

Abstract
The folding mechanism of the β-sheet protein CspA, the major cold shock protein of Escherichia coli, was previously reported to be a concerted, two-state process. We have reexamined the folding of CspA using multiple spectroscopic probes of the equilibrium transition and laser-induced temperature jump (T-jump) to achieve better time resolution of the kinetics. Equilibrium temperature-dependent Fourier transform infrared (1634 cm–1) and tryptophan fluorescence measurements reveal probe-dependent thermal transitions with midpoints (Tm) of 66 ± 1 and 61 ± 1 °C, respectively. Singular-value decomposition analysis with global fitting of the temperature-dependent infrared (IR) difference spectra reveals two spectral components with distinct melting transitions with different midpoints. T-Jump relaxation measurements of CspA probed by IR and fluorescence spectroscopy show probe-dependent multiexponential kinetics characteristic of non-two-state folding. The frequency-dependent IR transients all show biphasic relaxation with average time constants of 50 ± 7 and 225 ± 25 μs at a Tf of 77 °C and almost equal amplitudes. Similar biphasic kinetics are observed using Trp fluorescence of the wild-type protein and the Y42W and T68W mutants, with comparable lifetimes. All of these observations support a model for the folding of CspA through a compact intermediate state. The transient IR and fluorescence spectra are consistent with a diffuse intermediate having β-turns and substantial β-sheet structure. The loop β3–β4 structure is likely not folded in the intermediate state, allowing substantial solvent penetration into the barrel structure.
Concerted, two-state folding without populating intermediate states, either in an equilibrium or in a kinetic sense, is a remarkable feature of many small single-domain proteins.1,2 Apparently, these proteins fold in a highly cooperative process through a single high-energy transition state ensemble of structures.3β-Sheet proteins, however, do not usually fold with a two-state mechanism, perhaps because of their topological complexity and high contact order (the average separation in sequence between residues that are in contact in the native state).4 In many cases, intermediates are observed; for example, hydrophobic collapse is often the first step, followed by a slower conformational search to establish the long-range (in sequence space) contacts that make up the native state.5–9 Several groups have postulated that β-turns form early and facilitate the conformational search by establishing the proper registry of the strands.10–13 Recent work demonstrates that β-turns can form very rapidly, as fast as helices, and therefore can kinetically support the slower processes of β-strand registry search and sheet formation.14–16
Further support for this idea comes from the recent demonstration that the Trpzip β-hairpin folds through a transition state having a turn H-bond but not the cross-strand H-bonds. This study used a promising new approach to probe the influence of H-bonding strength on folding dynamics based on oxoamide-to-thioamide mutations.17 In the case of simple, two-state folding β-sheet proteins such as the src SH3 domain, protein L, and protein G, ϕ-value analysis has indicated the presence of a β-turn structure in the transition state ensemble.12,18–20 For larger proteins with more complex topologies, however, there is no direct evidence that β-turns form early in the folding process. Another mechanism for nucleating β-sheet formation may be the formation of specific hydrophobic side chain interactions that establish the strand registry.21 These mechanisms are not exclusive, and both are likely important for some proteins. Understanding how β-sheet proteins fold requires further work to clarify these issues.
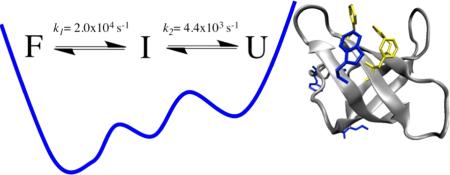
In contrast to the complex folding behavior observed for many β-sheet proteins, the major cold shock protein CspB of Bacillus subtilis was discovered to fold within milliseconds in a concerted, two-state manner.22 Subsequently, the closely related cold shock protein CspA of Escherichia coli was also proposed to fold by an apparent two-state mechanism via a compact transition state ensemble.23,24 These proteins are often cited as prototypical two-state folders, and they feature prominently in various attempts to correlate the folding rates of two-state systems with topology or other variables.1,2 CspA is an ideal model protein for the study of β-sheet formation because of its relative simplicity and fast folding rate. Its structure has been determined by both X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy.25–27 It is constructed of five β-strands arranged in an antiparallel barrel structure (Figure 1). It has a large, solvent-exposed cluster of aromatic amino acids that have been shown to be important for stability and nucleic acid binding function.28–31 NMR relaxation data for E. coli CspA suggest that Trp11 and the loop β3–β4 region are highly flexible with internal motions ranging from subnanosecond to millisecond time scales, which may also play a role in the nucleic acid binding function.27,32 The folding thermodynamics and kinetics of CspA have been examined previously using a variety of spectroscopic techniques with thermal and chemical denaturation methods and native state hydrogen exchange.23,33 The folding and unfolding kinetics of CspA, as measured by stopped-flow fluorescence spectroscopy at 25 °C, have been modeled by single-exponential kinetics, yielding folding and unfolding rates of ~200 and ~4 s–1, respectively. Subsequently, temperature-jump (T-jump) IR kinetics measurements with much better time resolution than the stopped-flow experiments revealed multiexponential kinetics characteristic of a non-two-state folding mechanism.34 Further evidence of non-two-state behavior was found in NMR measurements of hydrogen–deuterium exchange protection rates that are fast for residues within strands β2 and β3, suggesting the formation of an early intermediate having a hairpin turn between these two β-strands.33 These results suggest that a reexamination of the putative two-state mechanism for CspA is warranted.
Figure 1.

Ribbon representation of E. coli CspA (Protein Data Bank entry 1MJC)25 showing the relative locations of Trp11, Tyr42, Thr68, and the aromatic nucleic acid binding site.
In this study, the kinetics of folding and unfolding of wild-type (WT) E. coli CspA (Figure 1) and two CspA tryptophan (Trp) variants (Y42W and T68W) are measured using T-jump relaxation spectroscopy. The structural changes in the proteins are followed by either probing the changes in the amide I’ (amide I in D2O) infrared (IR) absorbance of the protein backbone or monitoring the fluorescence intensity changes of the Trp probes. The combination of laser-induced T-jump techniques for fast initiation of folding–unfolding events and spectroscopic approaches for probing structural changes provides the time resolution and structural specificity necessary for determining early protein folding events,35,36 which may otherwise be missed by conventional stopped-flow techniques because they are limited to a millisecond dead time. We observe biphasic relaxation behavior in response to a fast, laser-induced T-jump, regardless of whether the kinetics is measured with IR or fluorescence spectroscopy. The results of these experiments indicate that a simple two-state model cannot account for the observed relaxation dynamics of CspA.
MATERIALS AND METHODS
Sample Preparation
Recombinant, histidine-tagged wild-type CspA and the tryptophan variants were purified under denaturing conditions using Ni-NTA affinity resin (Qiagen, Valencia, CA).30,37 After dialysis to remove the denaturant and cleavage of the six-histidine affinity tag with TEV protease,30 the resulting purified proteins were dialyzed into buffer containing 50 mM potassium phosphate and 100 mM KCl at pH 7 and 4 °C. This solution was then dialyzed in deuterated buffer [50 mM potassium phosphate and 100 mM KCl (pH* 7)], filtered, and stored at –20 °C. The concentrations for wild-type CspA and the tryptophan variants used for all spectroscopic measurements were approximately 3.5 mg/mL. The protein samples used for the T-jump experiments were checked for aggregation, using the pair of sharp bands in the Fourier transform infrared (FTIR) spectrum near 1618 and 1685 cm–1, associated with the formation of extended β-structure aggregates.38 No aggregation was detected as long as the initial temperature for the T-jump measurements was kept below 65 °C.
Singular-Value Decomposition and Global Fitting
We have previously described our approach for using singular-value decomposition (SVD) to determine the number of spectral components required to describe the temperature dependence of the protein difference FTIR spectra.39 Briefly, temperature-dependent difference FTIR spectra are used to construct a data matrix, , in which each column represents the difference FTIR spectrum at a specific temperature. SVD analysis of this data matrix results in three matrices, A = U·S·VT, where the U, S, and VT matrices contain the basis spectra, the singular values, and the temperature evolution of the basis spectra, respectively. These matrices are truncated; the components containing the signal are retained, while the components containing only noise are removed. Two basis spectra are found to describe the original data matrix for CspA. A global fitting procedure is used to determine the individual spectral components and the temperature dependence of these basis spectra, as described previously.39 The model used here consists of two sigmoid functions because this represents the simplest way to globally fit the spectral components found for the temperature dependence of CspA. This analysis produces matrices D and FT that contain the individual spectral components and their temperature dependence, respectively, where D·FT equals . SVD analysis and global fitting were performed in IGOR Pro (WaveMetrics, Inc.).
Temperature-Jump Measurements
The methods developed by our group for studying fast protein folding and unfolding dynamics have been described in detail elsewhere.35,36 Our general approach is to use a fast, laser-induced T-jump to shift the equilibrium between the folded and unfolded ensemble of states. The relaxation dynamics are followed using IR and fluorescence spectroscopies to probe the structural changes induced by the T-jump. Each experiment has a dynamic range from ~20 ns (limited by the detector rise time and laser pulse width) to ~1 ms (where the solution starts to cool). Equilibrium IR and fluorescence spectra were obtained using a Bio-Rad (Cambridge, MA) model FTS-40 FTIR spectrometer and a Spex Fluorolog spectrofluorimeter (Instruments S.A., Edison, NJ), respectively. For both static and time-resolved measurements, a thermostated, dual-compartment sample cell with CaF2 windows and a Teflon spacer (50–150 μm) was employed to allow the separate measurement of sample and reference (D2O buffer or tryptophan solution) spectra under identical conditions. The reference measurements provide information needed both for T-jump calibration and for background subtraction.
RESULTS AND DISCUSSION
Thermal Denaturation of CspA
Fluorescence and IR spectroscopy provide complementary information about the specific structural changes that comprise protein folding. The IR amide I’ band is sensitive to the conformation and solvation of the polypeptide backbone and therefore reports on secondary structure formation as well as the global folding transition. Trp fluorescence is more sensitive to the local environment of the indole side chain, but it can also detect the global transition to the extent that this transition changes the local environment, such as the sequestration of the side chain from solvent in the formation of the hydrophobic core of the native state. Multiple Trp probes help to differentiate between local and global effects. A two-state folding reaction should give the same equilibrium and kinetics behavior, regardless of the probe.40 The thermal denaturation of wild-type CspA was mapped by measuring the temperature dependence of the IR and fluorescence spectra. The fluorescence spectrum of the folded protein at 20 °C has a peak near 350 nm due to the single tryptophan (Trp11) in CspA.23,41 Trp11 is partially exposed to solvent even in the folded structure (hence the red-shifted fluorescence maximum) and forms part of the aromatic and single-stranded nucleic acid binding cluster (Figure 1). The fluorescence intensity decreases, and the peak shifts slightly to a higher wavelength with an increasing temperature as a result of both the temperature-dependent fluorescence quantum yield and the protein conformational changes that increase the degree of solvent exposure of the tryptophan.42 The FTIR spectrum of the folded protein at 20 °C has several amide I’ components (caused by the loops, turns, and sheet), but the broad envelope is centered near 1634 cm–1 because of the predominantly β-sheet conformation of the polypeptide backbone.43,44 The amide I’ features lose intensity with an increase in temperature due to the loss of the secondary structure. Thus, both the IR and fluorescence spectra of the unfolded protein are consistent with the complete loss of the secondary structure (β-sheets and turns) and solvation of the indole side chains in the thermally denatured state of the protein.
The thermal denaturation transitions for wild-type CspA obtained by FTIR and fluorescence spectroscopy show different midpoints (Figure 2). The IR transition, measured near the peak of the main β-sheet component of the amide I’ IR absorbance (1634 cm–1), follows the global unfolding of the β-sheet structure. This melt curve is modeled by an apparent two-state transition, yielding a transition midpoint (Tm) of 66 ± 1 °C. The Trp fluorescence intensity measurements exhibit a more complex shape because of the intrinsic temperature dependence of the fluorescence quantum yield, which must be subtracted from the data to obtain the structural transition. Normally, it is difficult to separate the intrinsic thermal and structural effects, but in this case, because the Trp is partially solvent exposed in the folded protein, the intrinsic temperature dependence of its fluorescence can be approximated using free Trp in a D2O solution. Free Trp shows a quadratic dependence of fluorescence intensity on temperature. Therefore, the CspA fluorescence melt was modeled using an apparent two-state transition with a quadratic background (the inset of Figure 2 shows the background-subtracted data). The midpoint determined by this fitting procedure occurs at 61 ± 1 °C. Holding Tm at 66 °C during the fitting procedure results in a χ2 value that is 2.8 times larger. The difference in apparent Tm values measured by IR and fluorescence is clearly greater than the error of the measurement, meaning that the transition must involve more than two states.
Figure 2.

Thermal denaturation curves of wild-type CspA obtained by measuring the amide I’ absorbance (FTIR) at 1634 cm–1 (○) and the Trp fluorescence (△). The solid lines are fits to an apparent two-state model that yield apparent Tm values of 66 ± 1 and 61 ± 1 °C for IR and fluorescence, respectively. The inset shows an overlay of the background-corrected fluorescence data with the IR data and highlights the difference in apparent Tm values derived from the different probes.
Further evidence of non-two-state behavior was obtained from the temperature-dependent FTIR spectra (Figure 3). Figure 3A shows the FTIR difference spectra of CspA generated by subtracting the spectrum at 9 °C from each spectrum at the higher temperature. Two minima centered at 1625 and 1634 cm–1 are evident at the lower temperatures, and they appear to converge into a broader minimum with an increasing temperature. The 1625 cm–1 absorbance was previously assigned to loop structures.34,45–47 SVD analysis with global fitting yielded two D matrix spectral components from the FTIR difference spectra (Figure 3B). The first (largest amplitude) D matrix spectral component is dominated by a 1634 cm–1 band that is assigned to the β-sheet structure, and a broad positive absorbance at 1660 cm–1 that corresponds to the disordered polypeptide. The second D matrix spectral component is dominated by the 1625 cm–1 absorbance feature and is therefore assigned to loop structures. The inset of Figure 3B shows the corresponding temperature dependence of each of these spectral components, and the two are clearly different. The first spectral component has a more cooperative transition with an apparent Tm of 66 ± 1 °C, whereas the second spectral component has a broader transition with a lower apparent Tm. The best fit for the temperature dependence of the 1625 cm–1 spectral component is obtained with a model having a linear pretransition baseline and a cooperative transition. Such a model yields a sharper cooperative transition with an apparent Tm of 59 ± 1 °C.
Figure 3.

Temperature-dependent FTIR spectra. (A) FTIR difference spectra in 8 °C increments, generated by subtracting the lowest-temperature spectrum (9 °C) from higher-temperature spectra. The arrow shows the direction of increasing temperature. (B) D spectral components with the corresponding FT temperature profiles (shown as an inset) determined via SVD and global fitting analysis of the temperature-dependent FTIR difference spectra. The first spectral component (—) has a more cooperative transition with an apparent Tm of 66 °C (■), whereas the second spectral component (---) has a broader transition with a lower apparent Tm (□).
A previous study determined a thermal denaturation midpoint of 59 ± 1 °C by circular dichroism (222 nm),23,41 the same as the fluorescence and low-temperature IR transitions reported here within the uncertainty of the measurement. The native CD spectrum is dominated by a large positive signal in the far-UV region due to the aromatic cluster (six Phe residues, one Trp, and one Tyr). Because Trp11 is part of this aromatic cluster, it is not surprising that the Tm values from the fluorescence and CD probes are similar because both report on the same part of the protein structure. These results suggest that the lower apparent Tm observed by fluorescence, IR, and CD spectroscopy is due the formation of a partially unfolded intermediate state that involves the loops and the hydrophobic cluster, but without significant unfolding of the β-strands. To test this model, additional Trp fluorescence reporters were introduced into different parts of the CspA structure, in a Trp11 background (Figure 1). Single-point mutations were introduced at position 42 (Y42W) or 68 (T68W) to specifically label the β3–β4 loop that separates β-sheet 1 (strands β1–β3) from β-sheet 2 (strands β4 and β5) or the C-terminal strand β5 of sheet 2, respectively. The Y42W and T68W variants were previously characterized by CD and fluorescence spectroscopy, and the native structures, stabilities, and functions were found to be similar to those of wild-type CspA.37 The additional Trp in each of these variants was shown to contribute equally with Trp11 to the fluorescence intensities in both the native and chemically denatured states.37 Despite having these additional Trp probes that are remote from the aromatic cluster, the Y42W and T68W mutants have identical CD and fluorescence melts, with a Tm lower than that of the 1634 cm–1 IR melt, a clear indication that the lower-Tm process is not localized to the cluster.
SVD analysis of the IR melt provides additional insight into the nature of the low-temperature transition. The IR spectral signature of this transition (Figure 3B) is characteristic of a loop structure. Thus, we propose that the low-temperature transition involves melting of one or more of the surface loops of the protein, and an associated perturbation of the aromatic cluster. Previous NMR relaxation measurements indicate that the loop structures and the hydrophobic cluster are the most flexible parts of the CspA structure.27 These experiments reveal considerable internal motions within the nucleic acid binding epitope, especially the backbone amide protons within the β3–β4 and β4–β5 surface loops, the Trp11 side chain, and nearby side chains within the nucleic acid binding epitope. The flexibility of these structural regions was proposed to contribute to changes in entropy upon formation of the complex with the nucleic acid and thus to modulate the binding affinity and function of CspA. NMR titration experiments that monitor the binding of ssDNA to B. subtilis CspB show structural rearrangements that extend to the β3–β4 and β4–β5 loops.32
T-Jump Kinetic Measurements
Time-resolved IR and fluorescence spectroscopy were used to follow the relaxation of the protein to the new equilibrium point following a laser-induced T-jump. Figure 4A shows the IR relaxation kinetics of wild-type CspA in response to a T-jump from 60 to 77 (±1.0) °C. We emphasize that this T-jump occurs across the global unfolding transition (the transition with the higher apparent Tm in Figure 3B). The relaxation kinetics probed at 1634 cm–1 show a decrease in absorbance, corresponding to an overall loss of folded structure. This relaxation is modeled well by a double-exponential fit with a fast relaxation time constant (τ1) of 50 ± 2 μs (42%) and a slow time constant (τ2) of 228 ± 10 μs (58%). These two kinetic phases are separated well in time and easily resolved from one another, and both contribute almost equally to the observed relaxation. Clearly, these data are not consistent with a two-state kinetic model. Figure 4B compares the static FTIR difference spectrum with the time-resolved spectra at 50 μs and 1 ms following the laser-induced T-jump. Both the static and transient spectra correspond to the same change in temperature from an initial value of 60 °C to a final value of 77 °C. The static spectrum was computed by subtracting the FTIR spectrum at the initial temperature from the spectrum at the final temperature. The time-resolved spectra were constructed from the absorbance changes (the amplitude of the 50 μs kinetics phase and the total amplitude after 1 ms) derived from the double-exponential fits of the kinetics data in response to the same T-jump. The range of probe frequencies spanned the amide I’ band, corresponding to β-turn (1620 cm–1), β-sheet (1632–1635 cm–1), and disordered (1653–1658 cm–1) regions of the protein. The frequency-dependent IR transients all show biphasic relaxation with average time constants of 50 ± 7 and 225 ± 25 μs at a Tf of 77 °C. The transient spectrum at 1 ms matches the static spectrum within the error of the measurement. This observation is consistent with complete relaxation of the protein to the new equilibrium position on this time scale, meaning there is not a slower phase missed in the T-jump experiments. In contrast, the 50 μs transient spectrum is dominated by a bleach centered at 1634 cm–1 (characteristic of β-sheet and loop structures) with a minimal bleach at 1620 cm–1 (characteristic of β-turns), making it distinct from the transient spectrum at longer times. The 1634 cm–1 bleach has equal contributions from the fast and slow phases, whereas most (75%) of the amplitude of the 1620 cm–1 bleach appears in the slow phase. These data are consistent with the transient population of an intermediate state that has an IR spectrum distinct from that of the folded state. Furthermore, this transient intermediate likely has intact β-turns, based on the 50 μs IR spectrum, but the rest of the structure (including the loops, the aromatic cluster, and the β-sheet) is at least partly unfolded.
Figure 4.

Comparison of the wavelength-dependent time-resolved IR measurements with the static FTIR difference spectra. (A) IR transient at 1634 cm–1 of WT CspA in response to a T-jump from 60 to 77 (±1.0) °C. The relaxation is modeled well by a double-exponential fit (—). (B) Time-resolved IR spectra at 50 μs (○) and 1 ms (●) obtained from the frequency-dependent amplitudes of the T-jump transients. The transient spectra are mapped onto the static FTIR difference spectrum [ΔT = 77 – 60 °C (—)].
The T-jump relaxation kinetics of CspA was also probed using Trp fluorescence for the wild-type protein and two Trp mutants. The wild-type protein has a single Trp at position 11, located in the hydrophobic cluster (Figure 1). The transient fluorescence in response to a T-jump from 49 to 70 °C (Figure 5) fits well to a double-exponential function with the following relaxation times: τ1 = 56 ± 5 μs (15%), and τ2 = 327 ± 40 μs (85%). The relaxation times are the same as those observed for the IR transients with an equivalent T-jump. The fluorescence transient is dominated by the slow phase, however, and the relatively small amplitude of the fast phase is likely the reason it was not detected even as a burst phase in previous stopped-flow measurements. The T-jump Trp fluorescence transients for the Y42W and T68W variants are compared to that of the WT protein in Figure 5 (the intensities are normalized to facilitate comparison).
Figure 5.

Comparison of relaxation kinetics of WT and mutant CspA measured by Trp fluorescence in response to temperature jumps from 49 to 70 (±1.0) °C. The fluorescence decay signals are modeled well by double-exponential functions (—).
Previously, identical rates for the refolding of these two variants were observed by stopped-flow fluorescence spectroscopy and interpreted with an apparent two-state mechanism and a compact transition state.37 Here we find that all three proteins exhibit biphasic kinetics in response to a T-jump, as summarized in Table 1 for jumps to 74 °C. While the fast phase is nearly constant for all three proteins, the slow phase clearly becomes slower in the mutant proteins. The slower process represents the global unfolding of the protein, and therefore, its rate follows the expected trend for the increase in protein stability along the series: WT (ΔGf = 3.0 kcal/mol) < Y42W (ΔGf = 3.2 kcal/mol) < T68W (ΔGf = 3.7 kcal/mol). What is remarkable about these results is that regardless of where the Trp label is located within the structure (the aromatic cluster, the β3–β4 surface loop, or the C-terminal strand β5), biphasic kinetics are observed, with the same relative amplitude of the fast and slow phases.
Table 1.
Fluorescence T-Jump Results
| T-jump (°C) | Amp1 (%) | τobs1 (μs) | Amp2 (%) | τobs2 (μs) | |
|---|---|---|---|---|---|
| WT CspA | 58 to 74 ± 1 | 24 | 24 ± 5 | 76 | 262 ± 43 |
| Y42W | 58 to 74 ± 1 | 19 | 22 ± 5 | 81 | 303 ± 40 |
| T68W | 58 to 74 ± 1 | 23 | 30 ± 5 | 77 | 400 ± 50 |
Arrhenius plots of the fast and slow relaxation processes for WT CspA from both the IR and fluorescence T-jumps are shown in Figure 6. Both of the observed kinetic phases in the IR and fluorescence T-jump measurements exhibit a strong temperature dependence, thereby indicating that both of these processes are activated. The slow relaxation process has an apparent activation energy of 16.1 ± 1.3 kcal/mol. The observed rates for the fast kinetics process show considerable scatter; nevertheless, the rate clearly increases with temperature, yielding an apparent activation energy of 12.4 ± 3.2 kcal/mol. Also included in this graph are the relaxation rates derived from previous stopped-flow fluorescence measurements that report on the cooperative folding transition at lower temperatures.23,33 The T-jump relaxation rates are obtained at temperatures above the Tm for the global transition and are therefore dominated by the unfolding rate. In contrast, the stopped-flow measurements are obtained at temperatures below the Tm and therefore more closely reflect the folding rate. Extrapolated rates from both sets of measurements intersect at a temperature of 54.0 ± 1.0 °C, well below the nominal global melt temperature of 61 °C. A simple two-state process requires that the folding and unfolding branches intersect at Tm, so this result is additional evidence that CspA does not fold by such a simple mechanism.
Figure 6.

Arrhenius plot of relaxation rates for the fast (■) and slow (▲) processes from IR and fluorescence T-jumps. Relaxation rates derived from fluorescence stopped-flow measurements (●) at lower temperatures are also included.23,33
The IR and fluorescence kinetic measurements of WT CspA both detect two relaxation phases, regardless of the IR probe wavelength or the position of the Trp fluorescence probe. Furthermore, the equilibrium temperature dependence gives different Tm values depending on the IR probe frequency, or upon comparison of the IR and fluorescence melts. This probe dependence is characteristic of non-two-state behavior, even when an intermediate state is not sufficiently stable to be detected as a separate state in equilibrium experiments.4 These results indicate that CspA folds by a more complex mechanism than the previously proposed two-state kinetic model.23 The simplest model that explains these results is a three-state model:
The fast kinetic phase is observed in both the IR and fluorescence measurements of WT CspA and its mutants. The IR signature of the fast process is a bleach at 1634 cm–1 (the 50 μs spectrum in Figure 4), characteristic of the loss of some of the β-sheet structure, with possible contributions from one or more of the loop structures that have overlapping IR absorbance in this region. The intermediate state still has almost half of the native β-sheet structure content, and the β-turns are intact based on the 1620 cm–1 absorbance, evidence that this state is relatively compact. The fast process is also characterized by a reduction in the Trp fluorescence for all of the variants studied, although only ~20% of the total fluorescence change is part of this phase. The fluorescence intensity of Trp in proteins is sensitive to specific quenching interactions and to the solvent exposure of the indole ring, with an increased level of hydration causing a decrease and a red shift of the emission.42 The Trp probe is located in disparate parts of the structure of the different protein variants studied, including the surface aromatic cluster (W11, WT), the β3–β4 surface loop (Y42W), and the C-terminal β5 strand (T68W). Therefore, the fast process detected with Trp fluorescence is consistent with a global increase in the level of hydration of the structure that perturbs all of the labeled sites approximately equally. Because the fast fluorescence transient has a small amplitude, however, the degree of solvent penetration is likely relatively small, further evidence that the intermediate state retains a compact structure. It is possible that a specific structural change such as the melting of the β3–β4 surface loop disrupts the aromatic cluster and allows some surface hydration of the underlying β-strands, which would impact of all the labeled sites. In addition, the amide I’ absorbance band is sensitive to backbone hydration as observed in the distinct IR bands exhibited by surface-exposed amide carbonyls (1655–1660 cm–1) compared with solvent-protected amide carbonyls (1634 cm–1) of β-strands.14,48 Therefore, by analogy, β-strand “solvation” could shift the amide I’ band without breaking the β-strand hydrogen bond. Molecular dynamics (MD) simulations and 17O and 2H NMR relaxation experiments have shown that penetration of water into proteins is an activated process with activation enthalpies between 1.4 and 5.5 kcal/ mol.49,50 Our calculated apparent activation energy of 12.4 ± 3.2 kcal/mol for this fast kinetic process can certainly account for several water molecules penetrating into the structure of CspA.
We conclude that the first step in the unfolding of this protein is global in nature and produces an intermediate structure that retains a slightly expanded hydrophobic core and approximately half of the β-sheet structure. The subsequent slower process (I ↔ U) is the global unraveling of the hydrophobic core and the remaining β-sheet structure. This slower process is the rate-determining step as it represents the main barrier to unfolding. The Arrhenius plot (Figure 4) clearly shows that the unfolding branch has a steep temperature dependence with a large apparent activation energy for unfolding (16.1 ± 1.3 kcal/mol) because of the loss of the stabilizing interactions still present in the nativelike intermediate state. In comparison, the enthalpic barrier to folding (as shown in the folding arm of the plot) is much smaller.
We conclude that CspA folds through a compact intermediate state that has much of the native secondary structure and hydrophobic core already formed. Formation of this intermediate is likely initiated by rapid folding of the β-turns, which are already present in the intermediate state based on the transient IR spectrum. Previously, it has been shown that β-turns fold within hundreds of nanoseconds to microseconds, and for this reason, these structures have been postulated as nucleation sites for the folding of β-sheet structures.14,15,51 These results provide the first kinetic evidence of such a mechanism. The fast folding of the turns facilitates a rapid chain collapse to form a compact intermediate state that has substantial β-sheet structure. The intermediate state is slightly expanded, with some solvent penetration, on the basis of the global response of the IR and fluorescent probes. In support of this model, MD simulations of CspA by Brooks and colleagues suggest an early collapse of the protein with water-mediated hydrogen bonding interactions followed by final native structure rearrangements.52 Finally, CspB, a protein that is closely related to CspA structurally and in its folding behavior, has been found to fold through a compact transition state that is very diffuse, with only a few residues in strand β1 exhibiting Φ values of >0.5.53 The transient intermediate observed for CspA appears to be similar in nature and is likely close to the structure of the transition state.
A related hierarchical folding mechanism has been proposed for several β-sheet proteins in the intracellular lipid binding protein (iLBP) family that includes the rat intestinal fatty acid binding protein (IFABP), ileal lipid binding protein (ILBP), cellular retinoic acid binding proteins I and II (CRABP I and CRABP II, respectively), and cellular retinal binding protein I (CRBP I).7,8,54–57 Studies of the folding mechanisms of these various proteins all postulate the occurrence of a hydrophobic collapse that brings together residues that are distant in sequence as the initial event in the folding mechanism. Studies of CRABP I and ILBP I show that the early intermediates formed via hydrophobic collapse have nativelike secondary structures, suggesting that β-sheet formation is guided by incipient native side chain interactions in this compact state.7,9,55–58 For CRBP I and CRABP II, the β-turns have also been proposed to act as nucleation centers for β-strand propagation.8,55–57 These studies lend support to our hypothesis of a sequential model for the folding of CspA, which is initiated by rapid collapse of the hydrophobic core and formation of β-turns, followed by the slower formation of the β-sheet structure and the eventual squeezing out of water molecules to pack the hydrophobic core and form the β-barrel structure.
In summary, CspA clearly folds though a multistate kinetic pathway, and therefore, it is not an appropriate model system for predictions of two-state folding behavior. This result is somewhat unexpected because CspA appears to fit well within the range of chain length and contact order characteristic of two-state folding, a clear indication that there must be other factors influencing the folding mechanism. Finally, the detection of multistate folding of CspA required the use of multiple spectroscopic probes and sufficient time resolution for the kinetic measurements. These results emphasize the importance of using multiple probes and high time resolution to verify a two-state folding mechanism.
Acknowledgments
Funding
This work was supported by National Institutes of Health Grant GM53640 (R.B.D.).
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Bai YW, Zhou HY, Zhou YQ. Critical nucleation size in the folding of small apparently two-state proteins. Protein Sci. 2004;13:1173–1181. doi: 10.1110/ps.03587604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson SE. How do small single-domain proteins fold? Folding Des. 1998;3:R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 3.Fersht AR. Optimization of rates of protein-folding: The nucleation-condensation mechanism and its implication. Proc. Natl. Acad. Sci. U.S.A. 1995;92:10869–10873. doi: 10.1073/pnas.92.24.10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Plaxco KW, Simons KT, Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 5.Hamada D, Segawa S, Goto Y. Non-native α-helical intermediate in the refolding of β-lactoglobulin, a predominantly β-sheet protein. Nat. Struct. Biol. 1996;3:868–873. doi: 10.1038/nsb1096-868. [DOI] [PubMed] [Google Scholar]
- 6.Liu ZP, Rizo J, Gierasch LM. Equilibrium Folding Studies of Cellular Retinoic Acid-Binding Protein, a Predominantly β-Sheet Protein. Biochemistry. 1994;33:134–142. doi: 10.1021/bi00167a017. [DOI] [PubMed] [Google Scholar]
- 7.Clark PL, Lie Z-P, Rizo J, Gierasch LM. Cavity formation before stable hydrogen bonding in the folding of a β-clam protein. Nat. Struct. Biol. 1997;4:883–886. doi: 10.1038/nsb1197-883. [DOI] [PubMed] [Google Scholar]
- 8.Yeh S-R, Ropson IJ, Rousseau DL. Hierarchical folding of intestinal fatty acid binding protein. Biochemistry. 2001;40:4205–4210. doi: 10.1021/bi0155044. [DOI] [PubMed] [Google Scholar]
- 9.Rotondi KS, Gierasch LM. Role of local sequence in the folding of cellular retinoic acid binding protein I: Structural propensities of reverse turns. Biochemistry. 2003;42:7976–7985. doi: 10.1021/bi034304k. [DOI] [PubMed] [Google Scholar]
- 10.Grantcharova VP, Riddle DS, Santiago JV, Baker D. Important role of hydrogen bonds in the structurally polarized transition state for folding of the src SH3 domain. Nat. Struct. Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 11.Kortemme T, Kelly MJS, Kay LE, Forman-Kay J, Serrano L. Similarities between the spectrin SH3 domain denatured state and its folding transition state. J. Mol. Biol. 2000;297:1217–1229. doi: 10.1006/jmbi.2000.3618. [DOI] [PubMed] [Google Scholar]
- 12.McCallister EL, Alm E, Baker D. Critical role of β-hairpin formation in protein G folding. Nat. Struct. Biol. 2000;7:669–673. doi: 10.1038/77971. [DOI] [PubMed] [Google Scholar]
- 13.Matheson RR, Jr., Scheraga HA. A Method for Predicting Nucleation Sites for Protein Folding Based on Hydrophobic Contacts. Macromolecules. 1978;11:819–829. [Google Scholar]
- 14.Davis CM, Xiao S, Raleigh DP, Dyer RB. Raising the Speed Limit for β-Hairpin Formation. J. Am. Chem. Soc. 2012;134:14476–14482. doi: 10.1021/ja3046734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Y, Oyola R, Gai F. Infrared study of the stability and folding kinetics of a 15-residue β-hairpin. J. Am. Chem. Soc. 2003;125:15388–15394. doi: 10.1021/ja037053b. [DOI] [PubMed] [Google Scholar]
- 16.Chen RPY, Huang JJT, Chen HL, Jan H, Velusamy M, Lee CT, Fann WS, Larsen RW, Chan SI. Measuring the refolding of β-sheets with different turn sequences on a nanosecond time scale. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7305–7310. doi: 10.1073/pnas.0304922101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Culik RM, Jo H, DeGrado WF, Gai F. Using Thioamides To Site-Specifically Interrogate the Dynamics of Hydrogen Bond Formation in β-Sheet Folding. J. Am. Chem. Soc. 2012;134:8026–8029. doi: 10.1021/ja301681v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grantcharova VP, Riddle DS, Baker D. Long-range order in the src SH3 folding transition state. Proc. Natl. Acad. Sci. U.S.A. 2000;97:7084–7089. doi: 10.1073/pnas.97.13.7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riddle DS, Grantcharova VP, Santiago JV, Alm E, Ruczinski I, Baker D. Experiment and theory highlight role of native state topology in SH3 folding. Nat. Struct. Biol. 1999;6:1016–1024. doi: 10.1038/14901. [DOI] [PubMed] [Google Scholar]
- 20.Kim D, Fisher C, Baker D. A breakdown of symmetry in the folding transition state of protein L. J. Mol. Biol. 2000;298:971–984. doi: 10.1006/jmbi.2000.3701. [DOI] [PubMed] [Google Scholar]
- 21.Northey JG, Di Nardo AA, Davidson AR. Hydrophobic core packing in the SH3 domain folding transition state. Nat. Struct. Biol. 2002;9:126–130. doi: 10.1038/nsb748. [DOI] [PubMed] [Google Scholar]
- 22.Schindler T, Herrler M, Maraheil MH, Schmid FX. Extremely rapid protein folding in the absence of intermediates. Nat. Struct. Biol. 1995;2:663–673. doi: 10.1038/nsb0895-663. [DOI] [PubMed] [Google Scholar]
- 23.Reid KL, Rodriguez HM, Hillier BJ, Gregoret LM. Stability and folding properties of a model β-sheet protein, Escherichia coli CspA. Protein Sci. 1998;7:470–479. doi: 10.1002/pro.5560070228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perl D, Welker C, Schindler T, Schroeder K, Maraheil MA, Jaenicke R, Schmid FX. Conservation of rapid two-state folding in mesophilic, thermophilic, and hyperthermophilic cold shock proteins. Nat. Struct. Biol. 1998;5:229–235. doi: 10.1038/nsb0398-229. [DOI] [PubMed] [Google Scholar]
- 25.Schindelin H, Jiang W, Inouye M, Heinemann U. Crystal structure of CspA, the major cold shock protein of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1994;91:5119–5123. doi: 10.1073/pnas.91.11.5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newkirk K, Feng W, Jiang W, Tejero R, Emerson SD, Inouye M, Montelione GT. Solution NMR structure of the major cold shock protein (CspA) from Escherichia coli: Identification of a binding epitope for DNA. Proc. Natl. Acad. Sci. U.S.A. 1994;91:5114–5118. doi: 10.1073/pnas.91.11.5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng W, Tejero R, Zimmerman DE, Inouye M, Montelione GT. Solution NMR structure and backbone dynamics of the major cold-shock protein (CspA) from Escherichia coli: Evidence for conformational dynamics in the single-stranded RNA-binding site. Biochemistry. 1998;37:10881–10896. doi: 10.1021/bi980269j. [DOI] [PubMed] [Google Scholar]
- 28.Jiang W, Hou Y, Inouye M. CspA, the major cold-shock protein of Escherichia coli, is an RNA chaperone. J. Biol. Chem. 1997;272:196–202. doi: 10.1074/jbc.272.1.196. [DOI] [PubMed] [Google Scholar]
- 29.Ermolenko DN, Makhatadze GI. Bacterial cold-shock proteins. Cell. Mol. Life Sci. 2002;59:1902–1913. doi: 10.1007/PL00012513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hillier BJ, Rodriguez HM, Gregoret LM. Coupling of protein stability and protein function in E. coli CspA. Folding Des. 1998;3:87–93. doi: 10.1016/S1359-0278(98)00014-5. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez HM, Vu DM, Gregoret LM. Role of a solvent-exposed aromatic cluster in the folding of Escherichia coli CspA. Protein Sci. 2000;9:1993–2000. doi: 10.1110/ps.9.10.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeeb M, Balbach J. Single-stranded DNA binding of the cold-shock protein CspB from Bacillus subtilis: NMR mapping and mutational characterization. Protein Sci. 2003;12:112–123. doi: 10.1110/ps.0219703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez HM, Robertson AD, Gregoret LM. Native state EX2 and EX1 hydrogen exchange of Escherichia coli CspA, a small β-sheet protein. Biochemistry. 2002;41:2140–2148. doi: 10.1021/bi011347x. [DOI] [PubMed] [Google Scholar]
- 34.Leeson DT, Gai F, Rodriguez HM, Gregoret LM, Dyer RB. Protein folding and unfolding on a complex energy landscape. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2527–2532. doi: 10.1073/pnas.040580397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dyer RB, Gai F, Woodruff WH, Gilmanshin R, Callender RH. Infrared studies of fast events in protein folding. Acc. Chem. Res. 1998;31:709–716. [Google Scholar]
- 36.Callender R, Dyer RB. Probing protein dynamics using temperature jump relaxation spectroscopy. Curr. Opin. Struct. Biol. 2002;12:628–633. doi: 10.1016/s0959-440x(02)00370-6. [DOI] [PubMed] [Google Scholar]
- 37.Vu DM, Reid KL, Rodriguez HM, Gregoret LM. Examination of the folding of E. coli CspA through tryptophan substitutions. Protein Sci. 2001;10:2028–2036. doi: 10.1110/ps.16201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muga A, Mantsch HH, Surewicz WK. Membrane-binding induces destabilization of cytochrome-c structure. Biochemistry. 1991;30:7219–7224. doi: 10.1021/bi00243a025. [DOI] [PubMed] [Google Scholar]
- 39.Brewer SH, Tang Y, Vu DM, Gnanakaran S, Raleigh DP, Dyer RB. Temperature Dependence of Water Interactions with the Amide Carbonyls of α-Helices. Biochemistry. 2012;51:5293–5299. doi: 10.1021/bi3006434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oliveberg M, Wolynes PG. The experimental survey of protein-folding energy landscapes. Q. Rev. Biophys. 2005;38:245–288. doi: 10.1017/S0033583506004185. [DOI] [PubMed] [Google Scholar]
- 41.Chatterjee S, Jiang W, Emerson SD, Inouye M. The backbone structure of the major cold-shock protein CS7.4 of Escherichia coli in solution includes extensive β-sheet structure. J. Biochem. 1993;114:663–669. doi: 10.1093/oxfordjournals.jbchem.a124234. [DOI] [PubMed] [Google Scholar]
- 42.Eftink M. The use of fluorescence methods to monitor unfolding transitions in proteins. Biophys. J. 1994;66:482–501. doi: 10.1016/s0006-3495(94)80799-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haris PI, Lee DC, Chapman D. A Fourier transform infrared investigation of the structural differences between ribonuclease A and ribonuclease S. Biochim. Biophys. Acta. 1986;874:255–265. doi: 10.1016/0167-4838(86)90024-5. [DOI] [PubMed] [Google Scholar]
- 44.Olinger JM, Hill DM, Jakobsen RJ, Brody RS. Fourier transform infrared studies of ribonuclease in H2O and 2H2O solutions. Biochim. Biophys. Acta. 1986;869:89–98. doi: 10.1016/0167-4838(86)90314-6. [DOI] [PubMed] [Google Scholar]
- 45.Hilario J, Kubelka J, Keiderling TA. Optical spectroscopic investigations of model β-sheet hairpins in aqueous solution. J. Am. Chem. Soc. 2003;125:7562–7574. doi: 10.1021/ja030039e. [DOI] [PubMed] [Google Scholar]
- 46.Maness SJ, Franzen S, Gibbs AC, Causgrove TP, Dyer RB. Nanosecond temperature jump relaxation dynamics of cyclic β-hairpin peptides. Biophys. J. 2003;84:3874–3882. doi: 10.1016/S0006-3495(03)75115-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Honda S, Yamasaki K, Sawada Y, Morii H. 10 residue folded peptide designed by segment statistics. Structure. 2004;12:1507–1518. doi: 10.1016/j.str.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 48.Manas ES, Getahun Z, Wright WW, DeGrado WF, Vanderkooi JM. Infrared spectra of amide groups in α-helical proteins: Evidence for hydrogen bonding between helices and water. J. Am. Chem. Soc. 2000;122:9883–9890. [Google Scholar]
- 49.Denisov VP, Peters J, Horlein HD, Halle B. Using buried water molecules to explore the energy landscape of proteins. Nat. Struct. Biol. 1996;3:505–509. doi: 10.1038/nsb0696-505. [DOI] [PubMed] [Google Scholar]
- 50.Garcia AE, Hummer G. Water penetration and escape in proteins. Proteins. 2000;38:261–272. [PubMed] [Google Scholar]
- 51.Dyer RB, Maness SJ, Peterson ES, Franzen S, Fesinmeyer RM, Andersen NH. The mechanism of β-hairpin formation. Biochemistry. 2004;43:11560–11566. doi: 10.1021/bi049177m. [DOI] [PubMed] [Google Scholar]
- 52.Shea JE, Brooks C., III From folding theories to folding proteins: A review and assessment of the simulation studies of protein folding and unfolding. Annu. Rev. Phys. Chem. 2001;52:499–535. doi: 10.1146/annurev.physchem.52.1.499. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Mira MM, Boehringer D, Schmid FX. The folding transition state of the cold shock protein is strongly polarized. J. Mol. Biol. 2004;339:555–569. doi: 10.1016/j.jmb.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 54.Ropson IJ, Gordon JI, Frieden C. Folding of a predominantly β-structure protein: Rat intestinal fatty acid binding protein. Biochemistry. 1990;29:9591–9599. doi: 10.1021/bi00493a013. [DOI] [PubMed] [Google Scholar]
- 55.Burns LL, Dalessio PM, Ropson IJ. Folding mechanism of three structurally similar β-sheet proteins. Proteins. 1998;33:107–118. doi: 10.1002/(sici)1097-0134(19981001)33:1<107::aid-prot10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 56.Dalessio PM, Ropson IJ. β-Sheet proteins with nearly identical structures have different folding intermediates. Biochemistry. 2000;39:860–871. doi: 10.1021/bi991937j. [DOI] [PubMed] [Google Scholar]
- 57.Burns LL, Ropson IJ. Folding of intracellular retinol and retinoic acid binding proteins. Proteins. 2001;43:292–302. doi: 10.1002/prot.1040. [DOI] [PubMed] [Google Scholar]
- 58.Krishnan VV, Sukumar M, Gierasch LM, Cosman M. Dynamics of cellular retinoic acid binding protein I on multiple time scales with implications for ligand binding. Biochemistry. 2000;39:9119–9129. doi: 10.1021/bi000296l. [DOI] [PubMed] [Google Scholar]