

Summary
Csr is a conserved global regulatory system that controls expression of several hundred Escherichia coli genes. CsrA protein represses translation of numerous genes by binding to mRNA and inhibiting ribosome access. CsrA also activates gene expression, although an activation mechanism has not been reported. CsrA activates flhDC expression, encoding the master regulator of flagellum biosynthesis and chemotaxis, by stabilizing the mRNA. Computer modeling, gel mobility shift, and footprint analyses identified two CsrA binding sites extending from positions 1–12 (BS1) and 44–55 (BS2) of the 198-nt flhDC leader transcript. flhD'-'lacZ expression was reduced by mutations in csrA and/or the CsrA binding sites. The position of BS1 suggested that bound CsrA might inhibit 5' end-dependent RNase E cleavage of flhDC mRNA. Consistent with this hypothesis, CsrA protected flhDC leader RNA from RNase E cleavage in vitro and protection depended on BS1 and BS2. Primer extension studies identified flhDC decay intermediates in vivo that correspond to in vitro RNase E cleavage sites. Deletion of these RNase E cleavage sites resulted in increased flhD'-'lacZ expression. Data from mRNA decay studies and quantitative primer extension assays support a model in which bound CsrA activates flhDC expression by inhibiting the 5' end-dependent RNase E cleavage pathway.
Keywords: CsrA, RNase E, mRNA stability, gene regulation, motility, RNA binding protein
Introduction
Csr (carbon storage regulator) is a conserved global regulatory system that potentially regulates expression of several hundred genes in Escherichia coli (Edwards et al., 2011). The Csr system, also called Rsm (repressor of secondary metabolites) in some organisms, regulates virulence factors, quorum sensing, motility, carbon metabolism, peptide uptake, and biofilm development in various eubacterial species (reviewed in Babitzke and Romeo, 2007; Romeo et al., 2012). The major components of the E. coli Csr system include the homodimeric RNA binding protein CsrA (Liu and Romeo, 1997), two noncoding small RNAs (sRNA) CsrB and CsrC (Liu et al., 1997; Weilbacher et al., 2003), the BarA-UvrY two-component signal transduction system (TCS) that activates csrB and csrC transcription (Suzuki et al., 2002; Weilbacher et al., 2003), and CsrD, a protein that participates in RNase E-mediated degradation of the sRNAs (Suzuki et al., 2006). CsrB and CsrC contain several CsrA binding sites and antagonize CsrA binding to mRNA targets (Liu et al., 1997; Weilbacher et al., 2003; Babitzke and Romeo, 2007; Romeo et al., 2012).
Regulation of the E. coli Csr system is complex. Transcription of csrA is controlled by five promoters, two of which are σS-dependent. CsrA also indirectly activates its own transcription (Yakhnin et al., 2011a). The Csr system is also controlled by three negative feedback loops. In one case CsrA represses expression of CsrD, thereby stabilizing its own sRNA antagonists (Suzuki et al., 2006). In another regulatory loop CsrA indirectly stimulates transcription of csrB and csrC via the BarA-UvrY TCS, which responds to acetate and other short chain fatty acids (Suzuki et al., 2002; Chavez et al., 2010). Finally, CsrA represses its own translation, thereby ensuring that CsrA levels are tightly controlled (Yakhnin et al., 2011a).
CsrA represses translation initiation of a variety of genes, typically by binding to multiple sites in the leader region of target mRNAs, one of which overlaps the cognate Shine-Dalgarno (SD) sequence, such that bound CsrA blocks 30S ribosomal subunit binding (Babitzke and Romeo, 2007; Edwards et al., 2011; Yakhnin et al., 2011a; Pannuri et al., 2012). However, CsrA represses translation of hfq by binding to a single site that overlaps its SD sequence (Baker et al., 2007), as well as sdiA by binding exclusively to two sites in the amino terminal coding region (Yakhnin et al., 2011b). Translational repression often leads to rapid mRNA decay (Babitzke and Romeo, 2007). CsrA is a homodimer with two identical RNA-binding surfaces that simultaneously bind two sites within a transcript (Dubey et al., 2003; Mercante et al., 2006; 2009). Considerable sequence variation exists among the known CsrA binding sites; however, GGA is a highly conserved motif that is often present in the loop of short RNA hairpins (Dubey et al., 2005; Babitzke and Romeo, 2007).
RNase E is an essential endonuclease that cleaves RNA in single-stranded AU-rich regions (McDowall et al., 1994). This enzyme plays a central role in the degradation of mRNA and in a variety of RNA processing reactions (Kime et al., 2008; Steade et al., 2011). RNase E cleavage can occur by a 5' end-dependent mechanism in which 5' monophosphorylated transcripts interact with a 5' binding pocket on the enzyme (Callaghan et al., 2005). In addition to the 5' end-dependent pathway, RNase E is capable of cleaving transcripts by an internal entry pathway that does not involve interaction with the 5' end of the transcript (Mackie, 1998; Kime et al., 2010). When translation of mRNA is inhibited, transcripts are generally more susceptible to degradation by RNase E (Richards et al., 2012). The single-stranded RNA cleavage activity of RNase E is located in the N-terminal domain of the enzyme (Callaghan et al., 2005). Although the C-terminal domain of RNase E is not essential (Ow et al., 2000), it serves as a scaffold for assembly of a multisubunit complex called the degradosome, which also includes a processive 3' to 5' exoribonuclease (polynucleotide phosphorylase, PNPase), an RNA helicase (RhlB), and enolase. RNase E and PNPase can function independently, but association with the degradosome may coordinate RNA decay (reviewed in Carpousis, 2007; Arraiano et al., 2010). The composition and function of the degradosome may be altered by environmental conditions and modulated by other proteins including RraA and RraB, which inhibit RNase E activity (Gao et al., 2006; Carpousis, 2007).
Although CsrA-mediated translational repression is well documented, a molecular mechanism of CsrA-mediated activation has not been identified. CsrA activates expression of the flhDC operon (Wei et al., 2001), encoding a hexameric DNA binding protein (FlhD4C2) (Wang et al., 2006). FlhD4C2 plays a key role in activating flagellum synthesis and chemotaxis (reviewed in Smith and Hoover, 2009). Transcriptional regulation of flhDC expression occurs in response to many environmental factors including changes in temperature, osmolarity, pH, and the availability of carbon sources (Smith and Hoover, 2009). Examination of growth phase regulation of flhDC demonstrated that protein levels were highest at mid-exponential phase with a second peak occurring during the transition to stationary phase (Prüß and Matsumura, 1997). Previous results demonstrated that CsrA increases flhDC expression by stabilizing the transcript, although the underlying mechanism was not investigated (Wei et al., 2001). Here, we explored the mechanism of CsrA-mediated activation of flhDC expression. Two CsrA binding sites were identified in the flhDC leader transcript that are required for CsrA-dependent activation of flhDC expression. Our data further indicate that CsrA binding to these two sites stabilizes the flhDC transcript by interfering with the 5' end-dependent RNase E cleavage pathway.
Results
CsrA binds to two sites in the flhDC leader transcript
We previously found that expression of a flhDC'-'lacZ translational fusion was reduced in a csrA::kan strain, which correlated with a reduction in the flhDC mRNA half-life (Wei et al., 2001). In addition, csrA mutant cells were non-motile and lacked flagella (Wei et al., 2001). A 198-nt untranslated leader precedes the flhD coding sequence. RNA structure predictions using Mfold (Zuker, 2003) suggested that the flhDC untranslated leader was highly structured. Results from RNA structure mapping experiments using RNase T1 as a probe (single-stranded G specific) were consistent with five hairpin structures (HP1-HP5) that form in the leader transcript (Fig. 1A, 1B and 1C [0 μM CsrA]). As HP5 includes the flhD SD sequence in the loop of the structure, we tested whether bound CsrA altered the structure in such a way that the SD sequence would be more accessible for 30S ribosomal subunit binding, thereby providing an explanation for CsrA-mediated gene activation. The RNase T1 cleavage pattern of the G residues between positions 109 and 240, which includes the flhD SD sequence, was unaffected by bound CsrA; however, we did observe CsrA-dependent protection of G residues in the loops of HP1 and HP2 (Fig. 1C, 1 μM CsrA).
Fig. 1. flhDC leader RNA and CsrA-flhDC leader RNA footprint analysis.

A. Sequence of flhDC leader RNA. The CsrA binding sites (BS1 and BS2), the flhD Shine- Dalgarno (SD) sequence and translation initiation codon (Met) are in bold. Hairpins 1–5 (HP1-HP5) are shown with horizontal arrows. Residues protected by CsrA from RNase T1 or RNase T2 cleavage are indicated with a (–) below the residue. Arrowheads mark RNase E cleavage sites identified in vitro. Numbering is with respect to the start of transcription.
B. Secondary structure of flhDC leader RNA. Hairpins 1–5 (HP1-HP5) are shown. Positions of the CsrA binding sites (BS1 and BS2), the flhD Shine-Dalgarno (SD) sequence and translation initiation codon (Met) are in bold. Arrowheads mark RNase E cleavage sites identified in vitro. Binding sites for McaS sRNA (McaS1 and McaS2) are underlined. A long vertical arrow indicates the position of an engineered λtR2 terminator (Term). Numbering is with respect to the start of transcription.
C. RNA structure mapping of an flhDC transcript extending from +1 to +276. 5’ end-labeled RNA was treated with RNase T1. Experimental samples contained 0 or 1 μM CsrA. Partial alkaline hydrolysis (B) and RNase T1 digestion (T) ladders, and control lanes without Rnase treatment (C), are shown. The RNase T1 ladder was generated under denaturing conditions (3 M urea at 55°C) so that every G residue could be observed. Positions of the flhD Shine-Dalgarno (SD) sequence and HP1-HP5 are shown.
D. CsrA-flhDC RNA footprint analysis. 5’ end-labeled RNA (+1 to +99) was treated with RNase T1 or RNase T2 in the absence or presence of 0.25 or 0.5 μM CsrA. Partial alkaline hydrolysis (B) and RNase T1 digestion (T) ladders, and control lanes without RNase treatment (C), are shown. Residues in which bound CsrA reduced RNase cleavage are marked on the right of each gel (–). Positions of the CsrA binding sites (BS1 and BS2) are shown.
A position weighted matrix search tool (Baker et al., 2007) identified two potential CsrA binding sites in the first 55 nt of the flhDC leader transcript, both of which contained the conserved GGA motif (Fig. 1A). Importantly, these two predicted binding sites contained the protected G residues in the loops of HP1 and HP2 (Fig. 1B and 1C). To provide further evidence for CsrA binding in this region, footprint experiments were conducted with a transcript containing residues 1–99. CsrA protected each of the G residues in binding sites 1 and 2 (BS1 and BS2) from RNase T1 cleavage, but did not protect any of the other G residues. CsrA also protected several nucleotides in BS1 and BS2 from cleavage by RNase T2 (single-stranded A preference) (Fig. 1A, 1B and 1D). The cleavage pattern of A residues in BS1 suggests that bound CsrA protects HP1 directly by binding to the single stranded loop, and perhaps indirectly by stabilizing the hairpin structure (Schubert et al., 2007). HP2 was maintained in the presence or absence of CsrA. Thus, the footprinting results confirm that CsrA binds to both predicted sites. Furthermore, the footprinting data indicate that bound CsrA does not bind to nor does it alter the RNA structure in the region surrounding the flhD ribosome binding site (Fig. 1C), suggesting that CsrA may not affect translation of flhD.
Gel mobility shift assays were performed to characterize the interaction of CsrA with the same 99-nt RNA used in the footprinting experiments. CsrA bound to this transcript as a diffuse band between 1 and 32 nM CsrA, with a distinct band becoming prominent at the higher concentrations (Fig. 2). Quantification of these data yielded an apparent Kd value of 21 nM CsrA. Deletion of the GGA motif from BS1 (ΔBS1) resulted in a 5-fold decrease in binding affinity, whereas deletion of the GGA from BS2 (ΔBS2) resulted in a 20-fold binding defect. Importantly, deletion of both GGA motifs virtually eliminated CsrA binding (Fig. 2). The specificity of CsrA-flhDC leader RNA interaction was investigated by performing competition experiments with specific (flhD and pgaA) and non-specific RNA competitors (Fig. 2). Unlabeled E. coli flhD and pgaA were effective competitors for CsrA-flhD RNA interaction, whereas only minimal competition was observed with RNA derived from pTZ19R vector sequences, demonstrating that CsrA binds specifically to flhDC leader RNA.
Fig. 2. Gel mobility shift analysis of CsrA-flhDC leader RNA interaction.

5’ end-labeled RNA was incubated with the concentration of CsrA indicated at the bottom of each lane. Positions of bound (B) and free (F) flhDC leader RNA are marked. Kd values of CsrA interaction with wild type (WT), ΔBS1, ΔBS2 and ΔBS1 ΔBS2 mutant transcripts are shown. The simple binding curve for these data is shown at the right. For the RNA competition assay, labeled flhDC leader RNA was incubated with CsrA ± 10- or 100-fold excess of specific (flhD and pgaA) or non-specific (pTZ19R) competitor RNA.
Loss of CsrA binding to the flhDC leader transcript reduces flhDC expression
A csrA deletion was shown to be viable in a strain containing a mutation in the glycogen biosynthetic operon (glg) (Timmermans and Van Melderen, 2009). We constructed a similar strain and found that it grew poorly and appeared to acquire suppressor mutations. For this reason, we used a well-characterized csrA mutant allele (csrA::kan) containing a transposon insertion following the 50th codon in the 61 aa coding sequence (Romeo et al. 1993).
As CsrA binds to two sites in flhDC leader RNA, we tested the effect of csrA on expression of a plasmid-borne flhDC'-'lacZ translational fusion. Expression peaked in late exponential phase growth and was reduced in the csrA mutant (Fig. 3A), consistent with our previous finding that CsrA activates flhDC expression (Wei et al. 2001). Expression of a chromosomally integrated flhD'-'lacZ translational fusion was also tested. Although expression of this single-copy flhD'-'lacZ fusion was 4- to 5-fold lower than the plasmid-borne flhDC'-'lacZ fusion (Fig. 3B), expression of the integrated flhD'-'lacZ fusion was reduced in the csrA mutant to a similar extent as was observed for the plasmid-borne flhDC'-'lacZ fusion, indicating that CsrA-dependent activation of flhDC expression is unaffected by the flhDC copy number. We next tested the effect of the ΔBS1 and/or ΔBS2 mutations on expression of chromosomally integrated flhD'-'lacZ translational fusions in wild type (WT) and csrA mutant strains. Expression in the WT strain was reduced when the fusion carried a mutation in either BS1 or BS2, while mutating both binding sites resulted in a further reduction in expression (Fig. 3C). Expression levels in the csrA mutant background were similar for the WT, BS1 and BS2 mutant fusions, while expression was reduced when both binding sites were mutated (Fig. 3D).
Fig. 3. CsrA-dependent regulation of flhDC expression.

Cells were grown in LB at 30°C. Representative growth curves are shown with a dashed line in each panel.γ-galactosidase values are averages of at least two independent experiments.
A. Effect of csrA on expression of a plasmid-borne flhDC'-'lacZ translational fusion. Symbols are: WT, black circles; csrA, red squares.
B. Effect of csrA on expression of a chromosomally integrated flhD'-'lacZ translational fusion. Symbols are: WT, black circles; csrA, red squares.
C. Effect of CsrA binding sites on expression of chromosomally integrated flhD'-'lacZ translational fusions in WT (csrA+) strains. Symbols are: WT, black circles; ΔBS1, red squares;ΔBS2, blue diamonds; ΔBS1 ΔBS2, grey triangles.
D. Effect of CsrA binding sites on expression of chromosomally integrated flhD'-'lacZ translational fusions in csrA mutant strains. Symbols are: WT, black circles; ΔBS1, red squares;ΔBS2, blue diamonds; ΔBS1 ΔBS2, grey triangles.
The results in figure 3D were somewhat surprising because we did not expect that mutating the CsrA binding sites would cause a further reduction in expression in the csrA mutant. However, the mutant csrA allele contains a transposon insertion following the 50th codon in the 61 aa coding sequence, resulting in a 62 aa fusion protein. As L2, L4, R6, R7, V40, V42, R44 and I47 are critical for RNA interaction (Mercante et al., 2006), it was possible that the fusion protein retained some RNA binding activity. Thus, the mutant fusion protein was purified, and its activity was assessed using a 16-mer RNA target containing a single high-affinity CsrA binding site (Dubey et al., 2005; Mercante et al., 2009). Gel mobility shift results confirmed that the fusion protein retained significant binding activity; the binding affinity of the fusion protein was reduced 8-fold compared to WT CsrA (Fig. 4). Perhaps the reduction in flhD'-'lacZ expression caused by mutating BS1 and BS2 in the csrA::kan genetic background is explained by the partial RNA binding activity of the fusion protein.
Fig. 4. Gel mobility shift analysis of WT and mutant CsrA protein.

5’ end-labeled RNA was incubated with the concentration of WT or mutant CsrA shown at the bottom of each lane. Positions of bound (B) and free (F) RNA are shown.
Bound CsrA protects flhDC leader RNA from RNase E-mediated cleavage in vitro
The positions of BS1 and BS2, combined with the observation that bound CsrA does not cause structural rearrangements of flhDC leader RNA, suggested that the mechanism of flhDC activation and mRNA stabilization was restricted to the 5' end of the message. As RNase E possesses a 5' end-dependent cleavage mechanism with a strong preference for 5' monophosphorylated RNAs (Mackie, 1998; Kime et al., 2010), we hypothesized that bound CsrA could sequester the 5' end of flhDC leader RNA and inhibit RNase E-mediated cleavage. To test this hypothesis, 5' monophosphorylated RNAs containing WT or ΔBS1 ΔBS2 leaders were incubated with purified RNase E in vitro. In the absence of CsrA, cleavage occurred primarily between nucleotides 100 and 120, although minor cuts were also observed within BS2 and near position 145 (Fig. 1B and 5A). Note that the different sized cleavage products between the WT and mutant transcripts reflect the GGA deletions in BS1 and BS2. The cleavage sites between positions 100 and 120 were at the same nucleotides for both transcripts; however, there were qualitative differences in the cleavage pattern. Bound CsrA protected WT RNA from cleavage in the 100–120 and BS2 regions, the latter of which served as an internal control for CsrA binding. Importantly, CsrA-dependent protection from RNase E cleavage was eliminated in the ΔBS1 ΔBS2 RNA (Fig. 5A).
Fig. 5. RNase E cleavage of WT and ΔBS1 ΔBS2 flhDC leader RNA.
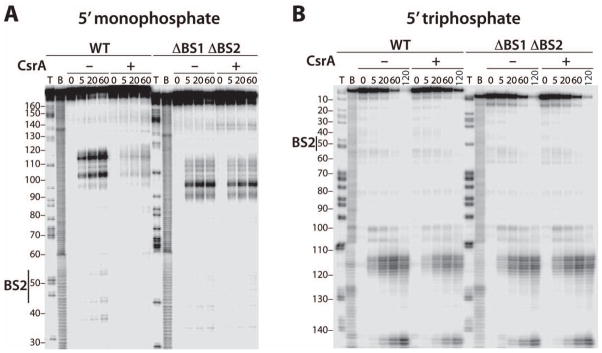
A. 5’ end-labeled WT or ΔBS1 ΔBS2 RNAs containing 5' monophosphate ends were treated with 8 nM RNase E for the indicated times in the absence (–) or presence (+) of 1 μM CsrA. Partial alkaline hydrolysis (B) and RNase T1 digestion (T) ladders are shown. Position of CsrA binding site 2 (BS2) is marked. Numbering is with respect to the start of transcription.
B. 3' end-labeled WT or ΔBS1 ΔBS2 RNAs containing 5' triphosphate ends were treated with 25 nM RNase E for the indicated times in the absence (–) or presence (+) of 1 μM CsrA. Partial alkaline hydrolysis (B) and RNase T1 digestion (T) ladders are shown. Position of CsrA binding site 2 (BS2) is marked. Numbering is with respect to the start of transcription.
Although RNase E exhibits a preference for 5' monophosphorylated RNA, cleavage can occur by an internal entry pathway in which the 5' end is bypassed (Mackie, 1998; Kime et al., 2010). Thus, we performed RNase E assays with flhDC leader transcripts that were labeled at the 3' end such that they retained 5' triphosphates and found that the cleavage rate was about 8-fold slower compared to 5' monophosphorylated RNA (data not shown). We also determined the effects of CsrA on RNase E cleavage of WT and ΔBS1 ΔBS2 RNAs containing 5' triphosphates. In this case CsrA-dependent protection of the WT transcript was greatly reduced relative to RNA containing a 5' monophosphate (Fig. 5B). As for the 5' monophosphorylated substrate, CsrA did not protect the ΔBS1 ΔBS2 transcript containing a 5' triphosphate. We conclude that bound CsrA protects the flhDC leader transcript by interfering with the 5' end-dependent RNase E cleavage pathway in vitro.
Bound CsrA stabilizes flhDC mRNA in vivo
As bound CsrA inhibits RNase E-mediated cleavage of the flhDC leader transcript in vitro, mRNA half-life experiments were conducted to examine the effect of CsrA on flhDC mRNA stability in vivo. Since we were unable to detect the flhDC transcript expressed from its native chromosomal locus by Northern blotting, experiments were performed with strains containing plasmids pCSB81 (WT flhDC operon) or pCSB83 (ΔBS1 ΔBS2 flhDC operon). Recall that the extent of CsrA-dependent activation of flhDC expression was unaffected by the flhDC copy number (Fig. 3). The influence of CsrA and the CsrA binding sites on the rate of flhDC mRNA decay was examined at 30°C, the same growth temperature used for the expression studies (Fig. 3). The half-life of the WT transcript was ~1 min in the WT strain and ~0.5 min in the csrA mutant strain. Importantly, the half-life of the ΔBS1 ΔBS2 flhDC transcript was ~0.5 min in both WT and csrA strains (Fig. 6). Recall that CsrA is unable to bind to this RNA (Fig. 2). We conclude that bound CsrA stabilizes the flhDC transcript twofold, which is similar to its threefold effect on expression of the flhD'-'lacZ fusion (Fig. 3).
Fig. 6. Northern blot analysis of flhDC mRNA half-life.
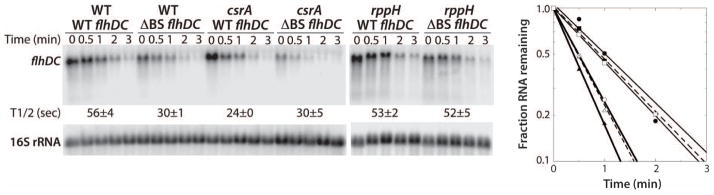
mRNA half-life experiments of wild type (WT) or ΔBS1 ΔBS2 mutant (ΔBS) flhDC mRNA were performed in wild type (WT), csrA or rppH strains. Cultures were grown at 30°C to late exponential phase prior to the addition of rifampicin. After 1 min, samples were harvested at the indicated times. mRNA half-lives (T1/2) are shown. 16S rRNA blots are shown as loading controls. The level of flhDC mRNA was normalized to the 16S rRNA level in each lane. Quantification of these data is shown at the right. Symbols are: WT flhDC, solid circles; ΔBS1 ΔBS2 flhDC, open circles; WT flhDC csrA, solid triangles; ΔBS1 ΔBS2 flhDC csrA, open triangles (dashed line); WT flhDC rppH, solid squares (dashed line); ΔBS1 ΔBS2 flhDC rppH, open squares.
As described above, RNase E preferentially acts on RNA containing a 5' monophosphate (Mackie, 1998). Since E. coli RppH is the 5' pyrophosphatase that converts 5' triphosphates to 5' monophosphates (Deana et al., 2008), we examined the effect of RppH on the stability of WT and ΔBS1 ΔBS2 flhDC transcripts. While the absence of RppH did not affect the stability of the WT transcript, the ΔBS1 ΔBS2 transcript was stabilized twofold in the absence of RppH (Fig. 6). We conclude that a 5' triphosphate can substitute for bound CsrA in stabilizing flhDC mRNA in vivo, which is consistent with bound CsrA interfering with the 5' end-dependent RNase E cleavage pathway.
As a test of this hypothesis, we compared expression levels of the WT and ΔBS1 ΔBS2 flhD'-'lacZ fusions in WT and rppH mutant strains. However, rather than the expected increase in expression, the absence of RppH caused a small decrease in expression of both fusions (data not shown). Because flhDC expression is activated and repressed by a variety of transcription factors (Shin et al., 1995; Soutourina et al., 1999; Lehnen et al., 2002; Sperandio et al., 2002; Smith and Hoover, 2009), while RppH affects the turnover of bulk mRNA, it is likely that the decrease in expression is due to indirect effects of RppH-deficiency on one or more of these transcription factors.
Deletion of the RNase E cleavage sites from the flhDC leader increases expression of the flhDC operon
Results from the in vitro RNase E cleavage and in vivo Northern analyses are consistent with bound CsrA protecting the flhDC leader transcript from cleavage between positions 100 and 120 (Figs. 5 and 6). Thus, primer extension mapping was used to determine if the leader transcript is cleaved in the same region in vivo. Each strain used in this analysis contained the pnp-200 and rnb-500 alleles, which encode thermolabile PNPase and RNase II, respectively. Use of this genetic background leads to stabilization of endonucleolytic cleavage intermediates after a shift to the non-permissive temperature by inactivating two exonucleases that play important roles in mRNA turnover (Yancey and Kushner, 1990; Arraiano et al., 2010). In addition, one of the strains contained the rne-1 allele, encoding thermolabile RNase E (Babitzke and Kushner, 1991). Several primer extension products were reduced or absent in the rne-1 strain following a shift to 44°C (Fig. 7). Five of the prominent bands (101, 113, 116, 142 and 145) correspond to RNase E cleavage sites that were observed in vitro. Moreover, CsrA protected positions 101, 113 and 116 from RNase E cleavage in vitro (Fig. 5). We conclude that RNase E cleaves flhDC leader RNA in vivo at some of the sites that CsrA protected from RNase E cleavage in vitro. We also examined the effect of CsrA on cleavage at these sites in vivo; however, the accumulation of cleavage intermediates was unaffected by CsrA deficiency or overexpression (data not shown).
Fig. 7. Primer extension analysis of RNase E cleavage sites in flhDC leader RNA.
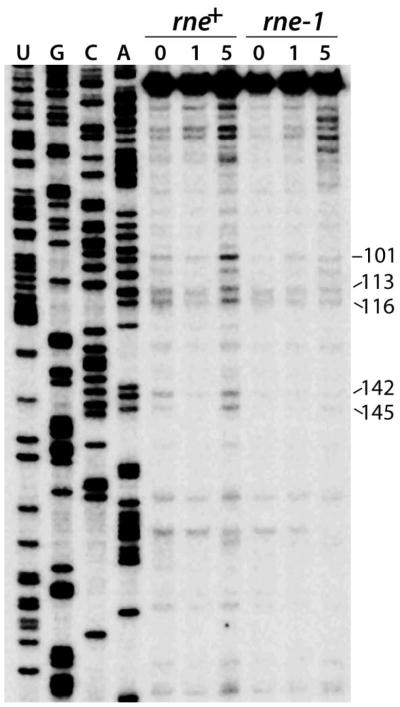
RNA was isolated from pnp rnb and pnp rnb rne mutant strains containing pCSB81 (flhDC) at 0, 1 or 5 min after a shift to the non-permissive temperature (44°C). A primer specific for lacZ was annealed to total cellular RNA and extended by reverse transcriptase. Positions of flhDC leader RNA 5' ends are marked on the right. DNA sequencing ladder is shown.
The data presented above provided compelling evidence that bound CsrA activates flhDC expression by protecting the mRNA from the 5' end-dependent RNase E cleavage pathway. As an additional test of this model, we deleted residues +97 through +122 from the flhDC leader, as the majority of the in vitro RNase E cleavage sites are within this region (Fig. 1B and 5). Deletion of this region eliminated RNase E cleavage between BS2 and positions 142 and 145 in vitro (Fig. 8A). We then compared expression of an integrated Δ97-122 (ΔE) flhD'-'lacZ fusion to WT and ΔBS1 ΔBS2 flhD'-'lacZ fusions. Consistent with CsrA-mediated protection of the region between 100 and 120 from RNase E cleavage in vitro, the ΔE fusion resulted in a substantial increase in expression (Fig. 8B). This result is consistent with a model in which CsrA-dependent protection from RNase E cleavage leads to increased expression of the flhDC operon. We next compared expression of the WT and ΔE fusions in WT and csrA mutant strains. Introduction of the csrA allele reduced expression of the ΔE fusion, but not to the level of the WT fusion (Fig. 8C). These latter results suggest that CsrA is still capable of protecting the flhDC transcript from cleavage at RNase E sites that remain in the ΔE fusion and/or from an alternative mRNA decay pathway.
Fig. 8. Effects of deleting RNase E cleavage sites on flhDC expression.

A. 5’ end-labeled WT orΔE RNAs were treated with 4 nM RNase E for 0 (lane 1), 5 (lane 2), 20 (lane 3) or 60 (lane 4) min in the absence (–) or presence (+) of 1 μM CsrA. Partial alkaline hydrolysis (B) and RNase T1 digestion (T) ladders, and control lanes without RNase treatment
(C), are shown. Positions of CsrA binding site 2 (BS2), as well as the flhD Shine-Dalgarno (SD) sequence and start codon are marked.
B. WT, ΔBS1 ΔBS2 and ΔE chromosomally integrated flhD'-'lacZ translational fusions in WT backgrounds were grown in LB at 30°C. Each experiment was performed at least twice and γ-galactosidase values from a representative experiment are shown. Symbols are: WT fusion, solid circles; ΔE fusion, solid triangles; ΔBS1 ΔBS2, solid squares. A representative growth curve is shown with a dashed line.
C. WT and ΔE chromosomally integrated flhD'-'lacZ translational fusions in WT and csrA mutant backgrounds were grown in LB at 30°C. Each experiment was performed at least three times and γ-galactosidase values from a representative experiment are shown. Symbols are: WT fusion, solid circles; WT fusion csrA, open circles; ΔE fusion, solid triangles; ΔE fusion csrA, open triangles. A representative growth curve is shown with a dashed line.
Our primer extension mapping experiments identified RNase E-dependent degradation intermediates, some of which arose from cleavage at sites that were protected from RNase E cleavage by bound CsrA in vitro (Figs. 5 and 7). To obtain direct evidence of CsrA-dependent protection of the flhDC leader transcript from RNase E cleavage in vivo, we carried out quantitative primer extension assays using a lacZ-specific primer and RNA purified from strains with integrated WT, ΔBS1 ΔBS2 (ΔBS), ΔE, or ΔBS ΔE flhD'-'lacZ fusions. Each fusion was tested in WT and rne-1 genetic backgrounds before and after a shift to the non-permissive temperature (44°C). Thus, the level of a full-length primer extension product would reflect the effects of bound CsrA and/or RNase E on flhDC mRNA turnover. The level of a primer extension product corresponding to tmRNA was used as an internal loading control for each sample; the level of tmRNA was not affected by the ΔBS, ΔE, or rne-1 mutations.
The shift to 44°C resulted in rapid degradation of the WT transcript in the WT (rne+) background but not in the rne-1 strain, which encoded thermolabile RNase E (Fig. 9A, first four lanes). Rapid degradation of flhDC mRNA at 44°C is consistent with a severe motility defect at this temperature (data not shown). The stability of the ΔBS transcript was twofold lower than the WT transcript in the rne+ genetic background at both temperatures (Fig. 9A). Importantly, the twofold reduction in transcript stability caused by loss of CsrA binding was essentially identical to what was previously observed in the Northern studies (Fig. 6). Of particular interest, the stability of the ΔBS transcript in the rne-1 strain at 44°C was similar to its stability in the rne+ strain at 30°C, indicating that destabilization of the flhDC transcript caused by the loss of CsrA binding was overcome when RNase E was inactivated (Fig. 9A). Compared to the WT and ΔBS transcripts, deletion of the RNase E sites from the flhDC leader (ΔE) led to considerable stabilization of the transcript in the rne+ background at 44°C (Fig. 9A), confirming that this region is normally cleaved by RNase E in vivo. Decay of the ΔBS ΔE transcript was intermediate between the ΔBS and ΔE transcripts. This latter finding suggests that bound CsrA protects the flhDC transcript from cleavage at sites in addition to those removed by the ΔE mutation. From the results of these experiments we conclude that bound CsrA protects the flhDC leader transcript from RNase E cleavage in vitro and in vivo.
Fig. 9. Quantitative primer extension analysis of WT, ΔBS, ΔE and ΔBS ΔE transcript decay.

RNA was isolated from WT and rne mutant strains containing chromosomally integrated flhD'-'lacZ fusions (A) or truncated flhD-tR2 constructs (B). Cells were grown at 30°C and harvested before or after a shift to the non-permissive temperature (44°C).
A. Primers specific for lacZ mRNA and tmRNA were annealed simultaneously to total cellular RNA and extended by reverse transcriptase. Levels of full-length flhDC transcripts were normalized to tmRNA levels. The level of RNA for the WT transcript in the WT (rne+) genetic background at 30°C was set to 100%. Normalized RNA levels are shown at the bottom of each lane. Control reactions with only the lacZ or tmRNA primers are marked.
B. Primers specific for flhDC leader and tmRNA were annealed simultaneously to total cellular RNA and extended by reverse transcriptase. Levels of full-length flhDC transcripts were normalized to tmRNA levels. The level of RNA for the WT transcript in the WT (rne+) genetic background at 30°C was set to 100%. Normalized RNA levels are shown at the bottom of each lane. Control reactions with only the flhDC leader or tmRNA primers are marked. Since the flhDC leader primer hybridizes upstream of the ΔE deletion, the length of the primer extension products for WT and ΔE, as well as for ΔBS and ΔBS ΔE, are the same.
It is well established that increasing or decreasing translation can lead to mRNA stabilization or destabilization, respectively (e.g. Babitzke and Romeo, 2007; Arraiano et al., 2010; Richards et al., 2012). Although the CsrA binding sites are about 190 (BS1) and 150 (BS2) nt upstream of the flhD translation initiation codon, while bound CsrA did not affect the flhDC leader RNA structure in the vicinity of the flhD SD sequence (Fig. 1), we had not ruled out the possibility that CsrA-dependent protection of the flhDC transcript is an indirect effect of CsrA-mediated translational activation. To test this possibility we performed quantitative primer extension studies using a flhDC leader-specific primer and RNA purified from strains with integrated WT, ΔBS, ΔE and ΔBS ΔE constructs in which the flhDC leader sequence downstream from position 146 was replaced with the strong λtR2 intrinsic terminator (Fig. 1B and 9B). Since none of these transcripts can be translated, any observed effect on RNA stability would be independent of translation. Decay of the WT, ΔBS and ΔE transcripts was similar to those observed for the corresponding translational fusion transcripts (Fig. 9A and 9B). Thus, we conclude that CsrA-dependent protection of flhDC mRNA from RNase E cleavage is independent of translation. A notable difference in the decay of these transcripts was observed after the shift to 44°C; stabilization due to RNase E deficiency (rne-1) was less pronounced than for the corresponding fusion transcripts. In contrast to the other three transcripts, decay of the ΔBS ΔE transcript was much more rapid (Fig. 9B), indicating that this transcript can be degraded by an alternative decay pathway (see Discussion).
Discussion
CsrA is a widely distributed global regulatory protein that binds to several hundred different E. coli transcripts (Edwards et al., 2011). A variety of related CsrA-mediated translation repression mechanisms have been identified in which CsrA binds near the translation initiation region and/or SD sequence of target transcripts, thereby blocking 30S ribosomal subunit binding (Babitzke and Romeo, 2007). Despite evidence for activation of several genes in E. coli (Sabnis et al., 1995; Wei et al., 2001), until now a CsrA-mediated activation mechanism had not been identified. We found that CsrA activates flhDC operon expression by binding to two sites in the leader transcript that are separated by 31 nt (Fig. 1–3). The level of activation is similar in magnitude to the level of CsrA-dependent stabilization of the flhDC transcript (Fig. 3, 6 and 9). The finding that the mutant CsrA fusion protein encoded by the csrA::kan allele retained significant RNA binding activity (Fig. 4) suggests that the reported level of CsrA-mediated regulation for all CsrA-controlled genes in E. coli is underrepresented.
CsrA binding to the extreme 5’ end of flhDC mRNA led to the hypothesis that sequestration of the 5' end by bound CsrA would inhibit 5' end-dependent cleavage by RNase E. Computer modeling and in vitro structure mapping revealed five hairpins (HP1-HP5) that likely form in the leader region of flhDC RNA (Fig. 1). The GGA motifs of BS1 and BS2 are located in the loops of HP1 and HP2, a common sequence arrangement of CsrA binding sites (Liu et al., 1997; Weilbacher et al., 2003; Dubey et al., 2005; Babitzke and Romeo, 2007). The predominant RNase E cleavage sites between positions 100 and 120 are located in an AU-rich region just downstream from HP2 (Fig. 5), a sequence arrangement that is similar to RNase E recognition sites found in other mRNA substrates (Carpousis, 2007; Kime et al., 2008). Deletion of the prominent RNase E cleavage sites between +97 to +122 (ΔE) resulted in increased flhD'-'lacZ expression (Fig. 8) and mRNA stabilization (Fig. 9). Furthermore, three of the flhDC decay intermediates that were identified in vivo by primer extension correspond to RNase E cleavage sites that CsrA protected from cleavage in vitro (Fig. 5 and 7). In conjunction with the finding that bound CsrA stabilizes the flhDC transcript (Fig. 6 and 9), we conclude that CsrA activates flhDC expression by protecting flhDC mRNA from endonucleolytic cleavage by RNase E.
Degradation of several mRNAs depends on a rate-limiting cleavage event by RNase E, resulting in a new 3' end that is degraded by 3' to 5' exonucleases (reviewed in Carpousis, 2007; Arraiano et al., 2010). RNase E cleavage also results in a new 5' monophosphorylated end that serves as a preferred RNase E substrate for cleavage further downstream (Deana et al., 2008). Thus sequential RNase E-mediated cleavage events, followed by exonucleolytic digestion of the resulting fragments from the 3' end, is thought to be a common mechanism for mRNA turnover in E. coli (Carpousis, 2007; Arraiano et al., 2010). RNase E cleaved several residues between positions 100 and 120 of the flhDC leader transcript that could serve as a rate-limiting cleavage event in vivo. RppH is responsible for converting the 5' terminal triphosphate to a monophosphate in vivo (Deana et al., 2008). Because RNase E has a preference for 5' monophosphate ends, the findings that a 5' triphosphate can compensate for bound CsrA in stabilizing the flhDC transcript in vitro (Fig. 5B) and in vivo (Fig. 6), indicate that bound CsrA protects the flhDC transcript from the 5' end-dependent RNase E cleavage pathway. In this mechanism, CsrA binding at BS1 would sequester the 5' end of the mRNA, thereby inhibiting RNase E action. CsrA bound at BS2 probably increases the affinity of CsrA interaction with the weaker BS1 due to its ability to bridge two RNA binding sites (Mercante et al., 2009). Our studies further imply that CsrA-mediated stabilization of the flhDC transcript leads to increased FlhD4C2 synthesis, which, in turn, activates flagellum synthesis and cell motility.
A recently published paper identified a 95 nt sRNA (McaS). This sRNA is expressed during late exponential and early stationary phase growth (Thomason et al., 2012). McaS is capable of activating flhDC expression ~1.4-fold by hybridizing to two sites in the flhDC leader transcript (positions 113 to 122 and 138 to 147, Fig. 1B). It was suggested that McaS might alter a predicted long-range RNA structure that sequesters the SD sequence, thereby relieving translational repression (Thomason et al., 2012). However, our structure mapping results are incompatible with this long-range structure and instead support the formation of five RNA hairpins (Fig. 1). In light of our data presented here, we offer an alternative hypothesis for the mechanism of McaS action. The upstream McaS hybridization site (McaS BS1) partially overlaps the RNase E cleavage sites between positions 100 and 120, while the downstream McaS hybridization site (McaS BS2) overlaps the RNase E cleavage sites at positions 142 and 145 (Fig. 1B). Thus the mechanism of McaS-mediated activation may involve protection of the flhDC leader from RNase E cleavage. Since our primer extension studies were performed with cultures grown to late exponential phase, this mechanism may explain the rapid decay observed for the ΔBS ΔE transcript (Fig. 9B). Deletion of the RNase E cleavage sites between positions 100 and 120 eliminated McaS BS1, while the engineered tR2 terminator sequesters the 3' end of McaS BS2. Thus, McaS may not interact with this transcript. Perhaps the lack of CsrA and McaS binding to this ΔBS ΔE transcript results in rapid RNA turnover. Note that all of the other transcripts described in figure 9 contained the CsrA binding sites and/or at least one of the McaS binding targets. While our results demonstrate that CsrA protects the flhDC transcript from the 5' end-dependent RNase E cleavage pathway, future studies will be required to identify the mechanism of McaS action and any potential connection with CsrA-dependent activation of flhDC expression.
Experimental procedures
Plasmids
The cloning vector pBR322 (Bolivar et al., 1977), the CRIM-based translational (pLFT) and transcriptional (pLFX) fusion vectors (Edwards et al., 2011), and pFDCZ6 containing a WT flhDC'-'lacZ translational fusion (Wei et al., 2001) have been described. Deletions of the GGA motifs from CsrA binding sites BS1 [G7, G8, A9 (ΔBS1)] and/or BS2 [G50, G51, A52 (ΔBS2)] were introduced into the flhDC leader in pFDCZ6 using the QuikChange protocol (Stratagene), resulting in plasmids, pCSB74 (ΔBS1), pCSB72 (ΔBS2) and pCSB75 (ΔBS1 ΔBS2). flhD'-'lacZ translational fusions were generated by PCR amplification of flhDC sequences extending from − 195 to +234 relative to the start of transcription using pFDCZ6, pCSB74, pCSB72 and pCSB75 as templates. The PCR products were digested with PstI and EcoRI and cloned into the same sites of pLFT. The resulting plasmids, pCSB76 (WT flhD'-'lacZ), pCSB77 (ΔBS1 flhD'-'lacZ), pCSB78 (ΔBS2 flhD'-'lacZ) and pCSB79 (ΔBS1 ΔBS2 flhD'-'lacZ), have the 12th codon of flhD fused in frame to the 10th codon of lacZ. Deletion of the RNase E cleavage sites between +97 and +122 (ΔE) was accomplished by the QuikChange protocol using pCSB76 and pCSB79 as DNA templates, resulting in plasmids pAY144 (ΔE flhD'-'lacZ) and pYH203 (ΔBS1 ΔBS2 ΔE flhD'-'lacZ), respectively. Plasmid pCSB81 contains the entire flhDC operon cloned into the BamHI and EcoRI sites of pBR322. Deletion of the GGA motifs from CsrA binding sites BS1 and BS2 were introduced into the flhDC leader in pCSB81 using the QuikChange protocol, resulting in plasmid pCSB83.
Plasmids with the flhDC promoter and a truncated leader region (−195 to +146 relative to the start of transcription) in which sequences downstream from +146 were replaced by the λtR2 intrinsic terminator were constructed as follows. PCR amplification of WT and mutant flhDC leader fragments followed by λtR2 was accomplished using plasmids pCSB76, pCSB79, pAY144 and pYH203 as DNA templates. The downstream primer in these reactions contained the λtR2 sequence. The resulting PCR products were digested with EcoRI and PstI and cloned into the same sites of pLFX, resulting in plasmids pYH207 (WT flhDldr-tR2), pYH208 (ΔBS1 ΔBS2 flhDldr-tR2), pYH209 (ΔE flhDldr-tR2) and pYH210 (ΔBS1 ΔBS2 ΔE flhDldr-tR2).
The amino-terminal hexahistidine-tagged RNase E expression vector p1VR122a was created by PCR amplification of the rne gene from E. coli strain MG1655. The resulting PCR product was digested with BamHI and PstI and cloned into the same sites of pCOLADuet-1 (Novagen).
Bacterial strains
E. coli strains used in this study are listed in Table 1. Details of the strain constructions are described in Supporting Information.
Table 1.
E. coli strains used in this study
| Strain | Descriptiona | Source |
| C43(DE3) | F− ompT hsdSB (rB- mB-) gal dcm (DE3) | Lucigen |
| CF7789 | F− λ−ΔlacI-lacZ (MluI) | Wei et al, 2001 |
| MG1655 | Prototrophic F- λ- | M. Cashel |
| PLB441 | MG1655/flhDldr::kan | This study |
| PLB442 | CF7789 uvrY::cam flhDldr::kan | This study |
| PLB931 | CF7789/pFDCZ6 (flhDC'-'lacZ) Apr | This study |
| PLB932 | CF7789/csrA::kan pFDCZ6 (flhDC'-'lacZ) Apr | This study |
| PLB966 | CF7789/uvrY::cam csrA::kan | This study |
| PLB1258 | CF7789/flhD'-'lacZ Apr | This study |
| PLB1259 | CF7789/ΔBS1 flhD'-'lacZ Apr | This study |
| PLB1260 | CF7789/ΔBS2 flhD'-'lacZ Apr | This study |
| PLB1262 | CF7789/ΔBS1 ΔBS2 flhD'-'lacZ Apr | This study |
| PLB1263 | CF7789/csrA::kan flhD'-'lacZ Apr | This study |
| PLB1264 | CF7789/csrA::kan ΔBS1 flhD'-'lacZ Apr | This study |
| PLB1265 | CF7789/csrA::kan ΔBS2 flhD'-'lacZ Apr | This study |
| PLB1266 | CF7789/csrA::kan ΔBS1 ΔBS2 flhD'-'lacZ Apr | This study |
| PLB1601 | CF7789/uvrY::cam rppH::kan | This study |
| PLB1618 | CF7789/uvrY::cam csrA::kan pCSB81 Apr | This study |
| PLB1619 | CF7789/uvrY::cam rppH::kan pCSB81 Apr | This study |
| PLB1620 | CF7789/uvrY::cam pCSB81 Apr | This study |
| PLB1625 | CF7789/uvrY::cam pCSB83 Apr | This study |
| PLB1626 | CF7789/uvrY::cam rppH::kan pCSB83 Apr | This study |
| PLB1627 | CF7789/uvrY::cam csrA::kan pCSB83 Apr | This study |
| PLB1646 | SK6632/pCSB81 (flhDC) Apr | This study |
| PLB1647 | SK6640/pCSB81 (flhDC) Apr | This study |
| PLB1759 | CF7789/ΔE flhD'-'lacZ Apr | This study |
| PLB1760 | CF7789/csrA::kan ΔE flhD'-'lacZ Apr | This study |
| PLB1762 | CF7789/rppH::kan | This study |
| PLB1768 | CF7789/uvrY::cam flhD'-'lacZ Apr | This study |
| PLB1770 | CF7789/uvrY::cam ΔBS1 ΔBS2 flhD'-'lacZ Apr | This study |
| PLB1771 | CF7789/uvrY::cam ΔE flhD'-'lacZ Apr | This study |
| PLB1773 | CF7789/uvrY::cam rppH::kan flhD'-'lacZ Apr | This study |
| PLB1774 | CF7789/uvrY::cam rppH::kan ΔBS1 ΔBS2 flhD'-'lacZ Apr | This study |
| PLB1775 | CF7789/uvrY::cam rppH::kan ΔE flhD'-'lacZ Apr | This study |
| PLB1776 | CF7789/rppH::kan flhD'-'lacZ Apr | This study |
| PLB1777 | CF7789/rppH::kan ΔBS1 ΔBS2 flhD'-'lacZ Apr | This study |
| PLB1778 | CF7789/rppH::kan ΔE flhD'-'lacZ Apr | This study |
| PLB1812 | CF7789/uvrY::cam pyrC::Tn10 flhD'-'lacZ Apr | This study |
| PLB1813 | CF7789/uvrY::cam pyrC::Tn10 ΔBS1 ΔBS2 flhD'-'lacZ Apr | This study |
| PLB1814 | CF7789/uvrY::cam pyrC::Tn10 ΔE flhD'-'lacZ Apr | This study |
| PLB1816 | CF7789/uvrY::cam rne-1 flhD'-'lacZ Apr | This study |
| PLB1817 | CF7789/uvrY::cam rne-1 ΔBS1 ΔBS2 flhD'-'lacZ Apr | This study |
| PLB1818 | CF7789/uvrY::cam rne-1 ΔE flhD'-'lacZ Apr | This study |
| PLB1832 | CF7789/uvrY::cam ΔBS1 ΔBS2 ΔE flhD'-'lacZ Apr | This study |
| PLB1845 | CF7789/uvrY::cam pyrC::Tn10 ΔBS1 ΔBS2 ΔE flhD'-'lacZ Apr | This study |
| PLB1846 | CF7789/uvrY::cam rne-1 ΔBS1 ΔBS2 ΔE flhD'-'lacZ Apr | This study |
| PLB1852 | CF7789/uvrY::cam flhDldr::kan flhD- tR2Apr | This Study |
| PLB1853 | CF7789/uvrY::cam flhDldr::kan ΔBS1 ΔBS2 flhD- tR2 Apr | This Study |
| PLB1854 | CF7789/uvrY::cam flhDldr::kan ΔE flhD- tR2 Apr | This Study |
| PLB1855 | CF7789/uvrY::cam flhDldr::kan ΔBS1 ΔBS2 ΔE flhD- tR2 Apr | This Study |
| PLB1856 | CF7789/uvrY::cam pyrC::Tn10 flhDldr::kan flhD- tR2 Apr | This Study |
| PLB1857 | CF7789/uvrY::cam pyrC::Tn10 flhDldr::kan ΔBS1 ΔBS2 flhD-tR2 Apr | This Study |
| PLB1858 | CF7789/uvrY::cam pyrC::Tn10 flhDldr::kan ΔE flhD- tR0 Apr | This Study |
| PLB1859 | CF7789/uvrY::cam pyrC::Tn10 flhDldr::kan ΔBS1 ΔBS2 ΔE flhD-tR2 Apr | This Study |
| PLB1860 | CF7789/uvrY::cam rne-1 flhDldr::kan flhD- tR2 Apr | This Study |
| PLB1861 | CF7789/uvrY::cam rne-1 flhDldr::kan ΔBS1 ΔBS2 flhD- tR2 Apr | This Study |
| PLB1862 | CF7789/uvrY::cam rne-1 flhDldr::kan ΔE flhD- tR2 Apr | This Study |
| PLB1863 | CF7789/uvrY::rne-1 flhDldr::kan ΔBS1 ΔBS2 Δ E flhD- tR2 Apr | This Study |
| SK4390 | thyA715 rph-1 rppH::kan | Mohanty and Kushner, 2010 |
| SK5664 | pyrC::Tn10 Tcr | Arraiano et al, 1988 |
| SK5665 | rne-1 | Babitzke and Kushner, 1991 |
| SK6632 | thyA715 rnb-500 pnp-200 Cmr | Yancey and Kushner, 1990 |
| SK6640 | thyA715 rnb-500 pnp-200 rne-1 Cmr | Yancey and Kushner, 1990 |
| TRCF7789 | CF7789/csrA::kan | Wei et al, 2001 |
| UYCF7789 | CF7789/uvrY::cam | Suzuki et al, 2002 |
All flhD'-'lacZ fusions and truncated promoter-leader regions were integrated into the λ att site via the CRIM system (Haldimann and Wanner, 2001). flhDldr::kan indicates that the chromosomal flhDC leader (−238 to +43 with respect to the start of flhD translation) was deleted. ΔBS1 corresponds to a deletion of G7, G8 and A9 from CsrA binding site 1, while ΔBS2 corresponds to a deletion of G50, G51 and A52 from CsrA binding site 2. The ΔBS1 BS2 fusion has all six of these nucleotides deleted. ΔE corresponds to a deletion of several RNase E cleavage sites between +97 to +122 of the flhDC leader. tR2 is an intrinsic terminator from bacteriophage λ.
β-Galactosidase assays
Bacterial cultures were grown in LB supplemented with 100 μg/ml ampicillin at 30°C. Culture samples (4 ml) were harvested at various times during growth, washed with 10 mM Tris-HCl (pH 7.5) and frozen as cell pellets at −20°C. Cell extracts were prepared by suspending frozen cell pellets in 0.5 ml of BugBuster (Novagen). Following 30 min of incubation at 37°C, 0.3 ml of Z buffer containing 0.2 mg/ml lysozyme was added, incubation was then continued for 30 min at 37°C. After removal of cell debris, protein concentrations were determined by the Bio-Rad protein assay. β-galactosidase assays were performed using the cell extracts as described previously (Baker et al., 2007).
RNA structure mapping and footprint assays
RNA was synthesized using the Ambion T7 MEGAscript kit and PCR-generated templates. Gel-purified RNA was dephosphorylated with calf intestinal alkaline phosphatase and then 5’ end-labeled using T4 polynucleotide kinase (NEB) and [γ 32P]ATP (7000 Ci/mmol). RNA suspended in Tris-EDTA buffer (TE) was renatured by heating to 85°C and cooling to room temperature. His-tagged E. coli CsrA (CsrA-H6) was purified as described (Mercante et al., 2006). Binding reactions (10 μl) contained 10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 100 mM KCl, 32.5 ng yeast RNA, 7.5% glycerol, 20 mM dithiothreitol (DTT), 4 U RNase inhibitor (Promega), 50 nM flhDC RNA, 200 μg/ml acetylated BSA, and various amounts of CsrA-H6. Reaction mixtures were incubated for 30 min at 37°C to allow CsrA–RNA complex formation. RNase T1 (0.025 U) or RNase T2 (0.03 U) was then added and incubation was continued for 15 min at 37°C. Reactions were terminated by adding 10 μl of stop buffer (95% formamide, 0.025% SDS, 20 mM EDTA, 0.025% bromophenol blue, 0.025% xylene cyanol, and 760 μg/ml yeast RNA) and placed on ice. Base hydrolysis and RNase T1 digestion ladders were prepared as described (Bevilacqua and Bevilacqua, 1998). Samples were fractionated through 6% sequencing gels. Radioactive bands were visualized using a phosphorimager (Molecular Dynamics).
Gel mobility shift assay
Quantitative gel mobility shift assays of flhDC leader RNA followed a published procedure (Baker et al., 2007). Binding reactions (10 μl) containing 0.1 nM 5’ end-labeled flhDC leader RNA, various amounts of CsrA-H6, and 0.1 mg/ml xylene cyanol, were otherwise identical to the footprint assay. Competition assays also contained unlabeled RNA competitors. Following incubation at 37°C for 30 min, samples were fractionated on native 15% polyacrylamide gels. Free and bound RNA species were visualized with a phosphorimager and quantified using ImageQuant 5.2 software (Molecular Dynamics), and the apparent equilibrium binding constants (Kd) of CsrA–RNA interaction was calculated as described (Yakhnin and Babitzke, 2010). Mutant CsrA fusion protein encoded by the csrA::kan allele was purified as described for CsrA- H6 (Mercante et al., 2006). Binding reactions contained 5’ end-labeled 16-mer RNA containing a single high-affinity CsrA binding site (Dubey et al., 2005; Mercante et al., 2009) and either WT or mutant CsrA.
RNase E assay
Hexahistidine-tagged RNase E was purified by immobilized metal affinity chromatography using the following procedure. E. coli C43(DE3) (Lucigen) carrying p1VR122a was grown at 37° C in 1 L of Luria broth containing 50 μg/ml kanamycin to an OD600 of 0.7, at which time 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) was added and the culture was incubated for an additional 3 h at 37°C. Bacteria were harvested by centrifugation and suspended in 10 ml of buffer A (20 mM Tris-HCl, pH 7.9, 6 M guanidine-HCl, and 500 mM NaCl) containing 10 mM imidazole. Cells were lysed via sonication on ice, and unbroken cells were removed by centrifugation. Lysates were applied to a 1-ml HisTrap column (GE Healthcare) previously equilibrated with buffer A containing 5 mM imidazole, washed with 10 ml buffer A containing 40 mM imidazole, and RNase E was eluted with a 30-ml linear imidazole gradient (30 to 500 mM) in buffer A. The elution peaks were established by SDS-PAGE. Purified, denatured RNase E was subjected to a series of refolding dialysis steps against buffer B (50 mM Tris-HCl, pH 7.9, 200 mM KCl, 10 mM MgCl2, 1 mM DTT, and 20% [vol/vol] glycerol) containing 4 M guanidine-HCl, 2 M guanidine-HCl, and no denaturant. Insoluble protein was removed via ultracentrifugation for 15 min (100,000 x g) after each dialysis step. The resulting material was stored in buffer B containing 50% glycerol (vol/vol) at –20° C.
RNase E assays were performed in vitro by modifying published procedures (Perwez and Kushner, 2006). WT, ΔBS1 ΔBS2 or ΔE flhDC leader RNA (+1 to +210) was 5’end-labeled with γ [32P]ATP and polynucleotide kinase. WT or ΔBS1 ΔBS2 flhDC leader RNA (+1 to +171) was 3’end-labeled using T4 RNA ligase (New England Biolabs) and [32P]pCp (Perkin Elmer) using previously published reaction conditions (England and Uhlenbeck, 1978). RNAs were renatured in TE buffer by heating for 3 min at 85°C and cooling to room temperature. RNase E assays (45 μl) contained 25 or 40 nM RNA, 25 mM Tris-Cl, pH 7.9, 5 mM MgCl2, 60 mM KCl, 100 mM NH4Cl, 15 mM DTT, 7.5% glycerol, 1.5 mg/ml acetylated BSA, 4 U RNasin, RNase E (various amounts), ± 1 μM CsrA. Reaction mixtures were incubated for 30 min at 37°C to allow CsrA-RNA complex formation before adding RNase E. Incubation was continued at 37°C and 10 μl aliquots were removed at various times and quenched with 10 μl of footprint stop buffer, frozen in dry ice, and stored at −80°C. RNA samples were fractionated through 6% sequencing gels. Radioactive bands were visualized using a phosphorimager and quantified with ImageQuant 5.2.
Primer extension analysis to identify RNase E cleavage sites
Strains SK6632 (rnb-500 pnp-200) and SK6640 (rnb-500 pnp-200 rne-1) both containing plasmid pCSB81 (flhDC operon) were grown in LB with 100 μg/ml ampicillin at 30°C to late exponential phase. Cultures were then shifted to 44°C for 0, 1 or 5 min, at which time RNAprotect Bacteria reagent (Qiagen) was added to culture samples. Total RNA was isolated using the RNeasy Mini kit (Qiagen). The resulting RNA (15 μg) was annealed to 50 pmol of a 5’ end-labeled oligo complementary to +256 to +277 of flhDC in TE buffer by heating to 70°C for 5 min and immediately placing on ice. Reverse transcription reactions (20 μl) were performed using the Verso cDNA synthesis kit (Thermo Scientific) according to the manufacturer's instructions. Reactions were incubated for 30 min at 42oC and stopped with an equal volume of loading buffer (95% formamide, 18 mM EDTA, 0.025% SDS, 0.025% xylene cyanol, and 0.025% bromophenol blue). Samples were fractionated through a taurine-buffered 6% sequencing gel. Sequencing reactions were performed using the same end-labeled primer and pCSB81 as template. Radioactive bands were visualized using a phosphorimager.
Quantitative analysis of flhDC transcript stability by primer extension
Strains containing chromosomally-integrated flhD’-‘lacZ translational fusions or flhD leaders truncated at position +146 followed by the strong λ tR2 intrinsic terminator (flhD-tR2) were grown in 20 ml of LB in a shaking water bath at 30°C. Eight ml aliquots were harvested at late exponential phase growth and the remaining cultures were shifted to 44°C for 15 min, at which time additional 8 ml aliquots were harvested. Culture samples were mixed with 6 ml of frozen killing buffer (12 mM Tris-HCl, pH 7.2, 6 mM MgCl2, 30 mM sodium azide, 15% ethanol, and 600 μg/ml chloramphenicol) and placed on ice. Following centrifugation, cell pellets were suspended in 1 ml of a 2:1 mixture of RNA Protect bacterial reagent (Qiagen):TE buffer, placed on ice for 10 min, and centrifuged. Cell pellets were frozen at −80 °C. Cells containing flhD’-‘lacZ fusions were lysed and total RNA was isolated using the RNeasy bacterial protocol (Qiagen). Cells containing truncated flhD-tR2 leaders were lysed as above and the short transcripts were isolated using the miRNeasy kit (Qiagen). Oligonucleotide primers were 5′ end-labeled with [γ-32P]ATP. A primer complementary to lacZ was used for the flhD'-'lacZ fusions, while a primer complementary to nt 69–91 of the flhD leader was used for the truncated flhD-tR2 leader transcripts. In each case, primer extension of tmRNA using a primer complementary to nt 95–115 of mature tmRNA was used as a loading control. Two μg of RNA was annealed to 0.3 pmol of the labeled lacZ or flhD leader primer. As tmRNA is considerably more abundant than flhD'-'lacZ or flhD-tR2 leader transcripts, a mixture of 0.15 pmol labeled and 3 pmol of unlabeled tmRNA primer was used to obtain primer extension signals that were similar in intensity to the flhD'-'lacZ or flhD-tR2 leader signals. Annealing was performed in TE buffer by heating to 80°C for 3 min, followed by slow cooling to room temperature. Reverse transcription reactions (4 μl) contained the hybridization mixture and 1x Superscript III reverse transcriptase buffer, 0.5 mM dNTP, 0.8 U RNasin, 20 μg/ml acetylated BSA, 1 mM DTT, and 8 U of Superscript III reverse transcriptase (Invitrogen). Reactions were incubated for 30 min at 42°C and then quenched by adding 2.5 μl of stop buffer (20 mM EDTA, 95% formamide, 0.1% SDS, 0.05% xylene cyanol, and 0.05% bromphenol blue). Samples were heated for 5 min at 95°C prior to fractionating through standard 5% sequencing gels. Radioactive bands were visualized using a phosphorimager and quantified with ImageQuant 5.2.
mRNA half-life assay
Cultures were grown at 30°C in tryptone broth supplemented with ampicillin (100 μg/ml) to late exponential phase prior to the addition of rifampicin (200 μg/ml) to prevent transcription initiation. After 1 min, 10 ml aliquots were removed at various times and added to an equal volume of frozen buffer (10 mM Tris-HCl, pH 7.2, 5 mM MgCl2, 25 mM sodium azide, 12.5% ethanol, and 500 μg/ml choramphenicol). Cells were collected by centrifugation and cell pellets were suspended in 1 ml of a 2:1 mixture of RNAProtect bacterial reagent:TE buffer and placed on ice for 10 min. Cells were then collected by centrifugation and cell pellets were frozen at −80°C. RNA was isolated using the RNeasy Mini kit. DNA was removed with 1 U of Turbo DNase (Ambion). The RNA was extracted with phenol, precipitated and suspended in water. Quantification by Northern blot analysis followed a published procedure (Oh et al., 2009). Ten micrograms of total RNA was mixed with at least two volumes of denaturing solution (66% formamide, 9% formaldehyde, 20 mM MOPS, pH 7.0, 5 mM NaOAc, and 2 mM EDTA) and then heated for 15 min at 65°C. RNA samples (8 μg) were separated by electrophoresis through a 1.2% denaturing formaldehyde agarose gel. RNA was transferred to Hybond N+ membranes (Amersham) by capillary blotting with 10x SSC (1.5 M NaCl and 150 mM sodium citrate, pH 7.0) and fixed to the membrane by heating at 80°C for 2 h under vacuum. Fixed membranes were prehybridized at 46°C for 3 hr in hybridization buffer (7% SDS, 0.5 M sodium phosphate, pH 7.2, 10 mM EDTA, and 0.0125% nonfat powdered milk). Hybridization was with a 5’ end-labeled probe complementary to the flhDC leader (2x106 cpm/mL final concentration) in hybridization buffer for 15 hr at 46°C. Membranes were washed three times for 20 min each with Buffer 1 (2x SSC and 0.1% SDS) at 37°C, followed by three 20-min washes with Buffer 2 (1x SSC and 0.1% SDS) at 42°C. mRNA levels were quantified with a phosphorimager and ImageQuant 5.2 software. Before reprobing, membranes were stripped by boiling in 0.2% SDS for 5 min and then hybridized with a 16S rRNA oligonucleotide probe at 52°C as described above. Each flhDC data point was normalized to the 16S rRNA value before calculating mRNA the half-live as described (Yakhnin et al., 2001).
Supplementary Material
Acknowledgments
We thank Sidney Kushner for bacterial strains. This work was supported by NIH grant GM059969 and CRIS project FLA-MCS-004949.
References
- Arraiano CM, Andrade JM, Domingues S, Guinote IB, Malecki M, Matos RG, et al. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol Rev. 2010;34:883–923. doi: 10.1111/j.1574-6976.2010.00242.x. [DOI] [PubMed] [Google Scholar]
- Arraiano CM, Yancey SD, Kushner SR. Stabilization of discrete mRNA breakdown products in ams pnp rnb multiple mutants of Escherichia coli K-12. J Bacteriol. 1988;170:4625–4633. doi: 10.1128/jb.170.10.4625-4633.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babitzke P, Kushner SR. The Ams (altered mRNA stability) protein and ribonuclease E are encoded by the same structural gene of Escherichia coli. Proc Natl Acad Sci USA. 1991;88:1–5. doi: 10.1073/pnas.88.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babitzke P, Romeo T. CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Curr Opin Microbiol. 2007;10:156–163. doi: 10.1016/j.mib.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Baker CS, Eöry LA, Yakhnin H, Mercante J, Romeo T, Babitzke P. CsrA inhibits translation initiation of Escherichia coli hfq by binding to a single site overlapping the Shine-Dalgarno sequence. J Bacteriol. 2007;189:5472–5481. doi: 10.1128/JB.00529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua JM, Bevilacqua PC. Thermodynamic analysis of an RNA combinatorial library contained in a short hairpin. Biochemistry. 1998;37:15877–15884. doi: 10.1021/bi981732v. [DOI] [PubMed] [Google Scholar]
- Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW, et al. Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene. 1977;2:95–113. [PubMed] [Google Scholar]
- Callaghan AJ, Marcaida MJ, Stead JA, McDowall KJ, Scott WG, Luisi BF. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature. 2005;437:1187–1191. doi: 10.1038/nature04084. [DOI] [PubMed] [Google Scholar]
- Carpousis AJ. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu Rev Microbiol. 2007;61:71–87. doi: 10.1146/annurev.micro.61.080706.093440. [DOI] [PubMed] [Google Scholar]
- Chavez RG, Alvarez AF, Romeo T, Georgellis D. The physiological stimulus for the BarA sensor kinase. J Bacteriol. 2010;192:2009–2012. doi: 10.1128/JB.01685-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deana A, Celesnik H, Belasco JG. The bacterial enzyme RppH triggers messenger RNA degradation by 5' pyrophosphate removal. Nature. 2008;451:355–358. doi: 10.1038/nature06475. [DOI] [PubMed] [Google Scholar]
- Dubey AK, Baker CS, Suzuki K, Jones AD, Pandit P, Romeo T, Babitzke P. CsrA regulates translation of the Escherichia coli carbon starvation gene, cstA, by blocking ribosome access to the cstA transcript. J Bacteriol. 2003;185:4450–4460. doi: 10.1128/JB.185.15.4450-4460.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey AK, Baker CS, Romeo T, Babitzke P. RNA sequence and secondary structure participate in high-affinity CsrA-RNA interaction. RNA. 2005;11:1579–1587. doi: 10.1261/rna.2990205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AN, Patterson-Fortin LM, Vakulskas CA, Mercante JW, Potrykus K, Camacho MI, et al. Circuitry linking the Csr and stringent response global regulatory systems. Mol Microbiol. 2011;80:1561–1580. doi: 10.1111/j.1365-2958.2011.07663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England TE, Uhlenbeck OC. 3'-terminal labelling of RNA with T4 RNA ligase. Nature. 1978;275:560–561. doi: 10.1038/275560a0. [DOI] [PubMed] [Google Scholar]
- Gao J, Lee K, Zhao M, Qiu J, Zhan X, Saxena A, et al. Differential modulation of E. coli mRNA abundance by inhibitory proteins that alter the composition of the degradosome. Mol Microbiol. 2006;61:394–406. doi: 10.1111/j.1365-2958.2006.05246.x. [DOI] [PubMed] [Google Scholar]
- Haldimann A, Wanner BL. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol. 2001;183:6384–6393. doi: 10.1128/JB.183.21.6384-6393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kime L, Jourdan SS, McDowall KJ. Identifying and characterizing substrates of the RNase E/G family of enzymes. Methods Enzymol. 2008;447:215–241. doi: 10.1016/S0076-6879(08)02212-X. [DOI] [PubMed] [Google Scholar]
- Kime L, Jourdan SS, Stead JA, Hidalgo-Sastre A, McDowall KJ. Rapid cleavage of RNA by RNase E in the absence of 5' monophosphate stimulation. Mol Microbiol. 2010;76:590–604. doi: 10.1111/j.1365-2958.2009.06935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnen D, Blumer C, Polen T, Wackwitz B, Wendisch VF, Unden G. LrhA as a new transcriptional key regulator of flagella, motility and chemotaxis genes in Escherichia coli. Mol Microbiol. 2002;45:521–532. doi: 10.1046/j.1365-2958.2002.03032.x. [DOI] [PubMed] [Google Scholar]
- Liu MY, Gui G, Wei B, Preston JF, III, Oakford L, Yuksel U, et al. The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J Biol Chem. 1997;272:17502–17510. doi: 10.1074/jbc.272.28.17502. [DOI] [PubMed] [Google Scholar]
- Liu MY, Romeo T. The global regulator CsrA of Escherichia coli is a specific mRNA-binding protein. J Bacteriol. 1997;179:4639–4642. doi: 10.1128/jb.179.14.4639-4642.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowall KJ, Lin-Chao S, Cohen SN. A+U content rather than a particular nucleotide order determines the specificity of RNase E cleavage. J Biol Chem. 1994;269:10790–10796. [PubMed] [Google Scholar]
- Mackie GA. Ribonuclease E is a 5'-end-dependent endonuclease. Nature. 1998;395:720–723. doi: 10.1038/27246. [DOI] [PubMed] [Google Scholar]
- Mercante J, Suzuki K, Cheng X, Babitzke P, Romeo T. Comprehensive alanine-scanning mutagenesis of Escherichia coli CsrA defines two subdomains of critical functional importance. J Biol Chem. 2006;281:31832–31842. doi: 10.1074/jbc.M606057200. [DOI] [PubMed] [Google Scholar]
- Mercante J, Edwards AN, Dubey AK, Babitzke P, Romeo T. Molecular geometry of CsrA (RsmA) binding to RNA and its implications for regulated expression. J Mol Biol. 2009;392:511–528. doi: 10.1016/j.jmb.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty BK, Kushner SR. Processing of the Escherichia coli leuX tRNA transcript, encoding tRNALeu5, requires either the 3' to 5' exoribonuclease polynucleotide phosphorylase or RNase P to remove the Rho-independent transcription terminator. Nucleic Acids Res. 2010;38:597–607. doi: 10.1093/nar/gkp997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh HS, Pathak HB, Goodfellow IG, Arnold JJ, Cameron CE. Insight into poliovirus genome replication and encapsidation obtained from studies of 3B-3C cleavage site mutants. J Virol. 2009;83:9370–9387. doi: 10.1128/JVI.02076-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ow MC, Liu Q, Kushner SR. Analysis of mRNA decay and rRNA processing in Escherichia coli in the absence of RNase E-based degradosome assembly. Mol Microbiol. 2000;38:854–866. doi: 10.1046/j.1365-2958.2000.02186.x. [DOI] [PubMed] [Google Scholar]
- Pannuri A, Yakhnin H, Vakulskas CA, Edwards AN, Babitzke P, Romeo T. Translational repression of NhaR, a novel pathway for multi-tier regulation of biofilm circuitry by CsrA. J Bacteriol. 2012;194:79–89. doi: 10.1128/JB.06209-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perwez T, Kushner SR. RNase Z in Escherichia coli plays a significant role in mRNA decay. Mol Microbiol. 2006;60:723–737. doi: 10.1111/j.1365-2958.2006.05124.x. [DOI] [PubMed] [Google Scholar]
- Prüß BM, Matsumura P. Cell cycle regulation of flagellar genes. J Bacteriol. 1997;179:5602–5604. doi: 10.1128/jb.179.17.5602-5604.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards J, Luciano DJ, Belasco JG. Influence of translation on RppH-dependent mRNA degradation in Escherichia coli. Mol Microbiol. 2012 doi: 10.1111/mmi.12040. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo T, Gong M, Liu MY, Brun-Zinkernagel AM. Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J Bacteriol. 1993;175:4744–4755. doi: 10.1128/jb.175.15.4744-4755.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo T, Vakulskas CA, Babitzke P. Posttranscriptional regulation on a global scale: Form and function of Csr/Rsm systems. Env Microbiol. 2012 doi: 10.1111/j.1462-2920.2012.02794.x. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabnis NA, Yang H, Romeo T. Pleiotropic regulation of central carbohydrate metabolism in Escherichia coli via the gene csrA. J Biol Chem. 1995;270:29096–29104. doi: 10.1074/jbc.270.49.29096. [DOI] [PubMed] [Google Scholar]
- Schubert M, Lapouge K, Duss O, Oberstrass FC, Jelesarov I, Haas D, Allain FH. Molecular basis of messenger RNA recognition by the specific bacterial repressing clamp RsmA/CsrA. Nat Struct Mol Biol. 2007;14:807–813. doi: 10.1038/nsmb1285. [DOI] [PubMed] [Google Scholar]
- Shin S, Park C. Modulation of flagellar expression in Escherichia coli by acetyl phosphate and the osmoregulator OmpR. J Bacteriol. 1995;177:4696–4702. doi: 10.1128/jb.177.16.4696-4702.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TG, Hoover TR. Deciphering bacterial flagellar gene regulatory networks in the genomic era. Adv Appl Microbiol. 2009;67:257–295. doi: 10.1016/S0065-2164(08)01008-3. [DOI] [PubMed] [Google Scholar]
- Soutourina O, Kolb A, Krin E, Laurent-Winter C, Rimsky S, Danchin A, Bertin P. Multiple control of flagellum biosynthesis in Escherichia coli: role of H-NS protein and the cyclic AMP-catabolite activator protein complex in transcription of the flhDC master operon. J Bacteriol. 1999;181:7500–7508. doi: 10.1128/jb.181.24.7500-7508.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio V, Torres AG, Kaper JB. Quorum sensing Escherichia coli regulators B and C (QseBC): a novel two-component regulatory system involved in the regulation of flagella and motility by quorum sensing in E. coli. Mol Microbiol. 2002;43:809–821. doi: 10.1046/j.1365-2958.2002.02803.x. [DOI] [PubMed] [Google Scholar]
- Stead MB, Marshburn S, Mohanty BK, Mitra J, Castillo LP, Ray D, et al. Analysis of Escherichia coli RNase E and RNase III activity in vivo using tiling microarrays. Nucleic Acids Res. 2011;39:3188–3203. doi: 10.1093/nar/gkq1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Wang X, Weilbacher T, Pernestig AK, Melefors Ö, Georgellis D, et al. Regulatory circuitry of the CsrA/CsrB and BarA/UvrY systems of Escherichia coli. J Bacteriol. 2002;184:5130–5140. doi: 10.1128/JB.184.18.5130-5140.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Babitzke P, Kushner SR, Romeo T. Identification of a novel regulatory protein (CsrD) that targets the global regulatory RNAs CsrB and CsrC for degradation by RNase E. Genes Dev. 2006;20:2605–2617. doi: 10.1101/gad.1461606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason MK, Fontaine F, De Lay N, Storz G. A small RNA that regulates motility and biofilm formation in response to changes in nutrient availability in Escherichia coli. Mol Microbiol. 2012;84:17–35. doi: 10.1111/j.1365-2958.2012.07965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans J, Van Melderen L. Conditional essentiality of the csrA gene in E. coli. J Bacteriol. 2009;191:1722–1724. doi: 10.1128/JB.01573-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Fleming RT, Westbrook EM, Matsumura P, McKay DB. Structure of the Escherichia coli FlhDC complex, a prokaryotic heteromeric regulator of transcription. J Mol Biol. 2006;355:798–808. doi: 10.1016/j.jmb.2005.11.020. [DOI] [PubMed] [Google Scholar]
- Wei BL, Brun-Zinkernagel AM, Simecka JW, Prüß BM, Babitzke P, Romeo T. Positive regulation of motility and flhDC expression by the RNA-binding protein CsrA of Escherichia coli. Mol Microbiol. 2001;40:245–256. doi: 10.1046/j.1365-2958.2001.02380.x. [DOI] [PubMed] [Google Scholar]
- Weilbacher T, Suzuki K, Dubey AK, Wang X, Gudapaty S, Morozov I, et al. A novel sRNA component of the carbon storage regulatory system of Escherichia coli. Mol Microbiol. 2003;48:657–670. doi: 10.1046/j.1365-2958.2003.03459.x. [DOI] [PubMed] [Google Scholar]
- Yakhnin AV, Babitzke P. Mechanism of NusG-stimulated pausing, hairpin-dependent pause site selection and intrinsic termination at overlapping pause and termination sites in the Bacillus subtilis trp leader. Mol Microbiol. 2010;76:690–705. doi: 10.1111/j.1365-2958.2010.07126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakhnin H, Babiarz JE, Yakhnin AV, Babitzke P. Expression of the Bacillus subtilis trpEDCFBA operon is influenced by translational coupling and Rho termination factor. J Bacteriol. 2001;183:5918–5926. doi: 10.1128/JB.183.20.5918-5926.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakhnin H, Yakhnin AV, Baker CS, Sineva E, Berezin I, Romeo T, Babitzke P. Complex regulation of the global regulatory gene csrA: CsrA-mediated translational repression, transcription from five promoters by Eσ70 and EσS, and indirect transcriptional activation by CsrA. Mol Microbiol. 2011a;81:689–704. doi: 10.1111/j.1365-2958.2011.07723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakhnin H, Baker CS, Berezin I, Evangelista MA, Rassin A, Romeo T, Babitzke P. CsrA represses translation of sdiA, which encodes the N-acylhomoserine-L-lactone receptor of Escherichia coli, by binding exclusively within the coding region of sdiA mRNA. J Bacteriol. 2011b;193:6162–6170. doi: 10.1128/JB.05975-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey SD, Kushner SR. Isolation and characterization of a new temperature-sensitive polynucleotide phosphorylase mutation in Escherichia coli K-12. Biochimie. 1990;72:835–843. doi: 10.1016/0300-9084(90)90193-k. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.