is that it assays replication timing in individual cells. Therefore, it has the ability to detect differences in replication timing between homologous chromosomes contained within the same cell. While there are other procedures, e.g. replication timing in situ hybridization (ReTiSH;11), that have the ability to detect differences in replication timing between alleles at specific loci on homologous chromosomes, the procedure described here can detect differences in the replication timing along the entire length of chromosomes. In addition, this procedure can assay differences in the replication timing of chromosomes that are present in only a fraction of cells of a population3. For example many cancer cell lines and primary tumor samples contain chromosome rearrangements that are present in less than 50% of cells. We are currently using this procedure to assay chromosomes in primary tumor samples, and have been able to detect asynchronous replication between chromosomes in multiple samples. However, given that primary tumor samples have a limited number of mitotic figures, about one third of the primary cultures failed to give sufficient numbers of mitotic spreads.
Another advantage that this procedure has over microarray or sequencing based assays is that individual chromosomes are assayed rather than immunoprecipitated DNA from pools of cells. In the immunoprecipitation-based assays polymorphisms must be identified and linked to specific alleles in order to distinguish the replication timing between alleles.
Furthermore, with the recognition that many cancer cells contain numerous chromosome rearrangements12, and the observation that DNA replication stress is associated with genomic instability in cancer cells13, we believe that this protocol is a useful and simple tool for the routine analysis of the replication timing of chromosomes in cancer cells.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by a grant from the National Cancer Institute, CA131967.
References
- Payer B, Lee JT. X chromosome dosage compensation: how mammals keep the balance. Annu. Rev. Genet. 2008;42:733–772. doi: 10.1146/annurev.genet.42.110807.091711. [DOI] [PubMed] [Google Scholar]
- Breger KS, Smith L, Turker MS, Thayer MJ. Ionizing radiation induces frequent translocations with delayed replication and condensation. Cancer Research. 2004;64:8231–8238. doi: 10.1158/0008-5472.CAN-04-0879. [DOI] [PubMed] [Google Scholar]
- Smith L, Plug A, Thayer M. Delayed Replication Timing Leads to Delayed Mitotic Chromosome Condensation and Chromosomal Instability of Chromosome Translocations. Proc. Natl. Acad. Sci. U.S.A. 2001;98:13300–13305. doi: 10.1073/pnas.241355098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffregen EP, Donley N, Stauffer D, Smith L, Thayer MJ. An autosomal locus that controls chromosome-wide replication timing and mono-allelicexpression. Hum. Mol. Genet. 2011;20:2366–2378. doi: 10.1093/hmg/ddr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breger KS, Smith L, Thayer MJ. Engineering translocations with delayed replication: evidence for cis control of chromosome replication timing. Hum. Mol. Genet. 2005;14:2813–2827. doi: 10.1093/hmg/ddi314. [DOI] [PubMed] [Google Scholar]
- Camargo M, Cervenka J. Patterns of DNA replication of human chromosomes. II. Replication map and replication model. Am. J. Hum. Genet. 1982;34:757–780. [PMC free article] [PubMed] [Google Scholar]
- Cohen SM, Cobb ER, Cordeiro-Stone M, Kaufman DG. Identification of chromosomal bands replicating early in the S phase of normal human fibroblasts. Exp. Cell Res. 1998;245(98):321–329. doi: 10.1006/excr.1998.4258. [DOI] [PubMed] [Google Scholar]
- Diaz-Perez S, et al. The element(s) at the nontranscribed Xist locus of the active X chromosome controls chromosomal replication timing in the mouse. Genetics. 2005;171:663–672. doi: 10.1534/genetics.105.043026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Perez SV, et al. A deletion at the mouse Xist gene exposes trans-effects that alter the heterochromatin of the inactive X chromosome and the replication time and DNA stability of both X chromosomes. Genetics. 2006;174:1115–1133. doi: 10.1534/genetics.105.051375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 2008;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger S, Selig S, Bergman Y, Cedar H. Allelic inactivation of rDNA loci. Genes Dev. 2009;23:2437–2447. doi: 10.1101/gad.544509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitelman Database of Chromosome Aberrations in Cancer. 2006. Available from: http://cgap.nci.nih.gov/Chromosomes/Mitelman.
- Branzei D, Foiani M. The checkpoint response to replication stress. DNA Repair (Amst) 2009;8:1038–1046. doi: 10.1016/j.dnarep.2009.04.014. [DOI] [PubMed] [Google Scholar]