

Abstract
An A-to-G transition at position 16247 in the human mtDNA genome denotes haplogroup B4a1a1a and its sublineages. Informally known as the “Polynesian motif,” this haplogroup has been widely used as a marker in Oceania of genetic affiliation with the Austronesian expansion. The 16247G allele has arisen only once in the human mtDNA phylogeny, about 7,000 thousand years ago, and is nearly fixed in Remote Oceania. We analyzed 536 complete mtDNA genome sequences from the Solomon Islands from haplogroup B4a1a1 and associated subhaplogroups and found multiple independent back mutations from 16247G to 16247A. We also find elevated levels of heteroplasmy at this position in samples with the 16247G allele, suggesting the ongoing occurrence of somatic back-mutations and/or transmission of heteroplasmy. Moreover, the G allele is predicted to introduce a novel stem-loop structure in the DNA sequence that may be structurally unfavorable, thereby accounting for the remarkable number of back-mutations observed at the 16247G allele in this short evolutionary time span. More generally, haplogroup-calling scripts result in inaccurate haplogroup calls involving the back-mutation and need to be supplemented with other types of analyses; this may be true for other mtDNA lineages because no other lineage has been investigated to the same extent (over 500 complete mtDNA sequences).
Main Text
Although other potential origins have been suggested,1–3 it is likely that the Austronesian expansion spread from Taiwan beginning about 5,000 years ago.4–7 The Austronesian expansion resulted in the spread not only of a new language family and culture but also the mtDNA B4a1 lineage.8,9 As the B4a1 lineage expanded across Island Southeast Asia, Near Oceania, and Remote Oceania, various subhaplogroups appeared along the migration route, often with signatures of specific homelands. For example, haplogroup B4a1a1a and the immediately descendent haplogroup B4a1a1a1 are found at very high frequency in Near Oceania and approach fixation in Remote Oceania.10–12 These two haplogroups are thought to have arisen in the Bismarck Archipelago due to the high diversity of lineages found in that region and their general scarcity further west.13
Haplogroup B4a1a1a is defined by a series of diagnostic mutations, many in the mtDNA coding region. The mutation that defines haplogroup B4a1a1a from its immediate predecessor B4a1a1 is an A-to-G transition at position 16247 in the noncoding control region (Figure 1). Because this position was an obvious marker for early mtDNA studies that sequenced only the hypervariable segments of the control region, and because B4a1a1a and its descendent haplogroups achieve near fixation in Remote Oceania, this mutation became known as the “Polynesian motif.”10 The derived 16247G allele occurs only once in the entire human mtDNA phylogeny (Phylotree Build 14) and has been previously estimated to have arisen 5,000–7,000 years ago.13,14 It is well known that some positions in the mtDNA genome are hypermutable, exhibiting independent forward and back-mutations across multiple lineages in the mtDNA phylogeny.15 We report here a type of mutational instability in that the derived 16247G allele has undergone multiple independent back-mutations to the ancestral allele since its single origin in the entire mtDNA phylogeny. Moreover, we detect high levels of heteroplasmy at this position, suggesting ongoing back-mutations within samples with the 16247G allele. We also infer that the 16247G allele induces a novel stem-loop structure that may account for the instability associated with this mutation.
Figure 1.

Schematic of the B4a1a1 Lineage and Sublineages
Haplogroups are named following current conventions (PhyloTree Build 14) and diagnostic mutations are indicated. Haplogroups connected by dashed lines are those now identified based on the back-mutation events at position 16247 and are left unnamed.
The sequencing of 536 complete mtDNA genomes from haplogroup B4a1a1 and associated subhaplogroups was undertaken as part of a larger ongoing population genetics study of the Solomon Islands.16 Samples were collected with written informed consent in the Solomon Islands in 2004 with the approval of the Solomon Islands Ministry for Educational Training and the Ministry of Health and Medical services. Ethical approval for sample collection and study was obtained from the Ethics Commission of the University of Leipzig Medical Faculty. Libraries were prepared for multiplex sequencing following the protocol of Meyer and Kircher17 with modifications for mtDNA target enrichment by in-solution capture after Maricic et al.18 Libraries were sequenced on the Illumina Genome Analyzer IIx platform with single-end, 76 base-pair reads to an average coverage of 430× per sample. The haplogroup of each sample was determined with a custom script by comparison to PhyloTree Build 14.19 Most of the sequences belonged to haplogroups B4a1a1a (47%) and B4a1a1a1 (39%).
An unusual trend was observed while verifying the haplogroup calls for some B4a1a1a1 sequences. Approximately 20% of the sequences that contained the diagnostic 6905G allele, indicating they belonged to haplogroup B4a1a1a1, had the 16247A allele instead of the expected 16247G allele that is diagnostic for the immediately ancestral haplogroup B4a1a1a (Figure 1). A network of all putative B4a1a1a1 samples (Figure 2) indicated that although the majority of samples with the ancestral A allele at position 16247 appeared to have arisen from a single back-mutation event, there were other samples that fell outside of this clade, indicating multiple independent back-mutations at 16247. These back-mutations at position 16247, which we denote as B4a1a1a1+16247!, appear to be widely prevalent because they are present in 14 of the 18 populations examined and account for up to 43% of the observed B4a1a1a1 haplotypes in these populations (Table 1).
Figure 2.
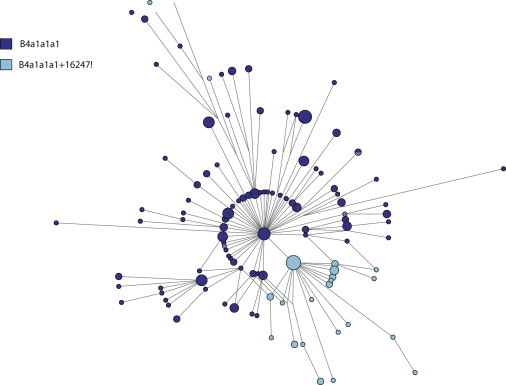
A Network of 211 Samples Identified as Belonging to Haplogroup B4a1a1a1
Samples in dark blue possess the 16247G allele and those in light blue possess the 16247A allele via back-mutation events. Networks were calculated with transversions weighted 3× greater than transitions and with Poly-C stretch and position 310 unweighted. The Reduced Median algorithm25 and then the Median Joining algorithm26 were used, followed by maximum parsimony postprocessing;27 the final network was edited using Network Publisher v1.3.
Table 1.
Frequency of Haplogroup B4a1a1a1, and of G16247A! Within B4a1a1a1, in Each Population
| Population | n | Percent B4a1a1a1 | Percent B4a1a1a1 with 16247! |
|---|---|---|---|
| Bellona | 38 | 32 | 8 |
| Choiseul | 33 | 21 | 14 |
| Gela | 40 | 20 | 38 |
| Guadalcanal | 50 | 32 | 25 |
| Isabel | 52 | 23 | 17 |
| Kolombangara | 18 | 39 | 0 |
| Makira | 17 | 65 | 9 |
| Malaita | 89 | 37 | 12 |
| Ontong Java | 32 | 16 | 20 |
| Ranongga | 46 | 24 | 9 |
| Rennell | 43 | 30 | 38 |
| Russell | 39 | 31 | 0 |
| Santa Cruz | 47 | 9 | 0 |
| Savo | 40 | 15 | 33 |
| Shortlands | 14 | 29 | 0 |
| Simbo | 22 | 45 | 20 |
| Tikopia | 46 | 50 | 43 |
| Vella Lavella | 51 | 33 | 18 |
To see whether other mtDNA sequences classified in the B4a1a1 lineage are also influenced by back-mutations from 16247G, we produced a network of all 536 sequences that fell in the B4a1a1 lineage and sublineages thereof (Figure 3). Indeed, a group of 16 sequences that are all from Polynesian Outlier populations and had been assigned to the B4a1a1 haplogroup instead fell as a terminal node on a long branch of the B4a1a1a haplogroup (Figure 3). This suggests that these sequences mark an additional back-mutation at position 16247 from G to A involving the B4a1a1a haplogroup (Figure 1), making them indistinguishable from B4a1a1 with respect to haplotyping algorithms.
Figure 3.

A Network of 536 Samples Belonging to the B4a1a1 Lineage with Haplogroup Affiliation Indicated
Heteroplasmic individuals are colored in white with cross-hatching. Independent back-mutation events at position 16247 are indicated by arrows; red arrows indicate clear back-mutations and black arrows indicate putative back-mutations identified either by heteroplasmy or reticulation between haplogroups (see Figure S1). The network was constructed and edited as described in the legend to Figure 2.
In order to determine the time span over which these back-mutations have occurred, we dated the age of the mutations of the B4a1a1a haplogroup to be approximately 7,200 years old, and the younger B4a1a1a1 haplogroup (from which most of the back-mutations arose) to be approximately 6,350 years old (Table 2). We also generated several trees both through maximum likelihood and Bayesian analyses. A recurrent observation in these trees is that many of the haplogroups do not appear as monophyletic clades, with individual or small groups of B4a1a1 or B4a1a1a1+16247! sequences falling within the larger B4a1a1a or B4a1a1a1 haplogroups (see Figure S1 available online). The B4a1a1 lineage network provides an explanation because all of the sequences (with 16247A) that are scattered across the tree are involved in reticulations with a second haplogroup with the 16247G allele (Figure 3). This raises the possibility that these sites of reticulation are yet further independent back-mutation events at position 16247, which were not obvious from the network as a result of their connection with two haplogroups.
Table 2.
Haplogroup Ages
| Complete Genomes Clock, ρ | Complete Genomes Clock, ρ from Soares et al.13 | |
|---|---|---|
| B4a1a1 | 8900 (4900, 13000) | 7900 (3450, 12450) |
| B4a1a1a | 7200 (4450, 9900) | 5850 (3850, 7800) |
| B4a1a1a+16247! | 1550 (0, 4600) | |
| B4a1a1a1 | 6350 (4700, 8100) | |
| B4a1a1a1+16247! | 4600 (2950, 6250) | |
| B4a1a1a4 | 419 (0, 954) |
Rho dating was calculated with the program Network using the whole mitochondrial clock rate and accounting for purifying selection using the method developed by Soares et al.28 Age of the B4a1a1a1+16247! haplogroup was calculated using only those individuals that fall into a large cluster and appear to be descendent from a single back-mutation event.
We then searched the literature for other whole mtDNA genome sequences that might harbor the B4a1a1a1 + 16247! motif and identified three such sequences. All are identified as B4a1a1a1; one comes from the Bismarck Archipelago,13 one from Bougainville,20,21 and the third from an individual from coastal Papua New Guinea.22 This back-mutation pattern therefore appears to be of high prevalence in the Solomon Islands, which may reflect the high incidence of the B4a1a1a lineage in the region and/or the fact that our current study is the most in-depth study of complete mtDNA genomes in Oceania. Further study of other regions where these haplogroups are highly prevalent, i.e., Remote Oceania, would be of interest.
These analyses have demonstrated multiple back mutations from the 16247G allele, which has arisen only once during human evolution. To investigate how unusual this is, we checked the mtDNA phylogeny for other occurrences of repeated back-mutations from a mutation that arose only once during human evolution. There are only three other positions that, like position 16247, have mutations that arose only once and then subsequently back-mutated on a terminal branch of the mtDNA phylogeny. However in all of these cases (forward mutations: position G92A defines haplogroup Q1, position C1703T defines haplogroup N1b, and position C3780T defines haplogroup M49, PhyloTree Build 14) both the forward and back-mutation events are much older than position 16247, with the forward mutations dated to 37.5, 20, and 30 Kya respectively.14 The back-mutations in these cases are 15, 9, and 7 Kya, respectively, and define haplogroups Q1b, N1b1a, and M49b14 (PhyloTree Build 14).
The ages of these forward mutations and their respective back-mutations would suggest that they are far more stable than the mutational pattern we have observed at position 16247: the forward mutations were stable for at least 10,000 years before the back-mutations arose. In contrast, the forward mutation at position 16247 and the multiple subsequent back-mutations have all occurred within the last 7,200 years (Table 2). Because our study sequenced the lineages within one haplogroup to such great depth and numbers, it has enabled us to discover these independent back-mutation events. Further sequencing may identify other back mutation events within the Q1, N1b, and M49 lineages or within other lineages. Nevertheless, the instability associated with position 16247 (namely, a single forward mutation followed by multiple independent back mutations within a few thousand years) appears to be unique in the human mtDNA genome.
Sequences inferred to have back-mutations were examined for evidence of contamination by inspecting the aligned sequence reads from the GAIIx; instead of contamination, we found evidence for potential heteroplasmy in some samples. We then examined the sequence reads for potential heteroplasmies at position 16247 and all other diagnostic positions for the B4a1 lineage in all 536 samples. Based on the criterion of requiring at least three forward and three reverse reads for each allele at position 16247, we identified 54 samples which were potentially heteroplasmic; additionally, requiring the minor allele to have a frequency of at least 20% reduced this to 11 potentially heteroplasmic samples.23 No other diagnostic positions showed an equivalent level of potential heteroplasmy, and many positions showed none at all, which suggests that the potential heteroplasmies observed are not the result of sample contamination (Table S1). To further investigate this, we examined the trace files from the mtDNA HVR-1 Sanger sequencing that had been previously performed on these samples16 and found that six of the nine samples of interest who had trace files of sufficient quality also clearly exhibited heteroplasmy at position 16247 (Figure S2). Thus, there seems to be an excess of heteroplasmy at position 16247 in samples with predominantly the 16247G allele, suggesting that there may be ongoing somatic mutations and/or transmission of heteroplasmy at this position.
We next investigated potential reasons for the apparent instability of the derived 16247G nucleotide. This position is located in the noncoding control region and therefore would not have any deleterious effect on any mitochondrial genes; moreover, a search of the literature does not indicate any role for this position in replication, transcription, or other regulatory processes. However, the 16247G nucleotide appears to have an effect on DNA secondary structure, with the derived G nucleotide associated with a 10 bp stem-loop structure, beginning at position 16247, that is not predicted to occur with the ancestral A nucleotide at position 16247 (Figure 4). Given the repeated and rapid back-mutation events associated with the derived G nucleotide at this position, we hypothesize that this stem-loop structure increases the instability of the mtDNA genome and/or influences the rate of replication or transcription.
Figure 4.
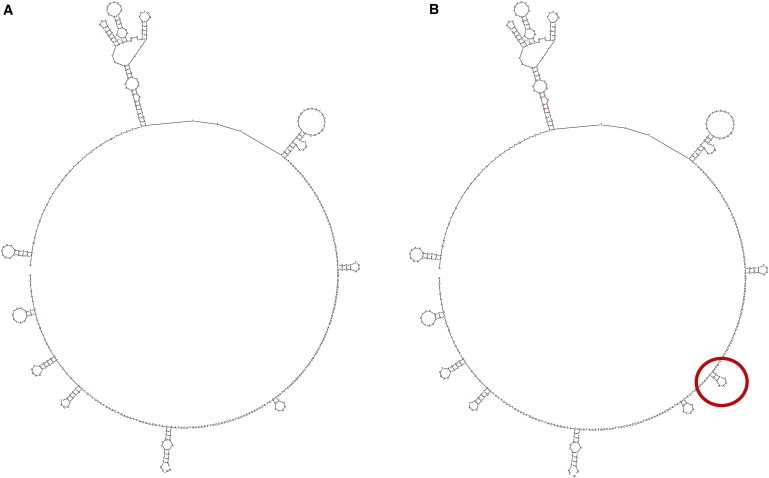
Predicted Secondary Structure Changes for the HVR-1 Region of the mtDNA Genome
With the ancestral A nucleotide at position 16247 (A) and with the derived G nucleotide at position 16247 (B). The additional 10 bp stem-loop structure predicted with the 16247G allele is circled.
Previously, the observation of a back-mutation at position 16247 in one sequence reported from Papua New Guinea22 was suggested to reflect recombination associated with heteroplasmy maintained over several generations.24 The multiple back-mutation events, identified because they occurred after several other mutations were acquired in various sequences, render recombination highly unlikely, because it is then not clear why recombination would influence only position 16247 and not any of the other mutations in these sequences.
This instability in position 16247 creates uncertainty for calling haplogroups within the B4a1 lineage. If the 16247G allele is present, haplogroup calling can simply follow established procedures (Figure 1). Indeed, even the back-mutation to the 16247A allele on the background of haplotype B4a1a1a1 is easy to classify, provided that whole mitochondrial genome sequencing has been completed and the state at position 6905 can be assessed. However, given the multiple independent back-mutation events at this position, classifying all of these as a single haplogroup (e.g., B4a1a1a1+16247!) has the undesirable effect of creating a paraphyletic haplogroup. Moreover, even more difficulty with haplogroup assignment arises with those sequences that appear to have back-mutated to 16247A from a background of B4a1a1a, because these sequences are indistinguishable from B4a1a1. For example, one heteroplasmic individual (VL08) falls into haplogroup B4a1a1 with no private mutations (Figure 3; Figure S2); without the existence of heteroplasmy it would be impossible to distinguish this sample from either B4a1a1 or B4a1a1a and indeed we cannot determine the direction of mutation in this sample (Figure 3; Figure S2).
The “Polynesian motif” is therefore still a useful marker, but its tendency to revert to the ancestral nucleotide underscores the importance of whole genome sequencing in place of control-region sequencing, and the further importance of investigating the relationship among individual sequences, for example in a network or tree, to identify back-mutation events that otherwise may be missed by haplotyping algorithms. This study also illustrates the value of high coverage sequencing of whole mtDNA genomes for detecting heteroplasmy and the benefits of sequencing large numbers of samples from single haplogroups for discovering unexpected and unusual mutation events. In particular, here we observed a different, and to our knowledge unique, phenomenon of a recent mutation that has occurred only once in the human mtDNA phylogeny and has subsequently undergone multiple independent back-mutations to the ancestral state.
Acknowledgments
We thank all sample donors for contributing to this study. We thank Sean Myles for providing access to original Sanger sequencing files. We thank Jing Li, Mingkun Li, and Gabriel Renaud for computational assistance and Sebastian Lippold and Alan McNally for helpful discussions. The sampling for this work, as part of the European Science Foundation EUROCORES Programme OMLL “Pioneers of Island Melanesia,” was supported by funds from the Max Planck Society and the EC Sixth Framework Programme under Contract no. ERAS-CT-2003-980409; the laboratory research was funded by the Max Planck Society.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Network and Network Publisher, http://fluxus-engineering.com
Phylotree, http://www.phylotree.org
Accession Numbers
The GenBank accession numbers for the 536 sequences reported in this paper are JX900327–JX900862.
References
- 1.Richards M., Oppenheimer S., Sykes B. mtDNA suggests Polynesian origins in Eastern Indonesia. Am. J. Hum. Genet. 1998;63:1234–1236. doi: 10.1086/302043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oppenheimer S.J., Richards M. Polynesian origins. Slow boat to Melanesia? Nature. 2001;410:166–167. doi: 10.1038/35065520. [DOI] [PubMed] [Google Scholar]
- 3.Hurles M.E., Nicholson J., Bosch E., Renfrew C., Sykes B.C., Jobling M.A. Y chromosomal evidence for the origins of oceanic-speaking peoples. Genetics. 2002;160:289–303. doi: 10.1093/genetics/160.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blust R. Subgrouping, circularity and extinction: some issues in Austronesian comparative linguistics. In: Zeitoun E., Li P.J., editors. Selected Papers from the Eight International Conference on Austronesian Linguistics. Academia Sinica; Taipei: 1999. pp. 31–94. [Google Scholar]
- 5.Moodley Y., Linz B., Yamaoka Y., Windsor H.M., Breurec S., Wu J.-Y., Maady A., Bernhöft S., Thiberge J.-M., Phuanukoonnon S. The peopling of the Pacific from a bacterial perspective. Science. 2009;323:527–530. doi: 10.1126/science.1166083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sykes B., Leiboff A., Low-Beer J., Tetzner S., Richards M. The origins of the Polynesians: an interpretation from mitochondrial lineage analysis. Am. J. Hum. Genet. 1995;57:1463–1475. [PMC free article] [PubMed] [Google Scholar]
- 7.Melton T., Clifford S., Martinson J., Batzer M., Stoneking M. Genetic evidence for the proto-Austronesian homeland in Asia: mtDNA and nuclear DNA variation in Taiwanese aboriginal tribes. Am. J. Hum. Genet. 1998;63:1807–1823. doi: 10.1086/302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gray R.D., Drummond A.J., Greenhill S.J. Language phylogenies reveal expansion pulses and pauses in Pacific settlement. Science. 2009;323:479–483. doi: 10.1126/science.1166858. [DOI] [PubMed] [Google Scholar]
- 9.Trejaut J.A., Kivisild T., Loo J.H., Lee C.L., He C.L., Hsu C.J., Lee Z.Y., Lin M., Lin M. Traces of archaic mitochondrial lineages persist in Austronesian-speaking Formosan populations. PLoS Biol. 2005;3:e247. doi: 10.1371/journal.pbio.0030247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melton T., Peterson R., Redd A.J., Saha N., Sofro A.S., Martinson J., Stoneking M. Polynesian genetic affinities with Southeast Asian populations as identified by mtDNA analysis. Am. J. Hum. Genet. 1995;57:403–414. [PMC free article] [PubMed] [Google Scholar]
- 11.Redd A.J., Takezaki N., Sherry S.T., McGarvey S.T., Sofro A.S., Stoneking M. Evolutionary history of the COII/tRNALys intergenic 9 base pair deletion in human mitochondrial DNAs from the Pacific. Mol. Biol. Evol. 1995;12:604–615. doi: 10.1093/oxfordjournals.molbev.a040240. [DOI] [PubMed] [Google Scholar]
- 12.Kayser M., Brauer S., Cordaux R., Casto A., Lao O., Zhivotovsky L.A., Moyse-Faurie C., Rutledge R.B., Schiefenhoevel W., Gil D. Melanesian and Asian origins of Polynesians: mtDNA and Y chromosome gradients across the Pacific. Mol. Biol. Evol. 2006;23:2234–2244. doi: 10.1093/molbev/msl093. [DOI] [PubMed] [Google Scholar]
- 13.Soares P., Rito T., Trejaut J., Mormina M., Hill C., Tinkler-Hundal E., Braid M., Clarke D.J., Loo J.H., Thomson N. Ancient voyaging and Polynesian origins. Am. J. Hum. Genet. 2011;88:239–247. doi: 10.1016/j.ajhg.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Behar D.M., van Oven M., Rosset S., Metspalu M., Loogväli E.-L., Silva N.M., Kivisild T., Torroni A., Villems R. A “Copernican” reassessment of the human mitochondrial DNA tree from its root. Am. J. Hum. Genet. 2012;90:675–684. doi: 10.1016/j.ajhg.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoneking M. Hypervariable sites in the mtDNA control region are mutational hotspots. Am. J. Hum. Genet. 2000;67:1029–1032. doi: 10.1086/303092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delfin F., Myles S., Choi Y., Hughes D., Illek R., van Oven M., Pakendorf B., Kayser M., Stoneking M. Bridging near and remote Oceania: mtDNA and NRY variation in the Solomon Islands. Mol. Biol. Evol. 2012;29:545–564. doi: 10.1093/molbev/msr186. [DOI] [PubMed] [Google Scholar]
- 17.Meyer M., Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010 doi: 10.1101/pdb.prot5448. Published online June 6, 2010. [DOI] [PubMed] [Google Scholar]
- 18.Maricic T., Whitten M., Pääbo S. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS ONE. 2010;5:e14004. doi: 10.1371/journal.pone.0014004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Oven M., Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009;30:E386–E394. doi: 10.1002/humu.20921. [DOI] [PubMed] [Google Scholar]
- 20.Hartmann A., Thieme M., Nanduri L.K., Stempfl T., Moehle C., Kivisild T., Oefner P.J. Validation of microarray-based resequencing of 93 worldwide mitochondrial genomes. Hum. Mutat. 2009;30:115–122. doi: 10.1002/humu.20816. [DOI] [PubMed] [Google Scholar]
- 21.Razafindrazaka H., Ricaut F.-X., Cox M.P., Mormina M., Dugoujon J.-M., Randriamarolaza L.P., Guitard E., Tonasso L., Ludes B., Crubézy E. Complete mitochondrial DNA sequences provide new insights into the Polynesian motif and the peopling of Madagascar. Eur. J. Hum. Genet. 2010;18:575–581. doi: 10.1038/ejhg.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ingman M., Gyllensten U. Mitochondrial genome variation and evolutionary history of Australian and New Guinean aborigines. Genome Res. 2003;13:1600–1606. doi: 10.1101/gr.686603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li M., Schönberg A., Schaefer M., Schroeder R., Nasidze I., Stoneking M. Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am. J. Hum. Genet. 2010;87:237–249. doi: 10.1016/j.ajhg.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White D.J., Wolff J.N., Pierson M., Gemmell N.J. Revealing the hidden complexities of mtDNA inheritance. Mol. Ecol. 2008;17:4925–4942. doi: 10.1111/j.1365-294X.2008.03982.x. [DOI] [PubMed] [Google Scholar]
- 25.Bandelt H.J., Forster P., Sykes B.C., Richards M.B. Mitochondrial portraits of human populations using median networks. Genetics. 1995;141:743–753. doi: 10.1093/genetics/141.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bandelt H.J., Forster P., Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 27.Polzin T., Daneshmand S.V. On Steiner trees and minimum spanning trees in hypergraphs. Oper. Res. Lett. 2003;31:12–20. [Google Scholar]
- 28.Soares P., Ermini L., Thomson N., Mormina M., Rito T., Röhl A., Salas A., Oppenheimer S., Macaulay V., Richards M.B. Correcting for purifying selection: an improved human mitochondrial molecular clock. Am. J. Hum. Genet. 2009;84:740–759. doi: 10.1016/j.ajhg.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.