

Abstract
Aims
Prion diseases are characterized by brain deposits of misfolded aggregated protease-resistant prion protein (PrP), termed PrPres. In humans and animals, PrPres is found as either disorganized non-amyloid aggregates or organized amyloid fibrils. Both PrPres forms are found in extracellular spaces of the brain. Thus, both might block drainage of brain interstitial fluid (ISF). The present experiments studied whether ISF blockage occurred during amyloid and/or non-amyloid prion diseases.
Methods
Various-sized fluorescein-labeled ISF tracers were stereotactically inoculated into the striatum of adult mice. At times from 5 min to 77 hours, uninfected and scrapie-infected mice were compared. C57BL/10 mice expressing wild-type anchored PrP, which develop non-amyloid PrPres similar to humans with sporadic CJD, were compared with Tg44+/+ mice expressing anchorless PrP, which develop amyloid PrPres similar to certain human familial prion diseases.
Results
In C57BL/10 mice, extensive non-amyloid PrPres aggregate deposition was not associated with abnormal clearance kinetics of tracers. In contrast, scrapie-infected Tg44+/+ mice showed blockage of tracer clearance and co-localization of tracer with perivascular PrPres amyloid.
Conclusions
Since tracer localization and clearance was normal in infected C57BL/10 mice, ISF blockage was not an important pathogenic mechanism in this model. Therefore, ISF blockage is unlikely to be a problem in non-amyloid human prion diseases such as sporadic CJD. In contrast, partial ISF blockage appeared to be a possible pathogenic mechanism in Tg44+/+ mice. Thus this mechanism might also influence human amyloid prion diseases where expression of anchorless or mutated PrP results in perivascular amyloid PrPres deposition and cerebral amyloid angiopathy (CAA).
Keywords: brain interstitial fluid, cerebral amyloid angiopathy, prion, glycophosphatidylinositol anchor, basement membrane
Introduction
Transmissible spongiform encephalopathy (TSE) diseases or prion diseases are a group of rare, slowly progressive, neurodegenerative conditions that affect both animals and humans. Misfolded aggregated partially protease-resistant host prion protein (PrPres) is a classic biochemical and histopathological marker of TSE disease. However, the pattern of PrPres accumulation in brain varies in different disease conditions. For example, in sporadic Creutzfeldt-Jakob disease (sCJD) of humans, PrPres is usually found in the CNS in a non-amyloid form which usually appears as extracellular diffuse/synaptic or perivacuolar deposits often associated with typical prion disease gray matter spongiosis [1, 2]. Similarly mice infected with mouse-adapted sheep scrapie strains have mostly extracellular non-amyloid PrPres [3, 4]. In contrast, both non-amyloid and amyloid extracellular PrPres deposits are prominent in brain pathology associated with familial human prion diseases [5, 6] and in certain animal prion disease models [7]. The amyloid form of PrPres is found in multicentric gray matter plaques and/or in perivascular sites as well as the vascular wall, resulting in cerebral amyloid angiopathy (CAA), which may be an important aspect of the pathogenic process [8–10].
The extracellular location of PrPres deposition in humans and animals is also the area of formation of brain interstitial fluid (ISF) which flows through extracellular spaces in both gray and white matter tracks along the basement membranes of capillaries and small arteries and finally drains into leptomeningeal arteries and cervical lymph nodes [11]. The ISF acts as an acellular lymph fluid in brain and is important for cell-to-cell communication, clearance of solutes from the extracellular spaces, and maintenance of ion homeostasis in the brain parenchyma [12]. Perturbation of ISF drainage by Aβ amyloid has been postulated to be a pathogenic mechanism in Alzheimer’s disease [13–16], and in a transgenic mouse model of Alzheimer’s disease, extracellular cerebral perivascular amyloid deposition has been shown to perturb brain interstitial fluid (ISF) drainage [17].
ISF drainage has not been previously studied in prion disease. We hypothesized that deposition of PrPres in brain at extracellular sites might cause alterations to the ISF pathway. In the present studies we compared C57BL/10 mice and transgenic Tg44+/+ mice with and without scrapie infection. C57BL/10 mice express PrP anchored to the plasma membrane by a glycophosphatidylinositol (GPI) linker, and after scrapie infection these mice die at 145–160 days post-infection (dpi) with severe gray matter spongiosis, gliosis and deposition of non-amyloid PrPres aggregates in many brain region mostly at extracellular sites. In contrast, Tg44+/+ mice express anchorless PrP which is secreted from cells, and after scrapie infection these mice die at 320–360 dpi with severe gliosis, but no gray matter spongiosis. However, they have extensive deposition of an amyloid form of PrPres at extracellular perivascular sites mostly associated with vascular basement membranes [18]. To study ISF drainage, we performed intracerebral stereotactic microinjections using several types of fluorescein-labeled ISF tracer molecules and followed distribution and clearance of tracers by microscopy at various times post-inoculation. In these experiments unexpected differences were observed between scrapie-infected mice expressing amyloid versus non-amyloid forms of PrPres.
Materials and Methods
Experimental Mice
All mice were housed at Rocky Mountain Laboratories (RML) in an AAALAC-accredited facility and experimentation followed NIH RML Animal Care and Use Committee (ACUC) approved protocols. Transgenic mice bearing the GPI anchorless prion protein were described previously [19]. All transgenic GPI anchorless mice (Tg44+/+) used in this study were bred at RML and expressed the transgene in homozygous form [18]. C57BL/10 mice were obtained from Harlan Sprague Dawley, Kent, WA. Young mice from 4–6 weeks of age were infected by the intracerebral (IC) route with 50 μl of a 1% brain homogenate of RML scrapie stock containing 4.0 × 104 ID50. One ID50 is the dose causing infection in 50% of the C57BL/10 mice. For IC infection, mice were anaesthetized with isoflurane and injected manually in the left parietal lobe with a 27 gauge, 0.5-inch needle. Uninfected groups of age-matched transgenic and C57BL/10 mice were used as controls.
Stereotaxic Surgery
Intracerebral injections of tracers were performed on adult male mice that weighed approximately 30 g. Scrapie-infected C57BL/10 mice and Tg44+/+ mice were injected at the time of early clinical disease when motor abnormalities were first noticeable. For C57BL/10 mice this time was 130–140 dpi, and for Tg44+/+ mice this time was 250–280 dpi. These times were chosen to insure that mice had significant brain pathology in multiple brain regions which might influence ISF flow if this were indeed a significant mechanism associated with the disease. Uninfected control mice of each strain were age-matched to scrapie-infected mice. The numbers of mice studied in each experimental group are presented in Table 1. Mice were anaesthetized with isoflurane and positioned on a stereotaxic frame (David-Kopf Instruments, Tujunga, CA). A 1cm midline incision was made in the skin over the dorsal surface of the skull, and the skull was exposed to allow positioning of the drill over the Bregma point of reference. Coordinates used from Bregma were, +1 mm anteroposterior, +1.7 mm lateral and −3 mm ventral to skull surface. These coordinates were selected to target the center of the striatum and avoid passing through any ventricles. All tracers were injected with NanoFil syringes (World Precision Instruments, Sarasota. FL) and steel bevel needles (32-gauge diameter) (World Precision Instruments) into the striatum at a rate of 0.25 μl/min with a total volume of 0.5 μl per mouse controlled with a pump (UltraMicroPump III with Micro4 pump controller, World Precision Instruments). The needle was kept in place for 2 min following injection to avoid the reflux of the tracer solution [13]. Skin incision was closed with either a suture or a wound clip. These conditions produced minimal brain damage and high reproducibility. Patency of needles was verified prior to and after injections.
Table 1.
Overview of experimental plan for study of different tracers of brain interstitial fluid (ISF) drainage.
| Tracera | Miceb | 5 minc | 30 min | 7 hours | 24 hours | 77 hours |
|---|---|---|---|---|---|---|
| FITC-ovalbumin (45 kDa) | Tg44+/+ (U) | 2 | 5 | 4 | 4 | 2 |
| Tg44+/+ (S) | 2 | 2 | 4 | 6 | 2 | |
| C57BL/10 (U) | 2 | 4 | 4 | 2 | 0 | |
| C57BL/10 (S) | 2 | 2 | 4 | 2 | 0 | |
| FITC-Dextran (70 kDa) | Tg44+/+ (U) | 0 | 2 | 0 | 0 | 0 |
| Tg44+/+ (S) | 0 | 2 | 3 | 0 | 0 | |
| C57BL/10 (U) | 0 | 1 | 2 | 0 | 0 | |
| FITC-Dextran (500 kDa) | C57BL/10 (U) | 0 | 1 | 2 | 0 | 0 |
| Tracer | Mice | 5 min | 30 min | 2 hours | 7 hours | 24 hours |
| FITC-Cadaverine (653 Da) | Tg44+/+ (U) | 0 | 2 | 3 | 2 | 4 |
| Tg44+/+ (S) | 0 | 3 | 3 | 3 | 4 |
FITC-labeled tracers of different molecular weight and composition were inoculated stereotactically into the striatum.
Mice were uninfected (U) or scrapie-infected (S) C57BL/10 or Tg44+/+.
Times shown are the time-points after tracer inoculation when mice were euthanized for study of tracer localization. Note different time-points were used for FITC-Cadaverine because of its smaller size.
Tracers
As tracers for the ISF fluid we used fluorescein-labeled ovalbumin (FITC-OVA) (45 kDa) (Invitrogen, Grand Island, NY), cadaverine (FITC-Cadaverine)(653 Da) (Invitrogen) and 500 kDa or 70 kDa dextran (FITC-dextran) (Invitrogen). Ovalbumin was chosen as it was used extensively in a landmark previous study of ISF [13]. Cadaverine is a smaller molecule, related to lysine, which was selected as a control for possible non-specific binding of the FITC group to PrPres as well as to see how small molecules were cleared in our mouse models. The dextrans were chosen as carbohydrate molecules to compare to the protein, ovalbumin. The 500 kDa dextran also provided information on the upper limit of molecular size for ISF drainage in our mice.
Histology
Mice were perfused intracardially with 0.1M sodium phosphate buffer, pH 7.4 with heparin (30 units/ml) followed by cold 4% paraformaldehyde, adjusted to pH 7.3 by addition of NaOH. Brains were removed, post-fixed overnight in the same fixative, and cryoprotected in 30% sucrose in phosphate buffered saline (PBS) without Mg+2 or Ca+2 for 48 h. Ten μm coronal sections were cut through the entire striatum region in a Leica cryostat, using a CryoJane® Tape-Transfer System (Leica, Richmond, IL). Four adjacent 10 μm sections were saved from each 110 μm linear region, and the remaining 70 μm of tissue was discarded. Approximately twelve 110 μm regions were required to cover the entire striatum. Sections to be saved were attached to separate gel-coated slides (Leica), which were then exposed to UV light to cross-link the tissue to the slide. The 4 adjacent sections from each region were separated into 4 groups (a,b,c,d) which formed 4 parallel or replicate tissue series of the striatum of each mouse which were used for different kinds of staining. Slides were then stored at −20°C until needed for histological analysis.
Immunofluorescence staining of sections
Slides were stained using the Dako Autostainer Plus (Dako, Carpentaria, CA) Sections were permeabilized with PBS without Mg+2 or Ca+2 with 0.1 % Triton X-100 for 15 min and blocked in 1% normal goat serum and 0.1% Triton X-100 in PBS for 15 min. Primary antibodies (described below) were diluted at the concentrations indicated in 1% normal goat serum and 0.1% Triton X-100 in PBS. Diluent without antibody was used as a negative control. Slides were exposed to antibody for 1 hour at room temperature, followed by 2 washes in PBS with 1% Triton X-100. Slides were next exposed to Alexa-Fluor 488 or 568-tagged secondary goat anti-rabbit Ig or anti-human Ig antibodies (specific for the species of the primary antibodies) (Molecular Probes, Eugene, OR) for 30 min at a 1:200 dilution. Prior to immunofluorescence staining for PrPres, frozen sections on slides were incubated for 15 min at room temperature in 4.0 M guanidine thiocyanate. For additional antigen retrieval, prior to laminin staining slides were pretreated with pepsin (Pepsin solution, Invitrogen) for 10 min at 37°C [20]. Nuclei were then stained with 0.01 % 4′,6-diamidino-2-phenylindole dilactate (DAPI, Invitrogen) for 5 min and rinsed with double-distilled H2O, and coverslips were applied to tissue sections with ProLong Gold Antifade reagent (Invitrogen). All images were acquired using an epifluorescent Olympus BX51 microscope (Olympus, Center Valley, PA) with Microsuite FIVE software (Olympus) or a confocal laser-scanning microscope (Zeiss SLM 510, Carl Zeiss, Germany). A Z-stack of 15–20 optical sections of 0.3–0.5 μm thickness was taken for all confocal imaging. Images were managed using Imaris software (Bitplane, So. Windsor, CT). Figures show individual optical sections as noted in legends. All images were obtained in sequential scanning laser mode to avoid fluorochrome cross-excitation.
In tracer studies using FITC-OVA, FITC-Dextran and FITC-cadaverine, non-adjacent slides from four of the twelve 110 μm linear regions (described above) with the highest area and intensity of tracer detection were chosen for each mouse. Slides were scored in the region of tracer inoculation for the relative amount of fluorescent tracer signal associated with the ISF system (capillaries and some slightly larger blood vessels) as well as with PrPres plaques. A scale from 0 to 4 was used as follows: 0 = no signal observed; 1 = faintly detectable signal on vessels or plaques visible only with 20 X objectives or higher; 2 = moderate signal visible with 10X or 20X objectives; 3 = strong signal on blood vessels or plaques with easily visible edges at sites of basement membrane; 4 = very intense signal associated mostly with larger plaque structures. Scoring was verified by two different observers. Data was analyzed statistically by a 2-way ANOVA with Bonferroni’s correction using Graph Pad software.
Primary antibodies
The following primary antibodies were used: rabbit anti-Iba1 (Wako, Richmond, VA) (dilution 1:4000), rabbit anti-glial fibrillary acidic protein (GFAP) polyclonal antibody (1:2000) (Dako), rabbit anti-pan-Laminin (Dako) (1:500), and human D13 anti-PrP recombinant antibody fragment (1:500) (InPro, San Francisco, CA). We also used D13 supernatants (1:100) made in our laboratory from CHO cells expressing the D13 antibody construct [21], which were kindly provided by Dr. R. Anthony Williamson, The Scripps Research Institute, La Jolla, CA.
Immunohistochemical staining
Immunohistochemical staining for detection of PrPres using monoclonal antibody D13 was done on frozen sections with PrPres antigen retrieval in a Ventana automated Discovery XT stainer (Ventana Medical Systems, Tucson, Arizona) using biotinylated anti-human IgG and avidin-horseradish peroxidase (HRP) with DAB as chromogen as previously described [22].
Results
Different patterns of OVA clearance in scrapie-infected C57BL/10 mice and Tg44+/+ mice
Initially we analyzed brain ISF drainage by following the clearance of FITC-labeled ovalbumin (FITC-OVA) (molecular weight 45 kDa) from the brain parenchyma in both C57BL/10 and Tg44+/+ mice. In each case age-matched uninfected mice were compared to scrapie-infected mice with early clinical disease. At 5 min and 30 min post-inoculation in uninfected C57BL/10 and Tg44+/+ mice, the tracer was observed in a smooth continuous pattern along capillaries and small vessels (Figure 1 A,C,E.G). At 7 h tracer was still faintly visible (Figure 1 I,K), but at 24 h tracer was no longer visible (Figure 1 M,O). In scrapie-infected C57BL/10 mice, FITC-OVA localization and clearance kinetics were indistinguishable from uninfected mice (Figure 1 B,F,J,N), in spite of the presence of abundant PrPres deposition (Figure 2). Therefore, the PrPres accumulation seen in scrapie-infected C57BL/10 mouse brain did not appear to interfere with the normal clearance of FITC-OVA via the ISF pathway.
Figure 1.

Brain microinjection of FITC-OVA in uninfected and scrapie-infected C57BL/10 mice and uninfected and scrapie-infected Tg44+/+ mice. At 5 and 30 min post-injection in uninfected (A,E) and infected (B,F) C57BL/10 mice and in uninfected Tg44+/+ mice (C,G), FITC-OVA was associated with walls of capillaries (arrows) and larger vessels (arrowheads). At 5 and 30 min after injection in scrapie-infected Tg44+/+ mouse (D,H), FITC-OVA was mainly concentrated around large plaque-like perivascular structures (arrowheads), but association with walls of capillaries (arrows) could also be seen. Panel D: Scale bar 100 μm; same scale for all panels. (I,J,K) At 7 hours post-injection in uninfected and infected C57BL/10 mice and in uninfected Tg44+/+ mice FITC-OVA was barely visible associated with small capillaries (arrows). (L) At 7 hours post-injection in infected Tg44+/+ mice, FITC-OVA remained associated with perivascular plaque-like structures (arrowheads), but intensity of staining was decreased for most plaques, and normal capillaries were no longer visibly stained. (M,N,O) At 24 h after injection in both uninfected and infected C57BL/10 mice, and uninfected Tg44+/+ mice FITC-OVA was no longer visible. (P) At 24 hours post-injection in Tg44+/+ scrapie-infected mice, FITC-OVA was detectable at low intensity in occasional blood vessels in the center of perivascular plaque-like structures (arrow).
Figure 2.
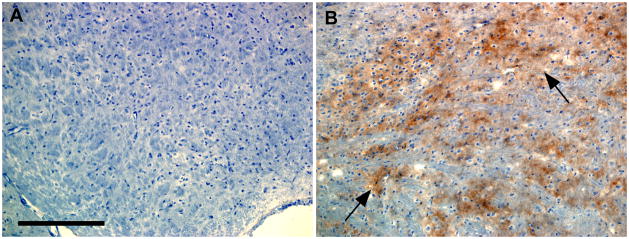
Immunohistochemical staining for PrPres in the striatum region of uninfected C57BL/10 (A) and scrapie-infected C57BL/10 mice with early clinical signs. Panel B shows abundant patchy diffuse brown staining of PrPres (arrows) in the infected mouse, whereas no staining of PrPres was seen in the uninfected mouse (A). Scale bar is 200μm. These sections were taken from the same mice studied at 30 min post-injection of FITC-OVA shown in Figure 1 E,F. PrPres amyloid with CAA in Tg44+/+ mice is not shown here because it was extensively documented in a previous paper [18].
In contrast, in scrapie-infected Tg44+/+ mice, a different pattern of tracer distribution and clearance was observed. At 5 and 30 min post-inoculation the tracer was concentrated in large rounded structures (Figure 1 D,H) often near blood vessels, which were morphologically similar to PrP amyloid plaques [18]. Tracer was also seen in association with the walls of capillaries within or near plaques (Figure 1 D,H). At 7 h less tracer was visible, but tracer was still observed near vessel walls at the center of a few plaques (Figure 1L). At 24h tracer was further decreased, but occasional capillary walls in the center of plaques were positive (Figure 1P). At 77 h tracer was rarely visible (not shown). In summary, in infected Tg44+/+ mice clearance of FITC-OVA tracer was transiently blocked, and this delay appeared to be due to association of the tracer with perivascular plaque-like structures.
To determine whether the above findings were reproducible, at each time-point 2–6 mice from each treatment group were scored for relative tracer intensity localized to blood vessels and plaques (Figure 3 A,B). The results showed that the findings of delay of clearance of FITC-OVA in scrapie-infected Tg44+/+ mice compared to other mice tested were consistent and significant.
Figure 3.
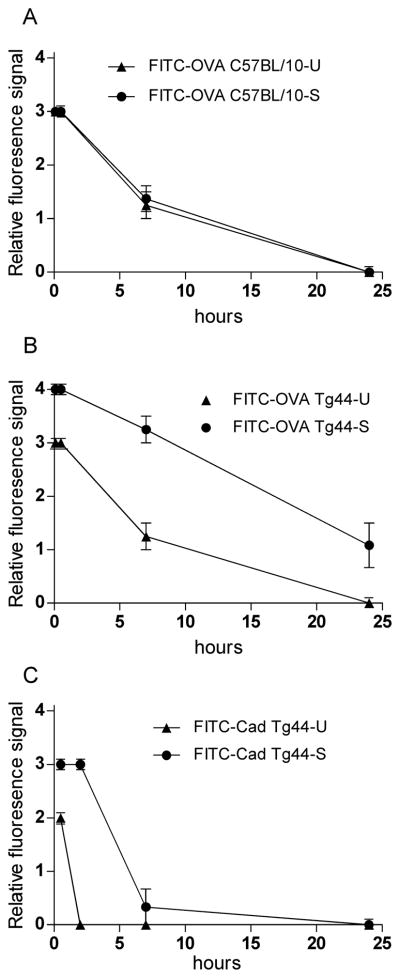
Comparison of relative fluorescence signal of tracers associated with blood vessels or PrPres plaques at various times after microinjection in replicate mice. (A) FITC-OVA-injected C57BL/10-U (uninfected) and C57BL/10-S (scrapie-infected) mice. (B) FITC-OVA-injected Tg44-U (uninfected) and Tg44-S (scrapie-infected) mice. (C) FITC-Cadaverine injected Tg44-U (uninfected) and Tg44-S (scrapie-infected) mice. Scale for relative fluorescence signal is defined in the Methods section. Points and error bars are means and standard errors. Each point has data from 2–6 mice. Data was analyzed statistically by a 2-way ANOVA with Bonferroni’s correction using Graph Pad software. For experiments studying FITC-OVA, Tg44-S in panel B was significantly different (P<0.001) from Tg44-U in same panel and from both C57BL/10-S and C57BL/10-U in panel A. In panel C showing data for FITC-Cadaverine, Tg44-S was significantly different (P<0.001) from Tg44-U.
Comparison of clearance of FITC-cadaverine in uninfected and infected Tg44+/+ mice
To control for possible non-specific binding of the FITC group to brain structures and to study the effect of the molecular size of the tracer on ISF clearance kinetics in this system, we injected a small molecule, FITC-cadaverine (653 Da). In uninfected mice at 30 min after injection, FITC-cadaverine was mostly associated with capillaries (Figure 4A). In scrapie-infected Tg44+/+ mice at 30 min the FITC-cadaverine was also associated with capillaries, but was also co-localized with plaque-like perivascular structures not seen in uninfected mice (Figure 4B). In uninfected mice after 2 and 7 hours FITC-cadaverine was no longer visible (Figure 4 C,E). However, in infected animals FITC-cadaverine was still visible at 2 hours (Figure 4D), but was cleared by 7 hours (Figure 4F). Thus, scrapie-infected Tg44+/+ mice showed a transient delay in clearance of FITC-cadaverine. However, the clearance of FITC-cadaverine was faster than the clearance of FITC-OVA (45 kDa) (Figure 3C), suggesting that the difference in size between OVA and cadaverine influenced the timing of tracer clearance.
Figure 4.

Brain microinjection of uninfected and scrapie-infected Tg44+/+ mice with FITC-cadaverine tracer (FITC-CDV) (green). (A) 30 min post-injection of uninfected Tg44+/+ mice showing tracer associated with capillaries (arrow) and a larger vessel (arrowhead). Scale bar = 100 μm; panels (B–F) are same scale. (B) 30 min post-injection of infected mouse showing tracer associated with large perivascular plaque-like structures (arrowhead), as well as small capillaries lacking plaques (arrow). (C) Clearance of tracer at 2 h post-injection in uninfected mouse. (D) At 2 h post-injection in infected mouse tracer is still visible associated with some perivascular plaques (arrowheads). Some faint staining of capillaries lacking plaques is also still visible (arrow). (E) and (F) At 7 h post-injection tracer is no longer visible in uninfected and infected mice.
Co-staining studies of FITC-OVA with PrPres, laminin, Iba-1 and GFAP
To demonstrate directly that the plaque-like structures associated with FITC-OVA and FITC-cadaverine in infected Tg44+/+ mice were PrPres plaques, staining for PrPres was done (Figure 5). At 30 min after injection of FITC-OVA the tracer was co-localized with PrPres in many perivascular areas (Figure 5A–C). However, in some PrPres areas, the tracer was not spread through the entire plaque, but instead was concentrated between the plaque and the vessel wall (Figure 5D–I). Anti-PrP staining of brain from infected Tg44+/+ mice 30 minutes post-inoculation with FITC-cadaverine showed similar staining of PrPres plaques, and FITC-cadaverine staining was visible on vessels within the plaques (Figure 5J–L). The eventual clearance of both OVA and cadaverine tracers at later times (Figures 1,2,3) indicated that accumulation of tracers at sites of perivascular PrPres plaques was transient, suggesting a perturbation of the ISF function in these areas by the amyloid form of PrPres in infected Tg44+/+ mice.
Figure 5.

Detection of FITC-OVA (green) and PrPres (red) at 30 min post-microinjection in scrapie-infected Tg44+/+ mice by confocal (A–I) and epifluorescent (J–L) microscopy. Panels show optical sections of 0.5 micron thickness. (A) FITC-OVA tracer in both perivascular (arrowhead) and plaque-like (arrows) distributions. (B) PrPres immunostaining with monoclonal antibody D13 in same section shows perivascular PrPres and a large PrPres plaque. (C) Merge of panels (A) and (B) shows partial co-localization of FITC-OVA with PrPres (yellow). (D) FITC-OVA distributed along the basement membrane of a blood vessel (arrow). Fine deposits of tracer are also seen outside the BM (arrowheads). Smaller capillaries associated with FITC-OVA are also visible in the background. (E) Immunodetection of perivascular PrPres in same area. The blood vessel itself appears as a dark shape surrounded by red PrPres staining. In this section the plaque is less dense than in panel (B). (F) Merge shows minimal co-localization of FITC-OVA and PrPres. DAPI channel was included in the merge, and this blue nuclear staining reveals FITC-OVA (green) ablumenal to the blue stained endothelial cell nuclei in the vessel (arrows). (G) In a different optical section of the same field, FITC-OVA accumulation is seen around vessels (arrowhead) and along BM (arrow). (H) PrPres in perivascular distribution around the same vessels (arrowheads). (I) Merge shows partial co-localization (yellow) of PrPres and FITC-OVA in perivascular and BM areas (arrows). (J,K,L) Detection of FITC-CDV (green) and PrPres (red) on vessels within plaques of infected Tg44+/+ mouse at 30 min post-injection. (J) Tracer is concentrated on vessel walls (arrows), but faint staining of larger plaques is also visible (arrowhead). (K) Immunostaining of PrPres plaques in same section. (L) Merge of previous panels shows FITC-CDV on vessels (green arrows) within PrPres plaques in same section. Scale bars: panels (A–C), 50 μm; panels (D–I), 30 μm; panels (J–L) 100 μm.
In previous studies of infected Tg44+/+ mice, microglia and astroglia were often detected in close proximity to PrP plaques [18]. However, in co-staining experiments in the present studies using FITC-OVA plus immunostaining with astroglial and microglial markers, significant tracer co-localization with astroglia and microglia was not detected in uninfected or infected Tg44+/+ mice (data not shown).
To confirm that the pathway of clearance of FITC-OVA was in the ISF drainage path near the BM of blood vessels, co-staining experiments were done with antibodies to laminin, an important component of BM which has been previously been identified with the pathway of clearance of solutes from the ISF [11]. In uninfected Tg44+/+ mice FITC-OVA was localized near laminin-stained BM at 30 min after tracer injection (Figure 6A–C). In infected Tg44+/+ mice with PrPres amyloid the FITC-OVA tracer was also localized to laminin-stained vascular BMs, indicating that the tracer appeared to be following the ISF drainage system, even though clearance was slower than normal. In addition, tracer was localized to PrPres plaques lacking laminin (Figure 6D–F), which might account for the delay observed in tracer clearance in infected Tg44+/+ mice.
Figure 6.
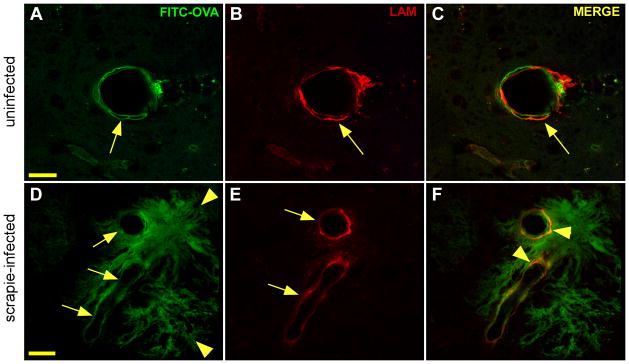
Detection of laminin and FITC-OVA by confocal microscopy in uninfected (A,B,C) and infected (D,E,F) Tg44+/+ mice at 30 min post-microinjection of FITC-OVA. Panels show optical sections of 0.25 μm. (A) FITC-OVA (green) associated with large 40 μm diameter blood vessel (arrow) in uninfected mouse. Scale bar = 20 μm; panels B and C have same scale. (B) Detection of laminin (red) in double basement membrane pattern in same section as panel (A). (C) Merge shows partial co-localization of laminin and FITC-OVA (orange) in same section (arrow). (D) Infected Tg44+/+ mouse showing FITC-OVA (green) in perivascular distribution around two large vessels (arrows) and adjacent PrPres plaques (arrowheads). Scale bar = 20 μm; panels (E and F) have same scale. (E) Laminin (red) around blood vessels in same section (arrowheads). (F) Merge of same section shows partial co-localization of FITC-OVA and laminin (orange) in basement membrane areas (arrowheads).
Studies of FITC-dextran tracer in uninfected and infected Tg44+/+ mice
In order to show that the properties seen for FITC-OVA in Tg44+/+ mice were not unique to the OVA protein, we did experiments with two carbohydrate tracers, FITC-dextran (70kDa and 500kDa). After stereotaxic injection of 500 kDa FITC-dextran, the tracer was not observed to be cleared by the ISF system at times from 30 min to 24 h in infected or uninfected mice (data not shown). In contrast, after injection of 70 kDa FITC-dextran, tracer clearance kinetics in uninfected and infected Tg44+/+ mice was similar to what was seen after injection of FITC-OVA (Figure 1). In PrPres co-staining studies of infected Tg44+/+ mice at 30 min post-injection, FITC-dextran was associated with capillary walls in the center of perivascular PrPres plaques (data not shown). Thus, FITC-OVA, FITC-cadaverine and FITC-dextran (70 kDa) were all transiently associated with PrPres amyloid, but were ultimately cleared from the brain by the ISF system. The lack of clearance of 500kDa FITC-dextran by the ISF was probably due to its large size, and was in agreement with previous reports by others [13].
Discussion
In our present experiments ISF tracers including ovalbumin, cadaverine and 70kDa dextran showed transient association with perivascular PrPres amyloid plaques in scrapie-infected Tg44+/+ mice, and clearance of the tracers was delayed compared to uninfected Tg44+/+ mice. Interestingly, tracer clearance was not delayed in scrapie-infected C57BL/10 mice where PrPres was deposited in a non-amyloid diffuse pattern mainly in extracellular sites throughout the neuropil. Similar results were seen in uninfected C57BL/10 mice and Tg44+/+ mice. Thus the presence of perivascular amyloid PrPres and the associated cerebral amyloid angiopathy (CAA) [18] strongly influenced the function of the brain ISF drainage system in the vicinity of PrPres plaques, whereas extracellular non-perivascular aggregated PrPres, which was not in an amyloid form, did not detectably influence ISF drainage. Based on these results, restriction of ISF drainage might be an important component of brain pathology in systems where perivascular amyloid PrPres or other molecules lead to CAA. However, such a mechanism would appear to be unlikely to play a role in the pathogenesis of typical prion diseases where non-amyloid PrPres is the predominant form observed.
Drainage of ISF solutes was also recently studied in the Tg2576 mouse Alzheimer’s disease model where parenchymal and perivascular deposition of Aβ amyloid is seen at 9 months of age and continues to progress with age [17]. In Tg2576mice 22 months of age, at 15 minutes post-injection of 10 kDa FITC-dextran, tracer was detected in a patchy and fragmented pattern on blood vessel walls and also in association with parenchymal amyloid deposits. These observations were similar to our present results; however, in the Tg2576 model only one time-point was analyzed, so it was unclear whether there was a delay in tracer drainage, such as we saw, or merely co-localization with amyloid and an irregular tracer distribution on vessels with amyloid.
The reasons for the different effects seen on ISF drainage in our amyloid and non-amyloid scrapie model systems are not clear at this time. Possibly the amyloid form of PrPres creates a denser filtration barrier due to the presence of numerous overlapping amyloid fibrils, which can be seen by electron microscopy [23–25]. In contrast, although the non-amyloid PrPres is known to consist of large aggregates when analyzed by ultracentrifugation, fibrils are only rarely seen by electron microscopy [24–26]. Thus these PrPres aggregates might be smaller than amyloid fibrils and might not create a barrier to the movement of molecules the size of ovalbumin or cadaverine in the ISF. In addition, the different locations of the PrPres deposits in these two models might also be important. Although most of the PrPres is extracellular in both models, in the amyloid model the PrPres is mostly perivascular and is located at the point where the ISF joins the capillary BMs where it might easily create a bottleneck to the ISF drainage. In contrast, in the non-amyloid model the PrPres is primarily perineuronal, located on the neuronal plasmalemma protruding into adjacent extracellular spaces. While such PrPres might cause localized changes to the extracellular space, in our studies there appeared to be no impedance to ISF drainage, suggesting that either the size of the PrPres aggregates was too small to block these spaces or that such blocks at this location might be circumvented by collateral flow pathways. Thus both the perivascular location and the amyloid form of the PrPres in the Tg44+/+ mice might account for the delay in ISF drainage observed.
When we compared FITC-OVA (45 kDa) with a smaller tracer, FITC-cadaverine (653 Da), there was a delay in clearance of both tracers in infected Tg44+/+ mice, but this delay was shorter with FITC-cadaverine than that seen with FITC-OVA. Thus molecular size appeared to be an important factor influencing the magnitude of delay seen. However, the delay in the clearance of the FITC-cadaverine tracer suggested that in addition to partial blockage of large molecules in the ISF, there was also a partial block of the flow of smaller solute molecules. Both these possibilities would be expected to have negative consequences for CNS function in the regions of PrPres plaque deposits, which would be consistent with extensive neuronal damage previously associated with PrPres plaques in Tg44+/+ mice [18].
In our studies, to better define the association of ISF tracers with BM components in the presence of PrP amyloid, immunostaining of laminin was done in infected Tg44+/+ mice shortly after microinjection of FITC-OVA. Tracer molecules were observed to accumulate near BM laminin even in vessels with abundant perivascular PrPres amyloid (Figure 6). In our previous electron microscopy studies of infected Tg44+/+ mice, BMs showed thickening with PrPres amyloid deposition in regions with abundant plaques [18]. However, thickening of laminin was difficult to substantiate in our present confocal light microscopy studies. In addition, in unpublished experiments with scrapie-infected C57BL/10 and Tg44+/+ mice, we found no evidence for blood brain barrier leakage using an Evans blue extravasation assay (data not shown). Possibly, infected Tg44+/+ mice which die at 300–400 dpi, did not survive long enough to develop late-stage CAA-associated abnormalities, such as microhemorrhages and blood brain barrier leakage, which have been seen in other mouse models [27, 28].
The scrapie pathology in Tg44+/+ mice [18] showed similarities with certain human familial prion diseases, where patients express PrP with an aberrant stop codon mutation which produces anchorless PrP [8–10]. In common with infected Tg44+/+ mice, those PrP genetic diseases have CAA pathology with perivascular amyloid PrPres plaques. In humans CAA has been associated with microvascular damage such as microhemorrhages, cerebral hypoperfusion, ischemia and edema [29–31].
In summary, based on our current and past results with the Tg44+/+ mouse model, CAA might contribute to PrP amyloid neuropathology in three ways: (1) alterations in effective ISF function, including partial blocking of fluid flow and/or filtration of macromolecules; (2) distortion of vascular BM and arterial walls by amyloid accumulation [18]; (3) displacement of normal brain cells by expanding PrPres amyloid [18]. The first two possibilities might also occur in human diseases associated with small perivascular amyloid deposits, such as Alzheimer’s disease, whereas all three possibilities might contribute to diseases with large amyloid deposits, such as familial prion diseases and genetic human amyloidosis of the CNS [10].
Acknowledgments
The authors thank Dan Long, Lori Lubke and Nancy Kurtz for histological technical support. We also thank Drs. Suzette Priola, Byron Caughey and John Portis for critical evaluation of the manuscript, Lynne Raymond for assistance in breeding the transgenic mice, Katie Phillips for assistance with the stereotactic surgeries, Jeffrey Severson for animal husbandry, Anita Mora for assistance with digital figures, and Drs. Gerald Baron and David Dorward for assistance with confocal microscopy. This research was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Abbreviations used
- PrP
prion protein
- PrPres
aggregated protease resistant prion protein
- CDV
cadaverine
- Tg44+/+
transgenic mice secreting anchorless PrP
- ISF
interstitial fluid
- BM
basement membrane
- FITC
fluorescein isothiocyanate
- CAA
cerebral amyloid angiopathy
- TSE
transmissible spongiform encephalopathy
- OVA
ovalbumin
Footnotes
Author contributions were as follows: Project was conceived by BC and AR; experiments were carried out by BR, AR and JS; paper was written by BC, AR, BR and JS.
References
- 1.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–33. [PubMed] [Google Scholar]
- 2.Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F, Haltia M, Hauw JJ, Ironside JW, Jellinger K, et al. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases) Brain Pathol. 1995;5:459–66. doi: 10.1111/j.1750-3639.1995.tb00625.x. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez L, Martin S, Begara-McGorum I, Hunter N, Houston F, Simmons M, Jeffrey M. Effects of agent strain and host genotype on PrP accumulation in the brain of sheep naturally and experimentally affected with scrapie. J Comp Pathol. 2002;126:17–29. doi: 10.1053/jcpa.2001.0516. [DOI] [PubMed] [Google Scholar]
- 4.Jeffrey M, Goodsir CM, Bruce ME, McBride PA, Fraser JR. In vivo toxicity of prion protein in murine scrapie: ultrastructural and immunogold studies. Neuropathol Appl Neurobiol. 1997;23:93–101. [PubMed] [Google Scholar]
- 5.Ghetti B, Piccardo P, Frangione B, Bugiani O, Giaccone G, Young K, Prelli F, Farlow MR, Dlouhy SR, Tagliavini F. Prion protein amyloidosis. Brain Pathol. 1996;6:127–45. doi: 10.1111/j.1750-3639.1996.tb00796.x. [DOI] [PubMed] [Google Scholar]
- 6.Piccardo P, Seiler C, Dlouhy SR, Young K, Farlow MR, Prelli F, Frangione B, Bugiani O, Tagliavini F, Ghetti B. Proteinase-K-resistant prion protein isoforms in Gerstmann- Straussler-Scheinker disease (Indiana kindred) J Neuropathol Exp Neurol. 1996;55:1157–63. doi: 10.1097/00005072-199611000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Jeffrey M, Goodsir CM, Bruce ME, McBride PA, Scott JR, Halliday WG. Infection specific prion protein (PrP) accumulates on neuronal plasmalemma in scrapie infected mice. Neurosci Lett. 1992;147:106–9. doi: 10.1016/0304-3940(92)90785-6. [DOI] [PubMed] [Google Scholar]
- 8.Ghetti B, Piccardo P, Spillantini MG, Ichimiya Y, Porro M, Perini F, Kitamoto T, Tateishi J, Seiler C, Frangione B, Bugiani O, Giaccone G, Prelli F, Goedert M, Dlouhy SR, Tagliavini F. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci U S A. 1996;93:744–8. doi: 10.1073/pnas.93.2.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jansen C, Parchi P, Capellari S, Vermeij AJ, Corrado P, Baas F, Strammiello R, van Gool WA, van Swieten JC, Rozemuller AJ. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol. 2010;119:189– 97. doi: 10.1007/s00401-009-0609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Revesz T, Holton JL, Lashley T, Plant G, Frangione B, Rostagno A, Ghiso J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009;118:115–30. doi: 10.1007/s00401-009-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 2009;117:1–14. doi: 10.1007/s00401-008-0457-0. [DOI] [PubMed] [Google Scholar]
- 12.Abbott NJ. Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int. 2004;45:545–52. doi: 10.1016/j.neuint.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, Weller RO. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2008;34:131–44. doi: 10.1111/j.1365-2990.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 14.Nicoll JA, Yamada M, Frackowiak J, Mazur-Kolecka B, Weller RO. Cerebral amyloid angiopathy plays a direct role in the pathogenesisof Alzheimer’s disease. Pro-CAA position statement. Neurobiol Aging. 2004;25:589–97. doi: 10.1016/j.neurobiolaging.2004.02.003. discussion 603–4. [DOI] [PubMed] [Google Scholar]
- 15.Weller RO. Pathology of cerebrospinal fluid and interstitial fluid of the CNS: significance for Alzheimer disease, prion disorders and multiple sclerosis. J Neuropathol Exp Neurol. 1998;57:885–94. doi: 10.1097/00005072-199810000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Weller RO, Boche D, Nicoll JA. Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s disease and their potential impact on therapy. Acta Neuropathol. 2009;118:87–102. doi: 10.1007/s00401-009-0498-z. [DOI] [PubMed] [Google Scholar]
- 17.Hawkes CA, Hartig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, Carare RO. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011;121:431–43. doi: 10.1007/s00401-011-0801-7. [DOI] [PubMed] [Google Scholar]
- 18.Chesebro B, Race B, Meade-White K, Lacasse R, Race R, Klingeborn M, Striebel J, Dorward D, McGovern G, Jeffrey M. Fatal transmissible amyloidencephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog. 2010;6:e1000800. doi: 10.1371/journal.ppat.1000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–9. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- 20.Ekblom P, Miettinen M, Rapola J, Foidart JM. Demonstration of laminin, a basement membrane glycoprotein, in routinely processed formalin-fixed human tissues. Histochemistry. 1982;75:301–7. doi: 10.1007/BF00496733. [DOI] [PubMed] [Google Scholar]
- 21.Matsunaga Y, Peretz D, Williamson A, Burton D, Mehlhorn I, Groth D, Cohen FE, Prusiner SB, Baldwin MA. Cryptic epitopes in N-terminally truncated prion protein are exposed in the full-length molecule: dependence of conformation on pH. Proteins. 2001;44:110–8. doi: 10.1002/prot.1077. [DOI] [PubMed] [Google Scholar]
- 22.Klingeborn M, Race B, Meade-White KD, Rosenke R, Striebel JF, Chesebro B. Crucial role for prion protein membrane anchoring in the neuroinvasion and neural spread of prion infection. J Virol. 2011;85:1484–94. doi: 10.1128/JVI.02167-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeffrey M, Goodsir CM, Bruce ME, McBride PA, Farquhar C. Morphogenesis of amyloid plaques in 87V murine scrapie. Neuropathol Appl Neurobiol. 1994;20:535–42. doi: 10.1111/j.1365-2990.1994.tb01007.x. [DOI] [PubMed] [Google Scholar]
- 24.Jeffrey M, Goodsir CM, Fowler N, Hope J, Bruce ME, McBride PA. Ultrastructural immuno- localization of synthetic prion protein peptide antibodies in 87V murine scrapie. Neurodegeneration. 1996;5:101–9. doi: 10.1006/neur.1996.0014. [DOI] [PubMed] [Google Scholar]
- 25.Jeffrey M, McGovern G, Siso S, Gonzalez L. Cellular and sub-cellular pathology of animal prion diseases: relationship between morphological changes, accumulation of abnormal prion protein and clinical disease. Acta Neuropathol. 2011;121:113–34. doi: 10.1007/s00401-010-0700-3. [DOI] [PubMed] [Google Scholar]
- 26.Jeffrey M, Goodsir CM, Bruce ME, McBride PA, Fowler N, Scott JR. Murine scrapie-infected neurons in vivo release excess prion protein into the extracellular space. Neurosci Lett. 1994;174:39–42. doi: 10.1016/0304-3940(94)90113-9. [DOI] [PubMed] [Google Scholar]
- 27.Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Sturchler-Pierrat C, Burki K, van Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–60. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- 28.Popa-Wagner A, Pirici D, Petcu EB, Mogoanta L, Buga AM, Rosen CL, Leon R, Huber J. Pathophysiology of the vascular wall and its relevance for cerebrovascular disorders in aged rodents. Curr Neurovasc Res. 2010;7:251–67. doi: 10.2174/156720210792231813. [DOI] [PubMed] [Google Scholar]
- 29.Breteler MM. Vascular risk factors for Alzheimer’s disease: an epidemiologic perspective. Neurobiol Aging. 2000;21:153–60. doi: 10.1016/s0197-4580(99)00110-4. [DOI] [PubMed] [Google Scholar]
- 30.Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm. 2002;109:813–36. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- 31.Chung YA, OJH, Kim JY, Kim KJ, Ahn KJ. Hypoperfusion and ischemia in cerebral amyloid angiopathy documented by 99mTc-ECD brain perfusion SPECT. J NuclMed. 2009;50:1969–74. doi: 10.2967/jnumed.109.062315. [DOI] [PubMed] [Google Scholar]