

Abstract
A number of histone-binding domains are implicated in cancer through improper binding of chromatin. In a clinically reported case of acute myeloid leukemia (AML), a genetic fusion protein between nucleoporin 98 and the third plant homeodomain (PHD) finger of JARID1A drives an oncogenic transcriptional program that is dependent on histone binding by the PHD finger. By exploiting the requirement for chromatin binding in oncogenesis, therapeutics targeting histone readers may represent a new paradigm in drug development. In this study, we developed a novel small molecule screening strategy that utilizes HaloTag technology to identify several small molecules that disrupt binding of the JARID1A PHD finger to histone peptides. Small molecule inhibitors were validated biochemically through affinity pull downs, fluorescence polarization, and histone reader specificity studies. One compound was modified through medicinal chemistry to improve its potency while retaining histone reader selectivity. Molecular modeling and site-directed mutagenesis of JARID1A PHD3 provided insights into the biochemical basis of competitive inhibition.
In the nucleus of eukaryotes, genes are organized and compacted into chromatin, which is achieved in part through the wrapping of DNA around histone proteins (1). Histones are enriched for sites of posttranslational modification, which include methylation, acetylation, and phosphorylation (2). Histone modifications both influence chromatin structure and provide ligands for protein domains that recruit gene regulatory complexes to specific loci within the genome (3). These histone “reader” domains are specialized for certain histone modifications. For example, subsets of the PHD finger domain class bind unmodified, methylated, or acetylated lysine side chains (4). These domains generally bind their preferred modification with a high degree of specificity; for example, the PHD finger of ING2 binds histone H3 trimethylated at lysine 4 (H3K4me3), with markedly reduced affinity as the methylation state decreases, and almost negligible binding to the unmodified side chain (5). In addition to PHD fingers, there are other classes of histone binding domains, including bromodomains, chromodomains, tudor and tandem tudor domains, and 14-3-3 domains (6). Altogether, there are hundreds of histone reader domains, which contribute to exquisite control over gene expression.
When misregulated, a number of histone-binding domains are linked to disease, including cancer, autoimmune and developmental conditions (7). For example, the third PHD finger of JARID1A (JARID1A PHD3), which binds H3K4me3, is implicated in acute myeloid leukemia (AML) (8). In a clinically reported case of AML, the patient expressed a genetic fusion protein containing nucleoporin protein 98 (NUP98) and the C-terminus of JARID1A, which includes its nuclear localization sequence and third PHD finger. A similar fusion protein was reported between NUP98 and the PHD finger of PHF23 in another AML patient (9). Later studies deduced that these genetic fusions caused aberrant transactivation of developmental genes required to maintain the myeloid progenitor state, which resulted in the onset of leukemia (10). The oncogenic properties of the NUP98-PHD finger fusion proteins are directly potentiated by the ability of the PHD finger to bind chromatin. Other examples of histone-binding proteins implicated in cancer include the overexpression of UHRF1 in lung cancer and TRIM24 in breast cancer (11, 12). Because of the role of JARID1A PHD3 and other histone readers in disease, identifying small molecules that inhibit histone binding by these domains is of paramount importance. We predict that epigenetic drugs targeting histone-binding domains represent a new paradigm for the development of cancer therapeutics, which has only recently begun to be explored (13, 14).
To target histone readers for small molecule inhibition, we developed a 96-well plate assay that uses a HaloTag fusion to the third PHD finger of JARID1A. HaloTag is a 34 kDa protein fusion tag that forms a specific covalent bond with its synthetic HaloTag ligand (15). HaloTag ligands can be attached to variety of surfaces to allow specific, irreversible, and oriented immobilization of a protein of interest fused to HaloTag (16–18). These features maximize functionality of the protein of interest while allowing stringent washing conditions. To leverage the advantages of HaloTag technology for small molecule screening of JARID1A PHD3, we developed an assay that uses 96-well polystyrene plates activated with HaloTag ligand for covalent and oriented capture of a HaloTag fusion to JARID1A PHD3. Small molecule inhibitors identified from screening were validated biochemically. Specificity studies allowed us to inform chemical modification of one hit compound to increase its potency as an inhibitor of JARID1A PHD3. Predictions made from molecular modeling allowed identification and biochemical analysis of residues within JARID1A PHD3 that contribute to competitive inhibitor binding.
EXPERIMENTAL PROCEDURES
General reagents
Dimethyl sulfoxide (DMSO), tetraethylthiuram disulfide (disulfiram), phenothiazine, and amiodarone HCl were purchased from Sigma Aldrich. Tegaserod maleate, di-N-desethylamiodarone, and desethylamiodarone were purchased from Santa Cruz Biotechnology. TMR HaloTag ligand was purchased from Promega. SuperSignal West Pico ELISA chemiluminescent substrate, high sensitivity streptavidin-HRP conjugate, and high capacity streptavidin-agarose beads were purchased from Pierce. StabilCoat buffer was purchased from Surmodics. AlphaScreen histidine detection kits (nickel chelate) and ½-area white 96-well plates were purchased from Perkin Elmer. Ni-NTA and glutathione sepharose 4B resins were purchased from GE Life Sciences.
Plasmids
Plasmids for the GST fusions of JARID1A PHD3 (1601–1660), AIRE PHD1 (293–354), ING2 PHD (201–281), BHC80 PHD (486–543), RAG2 PHD (414–487), and JMJD2A double tudor domain (895–1011) were kindly provided by D. Allis (Rockefeller Institute), G. Musco (Dulbecco Telethon Institute), T. Kutateladze (University of Colorado-Denver), Y. Shi (Harvard Medical School), W. Yang (US National Institutes of Health), and R.M. Xu (New York University), respectively. The (HQ)5-HaloTag N-terminal vector was kindly supplied by Dr. Mike Slater and Dr. Jim Hartnett of Promega Corporation (Madison, WI).
Peptide Synthesis
Histone H3 (1–11) peptides were synthesized using solid phase synthesis on the Intavis ResPep SL robot (Intavis, Koeln, Germany). Standard Fmoc/tBu amino acid couplings were used and premodified amino acid building blocks Kme3 and EPEG-biotin (Novabiochem) were incorporated into positions 4 and 12, respectively, depending on the peptide. Binding peptides contained EPEG-biotin at position 12 and “unlabeled” competing peptides were unmodified at this position. All peptides (except for fluorescein-labeled species) contained a C-terminal tryptophan for concentration determination by absorbance at 280 nm. Peptide mass was confirmed by MALDI and crude products purified by preparative HPLC (Beckman-Coulter).
Histone H3 (1–14) peptides were synthesized as above except for substitution of K(ivDde) (Novabiochem) at position 14. Following synthesis, this lysine was deprotected with 4% v/v hydrazine and labeled with 5-carboxyfluorescein (Novabiochem), followed by standard deprotection and purification.
Protein expression and purification
JARID1A PHD3 (1601–1660) was amplified from a GST plasmid and cloned into a bacterial expression vector encoding an N-terminal (HQ)5–HaloTag followed by a TEV site. Recombinant HQ5-HaloTag-JARID1A PHD3 was expressed in BL21DE3 pLysS cells under kanamycin and chloramphenicol resistance using a 3-hour ambient temperature induction period under 0.5 mM IPTG. Cell pellets were frozen at −80°C and thawed in 30 mM HEPES, 150 mM NaCl, pH 7.4 (HBS) supplemented with 0.5 mM TCEP, 0.01% v/v Triton X-100, 1 mM PMSF, 10 μg/mL leupeptin, 10 μg/mL aprotinin, and 1 mg/mL lysozyme, followed by two cycles of freeze-thawing and light sonication to lyse cells (5 s 20% amplitude, 5 s off, 1 minute total per sonication cycle, ¼″ probe, Thermo Fisher). Following clarification by centrifugation, the lysate supernatant was bound to Ni-NTA resin in batch for 2 hours at 4°C, washed in batch with HBS and HBS containing 20 mM imidazole, pH 7.4, followed by batch elution with 0.3 M imidazole in HBS, pH 7.4. Elutions were dialyzed in 2 steps into HBS supplemented with 3 mM DTT and glycerol up to 10% v/v. Protein was concentrated in 30 kDa MWCO amicons (Millipore), supplemented with 0.5 mM TCEP, quantitated by Bradford, and stored at −80°C. (HQ)5-HaloTag-AIRE PHD1, RAG2 PHD, BHC80 PHD, and JMJD2A double tudor domain were cloned, expressed, and purified in a similar manner.
His6-JARID1A PHD3 was cloned from the HQ-HaloTag vector into a pQE80L ampicillin-resistance vector and expressed in BL21DE3 pLysS cells. Expression and purification was performed as above except for the use of 3 kDa MWCO amicons (Millipore).
GST fusions to histone reader domains were expressed, lysed, and clarified as described for (HQ)5-HaloTag fusion proteins. The lysate supernatant was bound to glutathione sepharose 4B resin for 2 hours at 4°C, followed by washing in batch with HBS – 0.01% v/v Triton X-100 and elution with 25 mM glutathione in HBS. Elutions were dialyzed as above and protein concentrated in 10 kDa amicons.
HaloTag ligand 96-well plates
HaloTag plates were prepared at Promega by reacting an amine terminated long chain pegylated HaloTag ligand with amine reactive 96 well plates. HaloTag plates were made using white, black or clear base plates for chemiluminescence, fluorescence and absorbance readout respectively. These plates are available from Promega on special request (E.mail: cod@promega.com).
A sandwich immunoassay was used to determine the inter- and intra-plate sensitivity and reproducibility of the HaloTag plates. A dilution series of HaloTag-GST fusion protein (Promega) in PBS + 10 mg/mL BSA was made and added in triplicate to the HaloTag plate (50 μL/well). After a 1-hour incubation, the plate was washed and bound fusion protein detected by a sequential 1-hour incubation with anti HaloTag antibody and anti rabbit antibody conjugated to HRP. Colorimetric substrate for HRP was added and absorbance read on a plate reader at 450 nm. All binding steps occurred at ambient temperature. Microplates were sealed during binding steps to prevent evaporation. Results of these studies indicated an LOD of 10 ng/mL and excellent intra-plate reproducibility on clear HaloTag plates. Inter-plate reproducibility was determined using a similar assay on a clear, black and white plate (result not shown). For white and black plates, the substrate was transferred to a clear plate and absorbance read at 450 nm.
HaloTag assay for histone peptide binding
HaloTag fusion protein and peptide were diluted into StabilCoat buffer supplemented with 0.5 mM TCEP. High sensitivity streptavidin-HRP was diluted into SA-AP buffer (30 mM Tris, pH 7.6, 1 M NaCl, 20 mM K/PO4). All steps occurred at ambient temperature, with the microplate sealed to prevent evaporation.
The wells of a 96-well HaloLink microplate were rinsed with 0.2 mL 30 mM HEPES, 150 mM NaCl, pH 7.4, 0.01% v/v Triton X-100 (HBST). (HQ)5-HaloTag-JARID1A PHD3 (50 μL 40 μM) was added to each well and the plate shaken at 200–300 rpm for 2 hours at ambient temperature. Wells were aspirated and washed 3 times with 0.1 mL HBST. Biotinylated histone peptide (50 μL 250 nM) was added to each well and the plate shaken at 200–300 rpm for 1 hour. Wells were aspirated and washed 3 times with 0.1 mL HBST. Streptavidin-HRP (50 μL, 1:2,000 dilution) was added to each well and the plate shaken for 20 minutes at 300 rpm. Wells were aspirated and washed 6 times with 0.1 mL HBST. Immediately before detection, 0.1 mL chemiluminescent substrate was added to each well. Followed by 30 seconds shaking at slow speed, luminescence was measured (1 s integration time) on a Biotek Synergy™ H4 multimode plate reader. The initial concentration matrix assay was detected using a Tecan GENios Pro plate reader. These conditions were used for all (HQ)5-HaloTag-reader domain fusions except BHC80 PHD (1 μM biotinylated histone peptide).
IC50 values for the HaloTag assay, and all other peptide binding assays, were calculated by fitting relative binding/fraction bound versus log[Inhibitor] curves to the equation provided by GraphPad Prism curve fitting software:
| (1) |
where Fb is fraction bound/relative binding and [I] is inhibitor/competitor concentration. Z′ factor is calculated by the following equation:
| (2) |
where μ is the mean of the positive (p) and negative (n) controls, and σ is the standard deviation of the positive and negative controls (19).
AlphaScreen histone binding assay
Protein, peptides, and AlphaScreen beads were diluted into StabilCoat buffer supplemented with 0.5 mM TCEP. First, 13.5 μL 100 nM (HQ)5-HaloTag-reader domain followed by 13.5 μL 150 nM biotinylated histone H3K4 +/− me3 peptide, was added to the wells of a half-area white 96-well plate. The final concentration of protein and peptide was 50 and 75 nM, respectively. The sealed plate was shaken at ambient temperature for 30 minutes at 220 rpm. AlphaScreen streptavidin donor and nickel chelate acceptor beads were added to a final concentration of 15 μg/mL (3 μL 150 μg/mL to protein/peptide mixture) and the sealed plate shaken for another 20 minutes at 220 rpm at ambient temperature. Fluorescence was measured on a Biotek Synergy H4 multimode plate reader following a 5 minute delay, using a 680/30 nm excitation/570/100 nm emission filter set with a 635 nm top mirror, sensitivity set to 190. Filter switching to wheel plugs after excitation and once again before emission introduced a necessary time delay for measurement.
Small molecule screening
Small molecule screening was performed at the University of Wisconsin Carbone Cancer Center Small Molecule Screening Facility. The HaloTag assay was performed as above except for the addition of 1 μL 10 mM compounds dissolved in DMSO from the NIH Clinical Collection I by a Matrix Hydra liquid handler (Thermo-Fisher) immediately after addition of biotinylated peptide solution, for a final concentration of 200 μM compound. Wash buffer was dispensed by the Matrix Hydra, and all other additions were performed by multichannel pipetting. Screening was performed over 2 independent experiments with each plate containing DMSO controls for H3K4me3 peptide binding, unmethylated H3 peptide binding and 7.5 μM unlabeled H3K4me3 peptide competition.
Secondary screens, concentration dependence, and counterscreen studies
The HaloTag assay and AlphaScreen assays were performed as above, except for the addition of 1 μL compound dissolved in DMSO to each well immediately after addition of biotinylated peptide. Compounds were assayed at final concentrations between 0 and 200 μM, 2–3% v/v DMSO.
Affinity pull downs
Streptavidin-agarose beads were equilibrated in HBST, followed by immobilization of biotinylated histone peptides in 1% BSA (w/v)-HBST for 30 minutes in batch. In all experiments except amiodarone HCl and phenothiazine, approximately 300 pmol of biotinylated peptide per 6.6 μL bed volume resin was immobilized. Approximately 50 pmol biotinylated peptide was immobilized for studies on amiodarone HCl and phenothiazine due to the poor solubility of these compounds in order to maximize the sensitivity of the assay. Following peptide immobilization, beads were washed three times with 0.5 mL HBST followed by centrifugation for 1 minute at 800 x g, and resuspended in 1% BSA-HBST to form a 50% slurry. (HQ)5-HaloTag-JARID1A PHD3 labeled with TMR HaloTag ligand (10 μM PHD3 labeled with 10 μM TMR ligand in 1% BSA-HBST for 15 minutes at ambient temperature, covered from light) was dispensed to 0.6-mL microcentrifuge tubes, followed by 13.2 μL peptide-linked beads (6.6 μL bed volume) for a final concentration of 100 nM TMR-(HQ)5-HaloTag-JARID1A PHD3 in 1% BSA-HBST. Compounds at varying concentrations (0–500 μM) were added, with a final concentration of 5% v/v DMSO in all samples. Reactions were protected from light and mixed for 45 minutes at ambient temperature, followed by centrifugation and three 0.5-mL HBST washes. Beads were spun down and all remaining liquid aspirated. Beads were boiled in 20 μL SDS-sample buffer for 5–10 minutes before separation by SDS-PAGE (12%, 200 V). Gels were scanned on a Typhoon FLA9000 using the TAMRA setting at 100 μm resolution.
Fluorescence polarization binding studies
Increasing concentrations of compounds were added to solutions containing 1 μM GST-JARID1A PHD3 and 3 nM H3(1–14)K4me3K14fluo in 30 mM HEPES, 150 mM NaCl, pH 7.4, 0.01% v/v Triton X-100. Each condition was measured in triplicate in a black 384-well plate on a Biotek Synergy H4 multimode plate reader. Counterscreen studies utilized 4 μM GST-AIRE PHD1, 4 μM GST-JMJD2A DTD, 18 μM GST-UHRF1 TTD, 25 μM GST-RAG2 PHD, 60 μM GST-BHC80 PHD. For AIRE PHD1 and BHC80 PHD, 3 nM H3(1–14) K4unmodifiedK14fluo peptide was utilized. For UHRF1 TTD, 3 nM H3(1–19)K9me3K19fluo was utilized. The concentrations of protein for each GST fusion represent 90% fraction bound. From polarization values, fraction bound was calculated by
| (3) |
where Pobs is the observed polarization at a given concentration of compound, Pf is the polarization of free peptide probe, and Pb is the polarization of maximally bound peptide probe (20). Dissociation constants (Kd) were calculated using the equation:
| (4) |
where Fb is fraction bound ((21).
Zinc ejection studies (PAR assay)
Zinc released was monitored through the colorimetric reagent, 4-(2-pyridylazo) resorcinol (PAR). In a final volume of 300 μL, 5 μM His6-JARID1A PHD3 or (HQ)5-HaloTag-reader domain was treated with increasing concentrations of disulfiram, methyl methanethiosulfonate, tegaserod maleate, or amiodarone HCl in a solution of 10 mM PAR in 30 mM HEPES, 150 mM NaCl, pH 7.4, 5% v/v DMSO (includes DMSO from compounds). 90 μL of each solution was added in triplicate to a clear 96-well plate. The plate was shaken for 10 minutes; alternatively, 1 DMSO-only sample was boiled during this time to monitor zinc released due to structural destabilization. Absorbance at 500 nm was read on a Biotek Synergy H4 multimode plate reader. DMSO-only absorbance values were subtracted from each sample.
Molecular modeling
Protein structures were downloaded from RCSB PDB (2KGI, JARID1A PHD3; 2GFA, JMJD2A DTD; 2G6Q, ING2 PHD). Small molecule ligands were prepared using Sybyl (Tripos). Ligands were docked “blindly” onto protein receptors using the AutoGrid function within AutoDock 4, the entire histone-binding domain or lobe (in the case of JMJD2A) was accommodated within the grid.
Site directed mutagenesis of JARID1A PHD3 and Schild analysis
Aspartate residues within a GST fusion to JARID1A PHD3 at positions 1624 and 1629 were each mutated to alanine and asparagine, and tryptophan 1625 to alanine, using QuikChange Site-Directed Mutagenesis II reagents. Dissociation constants for each mutant were determined by fluorescence polarization, where increasing concentrations of GST fusion were titrated against 3 nM H3(1–14)K14fluo peptide, unmodified or trimethylated at lysine 4. EC50 values for each clone were determined in a similar manner, using fixed concentrations of either di-N-desethylamiodarone or tegaserod maleate (22). In these analyses, increasing concentrations of GST fusions to JARID1A PHD3 clones were titrated against solutions containing a fixed concentration of inhibitor and H3K4me3 probe. The fold change in EC50 with a fixed concentration of inhibitor relative to the EC50 in the absence of inhibitor is referred to as the dose ratio (22). A dose ratio greater than one suggests that the inhibitor is able to interact with JARID1A PHD3, causing a shift in apparent Kd for the H3K4me3 peptide. In contrast, a dose ratio equal to one indicates that the inhibitor does not interact with JARID1A PHD3 and consequently does not influence its ability to interact with the H3K4me3 probe.
RESULTS
Development of the HaloTag assay
To develop a small molecule screening assay for histone readers, we harnessed the properties of HaloTag. The schematic of the HaloTag assay for screening of small molecule inhibitors of binding of JARID1A PHD3 with histone peptide is shown in Figure 1A. An N-terminal (HQ)5-HaloTag fusion to JARID1A PHD3 is covalently immobilized to the wells of a HaloTag ligand-coated 96-well plate. JARID1A PHD3 then binds its ligand, a biotinylated histone H3 peptide trimethylated at lysine 4. The protein-ligand interaction is detected through a chemiluminescent reaction catalyzed by streptavidin-conjugated horseradish peroxidase (HRP), which binds the biotinylated peptide. The assay offers a number of advantages for assessing small molecule inhibitors. Compound interference is reduced through use of stringent washes and chemiluminescent detection. By utilizing the properties of HaloTag and the biotin-streptavidin interaction, the assay effectively isolates the protein-ligand binding step for small molecule inhibition. The HaloTag assay also achieves an exquisite degree of sensitivity since only femtomoles of streptavidin-HRP conjugate are required for sufficient signal to noise (Figure S1a). This property allowed interrogation of low micromolar affinity interactions with sub-picomole quantities of ligand, despite rigorous washing throughout the assay. Other groups have reported similar detection capabilities with ligand-binding assays (23).
Figure 1. HaloTag assay design and optimization.

A. HaloTag assay design. First, a HaloTag fusion to JARID1A PHD3 is covalently immobilized to the surface of a chloroalkane-coated 96-well plate. Second, biotinylated histone H3 peptides (+/− trimethylation at lysine 4) are allowed to bind the immobilized PHD finger. Finally, peptide binding is detected through a streptavidin conjugate to horseradish peroxidase (HRP) that binds biotinylated peptides and emits light when chemiluminescent substrate is added.
B. Initial concentration matrix. Increasing amounts of biotinylated histone H3K4me3 peptides (0.1, 0.5, 1, 10, 15 μM, circles; 15 μM unmethylated biotinylated H3 peptide, open circles) were allowed to bind increasing concentrations of (HQ)5-HaloTag-JARID1A PHD3 (0 to 40 μM).
C. Peptide displacement. Unlabeled (no biotin) H3K4me3 peptide (circles) specifically competed away binding of biotinylated H3K4me3 peptides, while a high concentration of unmethylated H3K4 peptide (open circle) did not significantly affect binding signal.
D. Other histone-binding modules within the HaloTag assay. RAG2 PHD, AIRE PHD1, BHC80 PHD, and JMJD2A double tudor domain distinguish the methylation status H3K4 by binding their preferred ligand within the HaloTag assay. Peptide binding can be specifically inhibited via peptide competition (H3K4-unmodified peptide, white; H3K4me3, black; peptide competition, grey).
HaloTag ligand-coated microtiter (HaloLink) plates for the HaloTag assay were generated by reacting amine-reactive 96-well plates with an amine-terminated long chain-pegylated chloroalkane ligand. Capture of HaloTag fusion protein to HaloLink plate is specific and achieves a 10 ng/mL limit of detection using an anti-HaloTag antibody (Figure S1b). JARID1A PHD3 binds the H3K4me3 peptide with a reported dissociation constant of 0.75 μM and the unmethylated H3 peptide with reduced affinity of 20 μM (10). Initial experiments addressed the dynamic range of the HaloTag assay, where concentrations of HaloTag fusion to JARID1A PHD3 and biotinylated histone peptide were simultaneously varied (Figure 1b). Half-maximal binding signal was observed with 1 μM biotinylated H3K4me3 peptide, which agreed well with the reported dissociation constant of JARID1A PHD3 (10). Sub-Kd concentrations of biotinylated histone peptide maximized the binding range between the methylated and unmethylated histone peptides while minimizing well-to-well error (Figure S1c)
Peptide binding within the HaloTag assay was specifically inhibited via peptide competition, yielding an IC50 value of 2.97 ± 0.35 μM for H3K4me3, while 25 μM unmethylated H3 peptide did not significantly inhibit JARID1A PHD3 (Figure 1c). The approximately 4-fold difference between the experimental IC50 and the reported Kd for H3K4me3 peptides may be due to an avidity effect caused by the high local concentration of immobilized JARID1A PHD3 (24). Competitive inhibition was unaffected by the order of addition of the competing H3K4me3 peptide, whether it was added concurrently with the biotinylated peptide or in a separate assay step following binding of the biotinylated peptide (Figure S1d).
The signal for JARID1A PHD3 binding biotinylated H3K4me3 peptide was 200-fold greater than signal observed for HaloTag alone incubated with biotinylated peptides (Figure S1e). The unmethylated histone H3 peptide served as the negative control for statistical calculations even though JARID1A PHD3 can bind it with weak affinity. Importantly, the unmethylated H3 peptide serves as an internal control for the expected binding range of JARID1A PHD3 and establishes greater stringency for statistical calculations. A 10-fold range in binding between the methylated and unmethylated peptides is typically observed within the optimized HaloTag assay, resulting in a Z′ factor of 0.65 ± 0.14 (n=18 independent experiments with JARID1A PHD3).
The HaloTag assay was readily adapted to other histone-binding proteins, including other PHD fingers (RAG2, BHC80, AIRE) and the double tudor domain (DTD) of JMJD2A (Figure 1d). RAG2 PHD and JMJD2A DTD bind H3K4me3 (25, 26). AIRE PHD1 and BHC80 PHD bind the unmodified form of lysine 4 on histone H3 (27, 28). All domains tested demonstrated specificity in binding their preferred form of H3K4 and sensitivity to peptide competition (Figure 1d). In some cases, increasing the concentration of biotinylated peptide to 1–2 μM for low affinity reader domains (e.g. BHC80 PHD, where Kd > 15 μM) improved assay signal.
Small molecule screening
The NIH Clinical Collection 1 was screened against JARID1A PHD3 in the HaloTag assay. It contains 446 compounds, all of which have undergone phase I–III clinical trials, and encompasses a broad range of therapeutic indications (29). The biosafety and bioavailability profiles of compounds within this collection are highly characterized, thus making hits identified from screening candidates for drug repositioning (30). Compounds were screened at 200 μM and plate-wide controls generated an average Z′ factor of 0.60. Raw screening data are shown sorted by relative binding, where the luminescence counts for the compound-treated well are divided by the luminescence counts for the DMSO-treated H3K4me3-binding control (Figure 2a). Compounds were selected for additional validation if they reduced relative binding by at least three standards of deviation (as calculated from the DMSO-treated H3K4me3-binding controls for individual plates).
Figure 2. Primary Screening against JARID1A PHD3 with a clinical drug library.

A. Primary screen sorted by relative binding. Compounds within the NIH Clinical Collection were screened at 200 μM against JARID1A PHD3 using the HaloTag assay.
B. HaloTag assay concentration dependence. Increasing concentrations of selected compounds (0, 50, 100, 200 μM) were added to the HaloTag binding assay. Within this assay, tegaserod maleate, amiodarone HCl, phenothiazine, and disulfiram had a significant effect (*p<0.05, **p<0.01) on JARID1A PHD3 binding to H3K4me3 peptides
C. Disulfiram, tegaserod maleate, amiodarone HCl, and phenothiazine were the only compounds identified from small molecule screening that significantly inhibited JARID1A PHD3 binding of H3K4me3 peptide within the HaloTag assay.
From primary screening, 23 compounds were selected for re-screening in triplicate with the HaloTag assay and an AlphaScreen®-based histone peptide binding assay (Perkin Elmer). Similar studies using AlphaScreen technology have been reported for a BET bromodomain and malignant brain tumor (MBT) repeat proteins, and representative binding data for JARID1A PHD3 are shown in Figure S2a (13, 14). Following secondary screening, compounds were chosen for additional validation based on their ability to reduce binding signal within both the HaloTag and AlphaScreen assays.
Concentration-dependence studies
Following small molecule screening, we evaluated compound dose dependence (0–200 μM) within the HaloTag assay. Disulfiram, phenothiazine, amiodarone, and tegaserod were the only compounds that significantly reduced signal (by p-value <0.05) (Figure 2b). The structures of these compounds are shown in Figure 2c. Before proceeding to additional studies, we confirmed that tegaserod was inhibiting JARID1A PHD3, and not maleate (Figure S2b).
Affinity pull downs were performed to detect loss of binding to histone peptides by fluorescently labeled (HQ)5-HaloTag-JARID1A PHD3. Biotinylated histone peptides were immobilized to streptavidin-conjugated agarose beads, followed by binding of tetramethylrhodamine (TMR)-labeled HaloTag-JARID1A PHD3 to histone peptides, and elution and resolution of bound protein by SDS-PAGE (Figure 3a). Competition with H3K4me3 peptide (Figure S2c) was greatly impaired within this assay (13.6 ± 4.5 μM experimental IC50 versus 0.75 μM expected Ki), so compounds were assayed at a high concentration range to detect inhibition. Only disulfiram and tegaserod maleate inhibited JARID1A PHD3 (Figure 3b). Amiodarone and phenothiazine were difficult to test by this strategy due to poor solubility (Figure S2d).
Figure 3. Dose dependence studies of hit compounds against JARID1A PHD3.

A. Affinity pull down design. Increasing concentrations of compound were added to a solution of TMR-labeled (HQ)5-HaloTag-JARID1A PHD3 binding to biotinylated histone peptides immobilized to streptavidin-agarose beads.
B. Disulfiram and tegaserod maleate only cause concentration-dependent loss of binding of JARID1A PHD3 to H3K4me3 peptides within the affinity pull down assay (representative data).
C. Tegaserod maleate only caused concentration-dependent losses in histone peptide binding within the fluorescence polarization assay.
Inhibition of histone binding was also evaluated using a fluorescence polarization binding assay, where a GST fusion to JARID1A PHD3 was allowed to bind fluorescein-labeled H3K4me3 peptide (Figure S2e). When assayed with disulfiram, tegaserod maleate, phenothiazine, and amiodarone, a concentration-dependent loss of polarization corresponding to loss of peptide binding was observed only for tegaserod, with an observed IC50 value of 74 ± 16 μM (Figure 3c). Amiodarone, disulfiram, and phenothiazine did not cause a significant loss in polarization, up to 200 μM (data not shown), consistent with weak inhibition observed in the HaloTag assay.
Next, we sought to evaluate the specificity of these compounds towards JARID1A PHD3. Disulfiram, amiodarone, and tegaserod were screened against AIRE PHD1, BHC80 PHD, RAG2 PHD, and JMJD2A DTD using the HaloTag assay. Significant (by p-value) loss of peptide binding was only observed for the PHD fingers when reader domains were treated with disulfiram, but not JMJD2A DTD (Figure 4a). This result suggested that disulfiram specifically inhibited PHD fingers. RAG2 PHD and JMJD2A DTD, but not AIRE PHD1 or BHC80 PHD, were significantly affected by amiodarone (Figure 4b). Modest reductions in binding were observed for all domains tested when treated with tegaserod, indicative of a nonspecific effect on the HaloTag assay (Figure S3a). When evaluated by fluorescence polarization, only JMJD2A DTD was inhibited by tegaserod, with an IC50 value of 95± 9.8 μM (Figure 4c). The IC50 values for RAG2 PHD, AIRE PHD1, and BHC80 PHD all exceeded the solubility limits of tegaserod.
Figure 4. Evaluating compound specificity towards other histone reader domains.
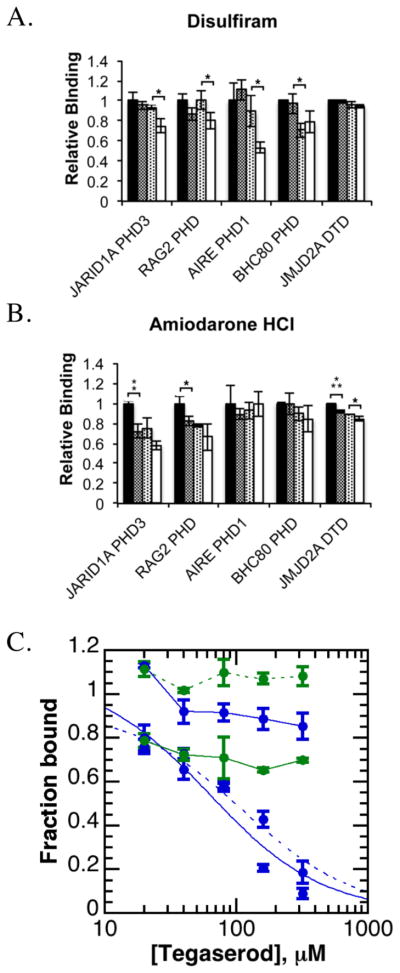
A. Disulfiram caused a concentration-dependent loss of histone peptide binding for PHD fingers in the HaloTag assay when assayed at 0, 50, 100, and 200 μM (*p<0.05). B. Amiodarone HCl caused significant (by p-value, *p<0.05, **p<0.01, ***p<0.001) reductions in histone peptide binding for only H3K4me3-binding domains in the HaloTag assay when tested at 0, 50, 100, and 200 μM.
C. Tegaserod maleate inhibited JMJD2A DTD (dotted blue curve) and JARID1A PHD3 (solid blue curve) within the fluorescence polarization assay, but not RAG2 PHD (solid blue line), ING2 PHD (dotted blue line), AIRE PHD1 (solid green line), or BHC80 (dotted green line).
Disulfiram modifies PHD fingers to cause zinc release
Disulfiram is a known cysteine alkylating agent, and was shown to eject zinc from a variety of enzymes and transcription factors (31). PHD fingers are classified by a Cys3-His-Cys4 motif used to engage two zinc ions, so rather than acting as a competitive ligand for PHD fingers, we hypothesized that disulfiram was inhibiting JARID1A PHD3 through modification of structural cysteine residues and subsequent zinc release (32). To monitor zinc release from JARID1A PHD3, the reagent 4-(2-pyridylazo) resorcinol (PAR) was added to solutions of His6-JARID1A PHD3 and increasing concentrations of disulfiram. Indeed, disulfiram ejected structural zinc from JARID1A PHD3 with a half-maximal zinc loss at 10 μM disulfiram (Figure 5a). The compound methyl methanethiosulfonate (MMTS), which methylates free cysteine side chains and also causes zinc ion ejection, resulted in a half-maximal zinc ion loss at 33 μM. Increasing concentrations of amiodarone and tegaserod added to solutions of JARID1A PHD3 and PAR reagent confirmed that these compounds were not causing zinc release through structural destabilization, suggesting that these compounds were noncovalent ligands for JARID1A PHD3 (Figure S3b).
Figure 5. Disulfiram ejects structural zinc from JARID1A PHD3.
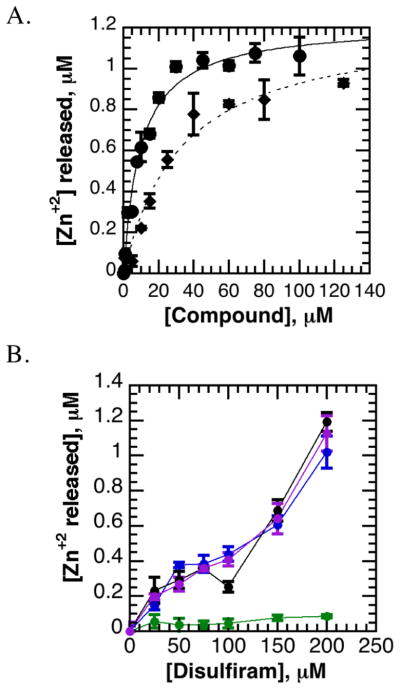
A. Disulfiram ejects zinc from JARID1A PHD3 in a dose-dependent manner. Zinc release from JARID1A PHD3 was monitored through PAR. Disulfiram (solid curve) causes zinc release at lower concentrations than methyl methanethiosulfonate (MMTS, dotted curve).
B. PHD fingers are differentially sensitive to zinc ejection by disulfiram. AIRE PHD1 (black) and BHC80 PHD (purple) were similarly susceptible to zinc loss as JARID1A PHD3 (blue) within the PAR assay, while RAG2 PHD (green) was more resilient to disulfiram.
We then investigated whether disulfiram was a general inhibitor of PHD fingers. Upon treatment with disulfiram, zinc release was measured from (HQ)5-HaloTag fusions to JARID1A PHD3, RAG2 PHD, AIRE PHD1, and BHC80 PHD (Figure 5b). AIRE PHD1 and BHC80 PHD, but not RAG2 PHD, were similarly susceptible to disulfiram-mediated zinc ejection as JARID1A PHD3. No zinc release was observed from the double tudor domain of JMJD2A, consistent with its lacking structural zinc (Figure S3c).
Chemical modification of amiodarone improves potency towards JARID1A PHD3
Histone reader specificity studies suggested that amiodarone (AMI) preferentially inhibited methyl-lysine readers. To test this hypothesis, we pursued a fragment-based approach to evaluate amiodarone (Figure 6a). Within the HaloTag assay, dronedarone, but not benzbromarone, inhibited JARID1A PHD3 (Figure S3d). These data suggested that the tertiary alkylated amine within amiodarone was an important determinant for binding inhibition. We therefore hypothesized that the alkylated amine chain of amiodarone was a ligand for the aromatic cage of JARID1A PHD3, thereby competitively inhibiting binding of H3K4me3.
Figure 6. Analogs of amiodarone inhibit JARID1A PHD3 binding of H3K4me3 more potently than amiodarone.

A. Amiodarone analogs studied included benzbromarone, dronedarone, metabolites of amiodarone, and four synthetic analogs.
B. Amiodarone analogs inhibit JARID1A PHD3 more potently than amiodarone. Of the compounds assayed, the metabolite di-N-desethylamiodarone (black curve) and the synthetic trimethyl-amiodarone analogs (blue curves; n=2 solid, n=3 dotted) inhibit JARID1A PHD3 within the fluorescence polarization assay, but not amiodarone (black, solid), desethylamiodarone (black, dotted) or dimethyl-amiodarone synthetic analogs (green; n=2 solid, n=3 dotted).
To test this hypothesis, we prepared a series of amiodarone derivatives built from 2-butyl-3-(3,5-diiodo-4-hydroxybenzoyl)benzofuran. Amiodarone is successively de-ethylated by cytochrome P450 in the liver to generate the major metabolite desethylamiodarone (desethyl-AMI) and minor metabolite di-N-desethylamiodarone (di-N-desethyl-AMI) (33). In addition to these derivatives, we synthesized four amiodarone analogs with varied “chain” length of the amine (2 or 3 carbons) and methylation states of the amine (dimethyl or trimethyl). Synthesis of the dimethyl-AMI compounds had been previously reported (34). As shown in Scheme 1, the synthesis of amiodarone derivatives 4–7 featured the alkylation of (2-butylbenzofuran-3-yl)(4-hydroxy-3,5-diiodophenyl)methanone 1 in the presence of 2-chloro-N,N-dimethylethanamine hydrochloride or 3-chloro-N,N-dimethylpropan-1-amine hydrochloride to afford the dimethyl-AMI analog 2, as well as homolog 3 with its chain extended by one methylene unit. With this series of amiodarone derivatives, a range of amine alkylation states could be evaluated.
Scheme 1. Synthesis of amiodarone analogs.

Synthesis of amiodarone derivatives 4–7. Reagents and conditions: (a) 1, K2CO3, toluene/H2O (2:1), 60 °C, then 2-chloro-N,N-dimethylethanamine hydrochloride, reflux 22 h, 66% (n = 1); (b) 1, K2CO3, toluene/H2O (2:1), 60 °C, then 3-chloro-N,N-dimethylpropan-1-amine hydrochloride, reflux 17 h, 92% (n = 2); (c) 2, conc. HCl, toluene, rt, 1 h, 63%; (d) 3, conc. HCl, toluene, rt, 1 h, 50%; (e) 2, MeI, CH2Cl2, rt, 17 h, 76%; (f) 3, MeI, CH2Cl2, rt, 2 h, 81%.
AMI-based inhibitors were evaluated via fluorescence polarization, utilizing a 90% fraction bound concentration of GST-JARID1A PHD3. Di-N-desethyl-AMI and both trimethyl-AMI analogs inhibited JARID1A PHD3 more potently than did AMI (Figure 6b), with similar IC50 values that ranged between 25–40 μM (Table 1). These studies suggest that di-N-desethylAMI and trimethyl-AMI analogs are at least 10-fold more potent than amiodarone. Neither dimethyl-AMI analog nor desethyl-AMI significantly inhibited JARID1A PHD3 within the concentration range tested. We performed anion exchange chromatography on trimethyl-AMI derivatives to generate sodium salts, which confirmed that the iodide counter-ion was not responsible for binding inhibition (data not shown).
Table 1.
IC50 values of amiodarone analogs towards JARID1A PHD3a
| Compound | IC50 |
|---|---|
| Amiodarone | NM |
| Desethylamiodarone | NM |
| Di-N-desethylamiodarone | 26 ± 15 μM |
| WAG-003 | 30 ± 14 μM |
| WAG-004 | NM |
| WAG-005 | 41 ± 16 μM |
| WAG-006 | NM |
IC50 values calculated from the fluorescence polarization assay, where GST-JARID1A PHD3 was treated with increasing concentrations of amiodarone and its analogs.
NM: Not measurable within solubility limit of compound
Amiodarone analogs were then assayed against a number of histone reader domains, including JMJD2A DTD, RAG2 PHD, ING2 PHD (H3K4me3), AIRE PHD1, BHC80 PHD (H3K4-unmmodified), and UHRF1 tandem tudor domain (TTD, H3K9me3) within the fluorescence polarization assay (5, 35). The results for trimethyl-AMI (n=2) and di-N-desethyl-AMI are shown in Figure 7 and Table 2. Trimethyl-AMI (n=2) and di-N-desethyl-AMI inhibited JARID1A PHD3 the most potently, followed closely by JMJD2A DTD. These data confirmed that amiodarone derivatives retain specificity for trimethyl-lysine reader domains. Interestingly, we observed a hyperpolarization effect when AIRE PHD1 or BHC80 PHD is treated with either compound, but the significance of these data is unclear since fluorescence intensity values were unaffected (data not shown).
Figure 7. Amiodarone analogs are specific for trimethyl-lysine reader domains.
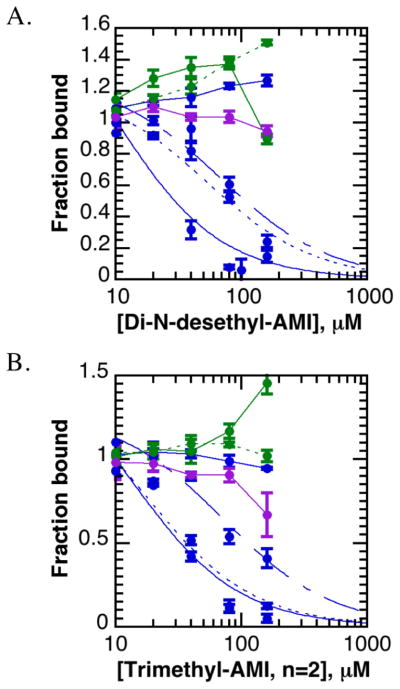
A–B. Di-N-desethylamiodarone (A) and trimethyl-amiodarone (n=2) (B) are most potent against specific H3K4me3-reader domains. JMJD2A DTD (dotted blue curve) is similarly inhibited as JARID1A PHD3 (solid blue curve), followed by ING2 PHD (dashed blue curve). AIRE PHD1 (solid green line), BHC80 PHD (dotted green line), RAG2 PHD (solid blue line), and UHRF1 TTD (purple line) were not inhibited by these compounds.
Table 2.
IC50 values of AMI analogs towards lysine reader domainsb
| Reader domain | WAG-003 | Di-N-desethylAMI |
|---|---|---|
| JARID1A PHD3 | 30 ± 14 μM | 26 ± 15 μM |
| JMJD2A DTD | 34 ± 14 μM | 72 ± 26 μM |
| ING2 PHD | 105 ± 33 μM | 97 ± 41 μM |
| RAG2 PHD | NM | NM |
| AIRE PHD1 | NM | NM |
| BHC80 PHD | NM | NM |
| UHRF1 TTD | NM | NM |
IC50 values calculated from the fluorescence polarization assay, where GST fusions to reader domains were treated with increasing concentrations of trimethyl-amiodarone (WAG-003) or di-N-desethylamiodarone. Fusion proteins were assayed at a 90% fraction bound protein concentration.
NM: Not measurable within solubility limit of compound
Fluorescence polarization studies suggested that amiodarone analogs were inhibiting JARID1A PHD3 independently of the aromatic cage. To identify possible sites on JARID1A PHD3 where inhibitors might be binding, molecular modeling was performed using AutoDock software (36), where the entire JARID1A PHD3 structure was included in the docking grid. The best docking hits for compounds that biochemically inhibit JARID1A PHD3 (tegaserod, di-N-desethylamiodarone, trimethyl-AMI) clustered almost exclusively to the peptide-binding groove of JARID1A PHD3. Amiodarone derivatives clustered to one of two positions (Figure 8). One abundant docking site positioned the amine chain of amiodarone analogs in an acidic pocket that normally engages H3R2 and the N-terminus of the histone H3 tail (Figure 8a) (10). Another abundant docking position for amiodarone-based molecules positioned the amine within hydrogen bonding distance of an aspartate side chain (D1624) (Figure 8b). Tegaserod docked most favorably to a similar position, interacting with D1629 (Figure 8c).
Figure 8. Molecular modeling of small molecule inhibitors reveals candidate residues important for interactions with JARID1A PHD3.

A–B. Di-N-desethylamiodarone docked favorably to two positions on JARID1A PHD3, where the amine interacts with either D1629 (A) or D1624 (B).
C. Tegaserod docked almost exclusively to this position, where the dibiguanidine group is coordinated in part by D1629.
D. H3K4me3 peptide binding in relation to D1624 and D1629 reveals probable hydrogen bonds (D1624) and ionic bonds (D1629) between JARID1A PHD3 and histone H3.
E. Affinity binding curves with WT, D1624, and D1629 mutants reveals defects in histone peptide binding via fluorescence polarization, consistent with structural predictions. WT JARID1A PHD3 is in black, D1624A in dark blue, D1624N in light blue, D1629A in dark green, D1629N in light green, and W1625A in purple. Binding of H3K4me3 peptide is shown by solid lines, and H3K4-unmethylated in dashed lines.
Modeling suggested that aspartate residues 1624 or 1629 contributed important interactions for binding amiodarone analogs and tegaserod. In order to test this hypothesis, we mutated these residues to asparagine and alanine. The ability of these mutants to bind histone H3 peptides was tested by fluorescence polarization (Table 3, Figure 8e). An aromatic cage mutant, W1625A, which was reported to completely abolish histone binding, was generated for comparison (10). As expected, this mutation severely impaired histone peptide binding. Based on the crystal structure of JARID1A PHD3, glutamine 5 on histone H3 forms a hydrogen bond to aspartate 1624, and the side chain of H3R2 forms an ionic bond with aspartate 1629 (Figure 8d). We therefore predicted that mutating either site would decrease the affinity of JARID1A PHD3 for the H3K4me3 peptide. Consistent with a hydrogen bond interaction, an alanine mutation at 1624 decreased the affinity of JARID1A PHD3 for H3K4me3 peptides by two-fold compared to an asparagine mutation, which would still permit hydrogen bonding (Table 3, Figure 8e). Compared to a wild-type affinity of approximately 0.3 μM for the H3K4me3 peptide, the D1624A and D1624N mutations decreased affinity by four- and two-fold, respectively (Table 3). Mutating D1629 was more damaging to histone binding and caused a greater than 10-fold defect in affinity for the H3K4me3 peptide, independent of the amino acid substitution at this site (Table 3, Figure 8e). These data are consistent with the formation of a charge-charge ionic bond between D1629 and the side chain of H3R2.
Table 3.
Dissociation constants of JARID1A PHD3 for H3K4me3 peptide.
| Clone | Peptide | Kd (m p | Error (mrr | R |
|---|---|---|---|---|
| WT | H3K4 | 9.35 | 1.57 | 0.954 |
| H3K4me3 | 0.294 | 0.046 | 0.97 | |
| D1624A | H3K4 | NM* | - | - |
| H3K4me3 | 1.15 | 0.145 | 0.988 | |
| D1624N | H3K4 | NM* | - | - |
| H3K4me3 | 0.634 | 0.08 | 0.986 | |
| W1625A | H3K4 | NM* | - | - |
| H3K4me3 | NM* | - | - | |
| D1629A | H3K4 | NM* | - | - |
| H3K4me3 | 4.05 | 0.691 | 0.98 | |
| D1629N | H3K4 | NM* | - | - |
| H3K4me3 | 3.81 | 0.551 | 0.984 |
NM: Not measurable within the concentration range utilized in the fluorescence polarization assay.
Because D1624 and D1629 mutants exhibited defects in histone binding, standard IC50 analyses would be influenced by protein concentration within fluorescence polarization binding studies. We therefore assayed the ability of amiodarone analogs and tegaserod to inhibit JARID1A PHD3 using a dose ratio analysis, as described in the methods. For both di-N-desethylamiodarone and tegaserod, increasing the fixed concentration of inhibitor caused an increase in EC50 values for wild-type (WT) JARID1A PHD3 (Figure 9a, g). The dose ratios values for WT JARID1A PHD3 increase linearly as the concentration of inhibitor is increased (Figure 9f, 9l). EC50 values for the D1624 mutants also increased with increasing concentrations of di-N-desethylamiodarone or tegaserod, though the dose ratios were less pronounced than for WT JARID1A PHD3. In contrast, the EC50 values for D1629 mutations increased very little with increasing either di-N-desethylamiodarone or tegaserod concentrations, resulting in dose ratios that remained near one. Our data therefore support a model where D1629 provides a major contribution to binding of amiodarone analogs and tegaserod to JARID1A PHD3. We predict that these interactions likely occur through electrostatic contacts between inhibitor amine groups and the carboxyl group of the aspartate side chain, as the D1629N mutation did not increase the dose response. Because D1624 forms part of the binding site for di-N-desethylamiodarone and tegaserod in modeling predictions, it is unsurprising that mutating this residue could impair inhibitor interactions with JARID1A PHD3, albeit to a lesser extent than D1629. In fact, the hydroxyl group extending from the indole ring of tegaserod may form a hydrogen bond with D1624. Weaker interactions with this site on JARID1A PHD3 may account for the reduced dose response in D1624 mutants relative to wild-type protein.
Figure 9. Analysis of JARID1A PHD3 mutants reveals that D1629 provides a major contribution to di-N-desethylamiodarone and tegaserod binding JARID1A PHD3.

A.–E. Di-N-desethylamiodarone induces a dose response for D1624, but not D1629 mutants. Increasing concentrations of GST-JARID1A PHD3 clones were titrated against solutions containing fixed concentrations of di-N-desethylamiodarone (0, 20, 40, 80 μM; solid to finer dashed curves with increasing inhibitor). Shifts in apparent Kd are observed for WT and D1624 mutants, but not D1629 mutants.
F. Dose ratio analysis for di-N-desethylamiodarone suggests that D1629 contributes to the interaction between di-N-desethylamiodarone and JARID1A PHD3, much more so than D1624.
G.–K. Tegaserod induces a dose response for D1624, but not D1629 mutants. Increasing concentrations of GST-JARID1A PHD3 clones were titrated against solutions containing fixed concentrations of tegaserod maleate (0, 80, 160, 320 μM; solid to finer dashed curves with increasing inhibitor). Shifts in apparent Kd are observed for WT and D1624 mutants, but not D1629 mutants.
L. Dose ratio analysis for tegaserod suggests that D1629 plays a major contribution to tegaserod maleate binding JARID1A PHD3, and less so D1624.
DISCUSSION
Utilizing a newly developed HaloTag assay, a small molecule screen identified disulfiram, amiodarone, and tegaserod as inhibitors of the interaction between JARID1A PHD3 and H3K4me3. Compounds were validated biochemically via dose dependent-binding inhibition using the HaloTag, affinity pull down, and fluorescence polarization assays. Structural destabilization and chemical reactivity towards JARID1A PHD3 were monitored by zinc release studies. The potency of amiodarone was improved by chemical modification, following fragment-based dose dependence studies that suggested that the alkylated amine chain played an important role in binding inhibition. Following molecular modeling and site-directed mutagenesis, we determined that aspartate 1629 in JARID1A PHD3 makes a significant contribution to amiodarone-derivatives and tegaserod binding to this PHD finger.
Our studies demonstrate the value of developing new assays for small molecule screening. Many fluorescence-based binding assays are prone to compound interference; the HaloTag assay described here appears to be very resilient to such effects and provided greater stringency for inhibitors during primary screening. It is important to note that even though the HaloTag assay incorporated several washing and incubation steps, low affinity peptide binding is readily detectable due to the high sensitivity of the HaloTag assay format. High sensitivity SPOT peptide and CADOR (chromatin associated domain) array formats that have been used for detection of weak peptide and histone binding domains (37, 38). Our ability to readily adapt the HaloTag assay to different histone reader domains demonstrated the flexibility and utility of the HaloTag assay for low to mid-range micromolar affinity interactions. Given the ease with which HaloTag fusion proteins and biotinylated ligands can be prepared, we anticipate that the assay would function well for a number of protein-ligand interactions.
Disulfiram inhibits JARID1A PHD3 and other PHD fingers not by acting as a ligand, but through ejection of structural zinc, thus revealing a general susceptibility specific to PHD fingers as a histone reader domain. Disulfiram is most well known for its use in the treatment of alcoholism, where it inhibits acetaldehyde dehydrogenase via modification of active site cysteine residues and subsequent zinc ejection (31). In addition to acetaldehyde dehydrogenase, disulfiram inhibits a number of proteins with diverse functions, including dopamine beta-hydroxylase, viral nucleocapsin protein, DNA methyltransferase I, and histone demethylases (31, 39–41) It is interesting to note that disulfiram exhibits anti-tumor activity, demonstrating its potential for a broad range of therapeutic indications (42).
Amiodarone was initially identified from screening as a weak inhibitor of JARID1A PHD3. We hypothesized that the alkylated amine chain inhibited histone binding by acting as a competitive ligand for JARID1A PHD3. By preparing a series of derivatives that probed the role of the amine chain, we were able to identify inhibitors that were approximately 10-fold more potent than amiodarone. Our data suggest that amiodarone inhibits JARID1A PHD3 independently of the aromatic cage, as the unmethylated and trimethylated forms of the amine chain were equally potent inhibitors against histone binding.
Informed by molecular modeling studies, we identified two probable sites where amiodarone analogs appeared to bind JARID1A PHD3, and one site for tegaserod. Sites for both inhibitors contain aspartate residues that contribute to histone binding. To test our modeling predictions, we performed site-directed mutagenesis on JARID1A PHD3. Dose ratio analyses with D1624 and D1629 mutants support a model where amiodarone analogs and tegaserod bind near aspartate 1629, which interacts with arginine 2 on histone H3.
Amiodarone was developed for the treatment of arrhythmias for patients with atrial fibrillations (43). Like disulfiram, amiodarone may be a candidate for drug repositioning, since a recent preclinical mouse model study demonstrated that amiodarone could treat leukemia (44). Another study performed in Saccharomyces cerevisiae found that amiodarone significantly down-regulated the transcription of genes involved in cell cycle and DNA processing (45). To date, the mechanisms of action underlying these activities are unclear, but suggest amiodarone could influence a number of cellular processes. Tegaserod was developed as a partial agonist for 5-HT4 receptors in the treatment of irritable bowel syndrome (46, 47). Our studies demonstrate novel activities for amiodarone derivatives and tegaserod through inhibition of histone reader domains.
In conclusion, identifying inhibitors of JARID1A PHD3 adds to the growing body of studies that target histone readers as a strategy for drug development efforts. The development of JQ1 as a small molecule inhibitor of the bromodomain of BRD4 shows great promise in mouse cancer models, and others have developed in vitro inhibitors of MBT methyl-lysine readers (14, 48). PHD fingers and other histone reader domains play critical roles in gene regulation by recruiting protein complexes to specific regions of the genome (6). By disrupting histone reader proteins from chromatin via small molecule inhibitors, it may be possible to alter transcriptional programs that are misregulated in cancer cells and thereby develop novel therapeutics.
Supplementary Material
Acknowledgments
We are grateful to Dr. Danette Daniels, Dr. Marjeta Urh, Dr. Mike Slater, Dr. Tetsuo Uyeda, Dr. Thomas Lubben and Dr. Keith Wood of Promega Corporation for invaluable suggestions and advice during the development of the HaloTag assay. We are grateful to Dr. Ken Satyshur of the University of Wisconsin Small Molecule Screening and Synthesis Facility for assistance with molecular modeling studies, as well as Noël Peters and Song Guo of the same facility for invaluable assistance with small molecule screening. We acknowledge the University of Wisconsin – Madison School of Pharmacy Analytical Instrumentation Center for use of NMR, IR, and mass spectrometry instruments for the characterization of synthetic analogs of AMI. We thank Dr. Martha Vestling of the University of Wisconsin – Madison Chemistry Department Mass Spectrometry Facility for training and use of mass spectrometry instruments, Dr. Sam Oliver for assistance in peptide synthesis, and Dr. Brittany Albaugh for providing GST-UHRF1 TTD fusion protein and fluorescein-labeled H3K9me3 peptide.
Abbreviations used in this work
- PHD finger
Plant homedomain finger
- JARID1A
Jumonji A/T rich interaction domain
- NUP98
nucleoporin 98
- AML
acute myeloid leukemia
- ING
inhibitor of growth
- DTD
double tudor domain
- TTD
tandem tudor domain
- H3K4me3
histone H3 trimethylated at lysine 4
- H3K9me3
histone H3 trimethylated at lysine 9
- HRP
horseradish peroxidase
- DMSO
dimethyl sulfoxide
- TMR
tetramethylrhodamine
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide electrophoresis
- GST
glutathione S-transferase
- IC50
half-maximal inhibitory concentration
- PAR
4-(2-pyridylazo) resorcinol
- MMTS
methyl methanethiosulfonate
- AMI
amiodarone
- desethyl-AMI
desethylamiodarone
- di-N-desethyl-AMI
di-N-desethylamiodarone
- UHRF1
ubiquitin-like, containing PHD and RING finger domains
- RAG2
recombination activating gene 2
- JMJD2A
Jumonji domain protein 2A
- AIRE
autoimmune regulator
- BHC80
BRAF-HDAC complex protein
- MBT
malignant brain tumor
- TCEP
tris(2-carboxyehyl)phosphine
- LOD
limit of detection
- HPLC
high performance liquid chromatography
- MeI
methyl iodide
- rt
room temperature
Footnotes
This work was supported by a National Institutes of Health (NIH) Grant GM059785 (to J.M.D.) and by NIH Training Grant in Molecular Biosciences GM07215 (to E.K.W.)
SUPPORTING INFORMATION AVAILABLE
Additional data for assay optimization, inhibitor validation, and molecular modeling, as well as compound synthesis methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 2.Jenuwein T. Translating the Histone Code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 3.Kouzarides T. Chromatin Modifications and Their Function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez R, Zhou MM. The PHD finger: a versatile epigenome reader. Trends Biochem Sci. 2011;36:364–372. doi: 10.1016/j.tibs.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peña PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006 doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker LA, Allis CD, Wang GG. PHD fingers in human diseases: disorders arising from misinterpreting epigenetic marks. Mutat Res. 2008;647:3–12. doi: 10.1016/j.mrfmmm.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Zutven LJ, Onen E, Velthuizen SC, van Drunen E, von Bergh AR, van den Heuvel-Eibrink MM, Veronese A, Mecucci C, Negrini M, de Greef GE, Beverloo HB. Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Gene Chromosome Canc. 2006;45:437–446. doi: 10.1002/gcc.20308. [DOI] [PubMed] [Google Scholar]
- 9.Reader JC, Meekins JS, Gojo I, Ning Y. A novel NUP98-PHF23 fusion resulting from a cryptic translocation t(11;17)(p15;p13) in acute myeloid leukemia. Leukemia. 2007 doi: 10.1038/sj.leu.2404579. [DOI] [PubMed] [Google Scholar]
- 10.Wang GG, Song J, Wang Z, Dormann HL, Casadio F, Li H, Luo J-L, Patel DJ, Allis CD. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature. 2009;459:847–851. doi: 10.1038/nature08036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Unoki M, Daigo Y, Koinuma J, Tsuchiya E, Hamamoto R, Nakamura Y. UHRF1 is a novel diagnostic marker of lung cancer. Br J Cancer. 2010;103:217–222. doi: 10.1038/sj.bjc.6605717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsai WW, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S, Tsai CY, Shi X, Schwarzer D, Plunkett W, Aronow B, Gozani O, Fischle W, Hung MC, Patel DJ, Barton MC. TRIM24 links a non-canonical histone signature to breast cancer. Nature. 2010;468:927–932. doi: 10.1038/nature09542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wigle TJ, Herold JM, Senisterra GA, Vedadi M, Kireev DB, Arrowsmith CH, Frye SV, Janzen WP. Screening for Inhibitors of Low-Affinity Epigenetic Peptide-Protein Interactions: An AlphaScreenTM-Based Assay for Antagonists of Methyl-Lysine Binding Proteins. J Biomol Screen. 2009;15:62–71. doi: 10.1177/1087057109352902. [DOI] [PubMed] [Google Scholar]
- 14.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 16.Hurst R, Hook B, Slater MR, Hartnett J, Storts DR, Nath N. Protein-protein interaction studies on protein arrays: effect of detection strategies on signal-to-background ratios. Anal Biochem. 2009;392:45–53. doi: 10.1016/j.ab.2009.05.028. [DOI] [PubMed] [Google Scholar]
- 17.So MK, Yao H, Rao J. HaloTag protein-mediated specific labeling of living cells with quantum dots. Biochem Biophys Res Commun. 2008;374:419–423. doi: 10.1016/j.bbrc.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Motejadded H, Kranz B, Berensmeier S, Franzreb M, Altenbuchner J. Expression, one-step purification, and immobilization of HaloTag(TM) fusion proteins on chloroalkane-functionalized magnetic beads. Appl Biochem Biotechnol. 2010;162:2098–2110. doi: 10.1007/s12010-010-8985-1. [DOI] [PubMed] [Google Scholar]
- 19.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 20.Albaugh BN, Kolonko EM, Denu JM. Kinetic mechanism of the Rtt109-Vps75 histone acetyltransferase-chaperone complex. Biochemistry. 2010;49:6375–6385. doi: 10.1021/bi100381y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Jong LA, Uges DR, Franke JP, Bischoff R. Receptor-ligand binding assays: technologies and applications. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2005;829:1–25. doi: 10.1016/j.jchromb.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Wyllie DJ, Chen PE. Taking the time to study competitive antagonism. British journal of pharmacology. 2007;150:541–551. doi: 10.1038/sj.bjp.0706997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang L, Rissin DM, Fournier DR, Piech T, Patel PP, Wilson DH, Duffy DC. Single molecule enzyme-linked immunosorbent assays: Theoretical considerations. J Immunol Methods. 2012;378:102–115. doi: 10.1016/j.jim.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cer RZ, Mudunuri U, Stephens R, Lebeda FJ. IC50-to-Ki: a web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic acids Res. 2009;37:W441–445. doi: 10.1093/nar/gkp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramon-Maiques S, Kuo AJ, Carney D, Matthews AGW, Oettinger MA, Gozani O, Yang W. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. PNAS. 2007;104:18993–18998. doi: 10.1073/pnas.0709170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Y. Recognition of Histone H3 Lysine-4 Methylation by the Double Tudor Domain of JMJD2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 27.Org T, Chignola F, Hetényi C, Gaetani M, Rebane A, Liiv I, Maran U, Mollica L, Bottomley MJ, Musco G, Peterson P. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO reports. 2008;9:370–376. doi: 10.1038/embor.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X, Gozani O, Cheng X, Shi Y. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.http://www.nihclinicalcollection.com.
- 30.Wermuth CG. Selective optimization of side activities: the SOSA approach. Drug Discov Today. 2006;11:160–164. doi: 10.1016/S1359-6446(05)03686-X. [DOI] [PubMed] [Google Scholar]
- 31.Barth KS, Malcolm RJ. Disulfiram: an old therapeutic with new applications. CNS Neurol Disord Drug Targets. 2010;9:5–12. doi: 10.2174/187152710790966678. [DOI] [PubMed] [Google Scholar]
- 32.Bienz M. The PHD finger, a nuclear protein-interaction domain. Trends Biochem Sci. 2006;31:35–40. doi: 10.1016/j.tibs.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 33.Shayeganpour A, El-Kadi AO, Brocks DR. Determination of the enzyme(s) involved in the metabolism of amiodarone in liver and intestine of rat: the contribution of cytochrome P450 3A isoforms. Drug Metab Dispos. 2006;34:43–50. doi: 10.1124/dmd.105.006742. [DOI] [PubMed] [Google Scholar]
- 34.Bigler L, Spirli C, Fiorotto R, Pettenazzo A, Duner E, Baritussio A, Follath F, Ha HR. Synthesis and cytotoxicity properties of amiodarone analogues. Eur J Med Chem. 2007;42:861–867. doi: 10.1016/j.ejmech.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 35.Nady N, Lemak A, Walker JR, Avvakumov GV, Kareta MS, Achour M, Xue S, Duan S, Allali-Hassani A, Zuo X, Wang YX, Bronner C, Chedin F, Arrowsmith CH, Dhe-Paganon S. Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J Biol Chem. 2011;286:24300–24311. doi: 10.1074/jbc.M111.234104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nady N, Min J, Kareta MS, Chédin F, Arrowsmith CH. A SPOT on the chromatin landscape? Histone peptide arrays as a tool for epigenetic research. Trends Biochem Sci. 2008;33:305–313. doi: 10.1016/j.tibs.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 38.Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin LZ, Lin J. Antabuse (disulfiram) as an affordable and promising anticancer drug. Int J Cancer. 2011;129:1285–1286. doi: 10.1002/ijc.25780. author reply 1286–1287. [DOI] [PubMed] [Google Scholar]
- 40.Boukhvalova MS, Prince GA, Blanco JC. Inactivation of respiratory syncytial virus by zinc finger reactive compounds. Virol J. 2010;7:20. doi: 10.1186/1743-422X-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sekirnik R, Rose NR, Thalhammer A, Seden PT, Mecinovic J, Schofield CJ. Inhibition of the histone lysine demethylase JMJD2A by ejection of structural Zn(II) Chem Commun (Camb) 2009:6376–6378. doi: 10.1039/b916357c. [DOI] [PubMed] [Google Scholar]
- 42.Yip NC, Fombon IS, Liu P, Brown S, Kannappan V, Armesilla AL, Xu B, Cassidy J, Darling JL, Wang W. Disulfiram modulated ROS-MAPK and NFkappaB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br J Cancer. 2011;104:1564–1574. doi: 10.1038/bjc.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh BN. Amiodarone as paradigm for developing new drugs for atrial fibrillation. J Cardiovasc Pharmacol. 2008;52:300–305. doi: 10.1097/FJC.0b013e31818914b6. [DOI] [PubMed] [Google Scholar]
- 44.Papageorgiou AD, Dalezis P, Mourelatos C, Lioutas K, Sahpazidou D, Geromichalou E, Geromichalos G, Lialiaris T, Athanasiadou P, Athanasiadis P. Preclinical evaluation of amiodarone for the treatment of murine leukemia P388. In vivo and in vitro investigation. J BUON. 2010;15:568–571. [PubMed] [Google Scholar]
- 45.Zhang YQ, Rao R. Global disruption of cell cycle progression and nutrient response by the antifungal agent amiodarone. J Biol Chem. 2007;282:37844–37853. doi: 10.1074/jbc.M707593200. [DOI] [PubMed] [Google Scholar]
- 46.Beglinger C. Tegaserod: a novel, selective 5-HT4 receptor partial agonist for irritable bowel syndrome. Int J Clin Pract. 2002;56:47–51. [PubMed] [Google Scholar]
- 47.Sloskey GE. Amiodarone: a unique antiarrhythmic agent. Clin Pharm. 1983;2:330–340. [PubMed] [Google Scholar]
- 48.Herold JM, Wigle TJ, Norris JL, Lam R, Korboukh VK, Gao C, Ingerman LA, Kireev DB, Senisterra G, Vedadi M, Tripathy A, Brown PJ, Arrowsmith CH, Jin J, Janzen WP, Frye SV. Small-molecule ligands of methyl-lysine binding proteins. J Med Chem. 2011;54:2504–2511. doi: 10.1021/jm200045v. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.