

Abstract
Venous thrombosis is a major medical disorder caused by both genetic and environmental factors. Little is known about the genetic background of venous thrombosis in the Chinese population. A total of 1,304 individuals diagnosed with a first venous thrombosis and 1,334 age- and sex-matched healthy participants were enrolled in this study. Resequencing of THBD (encoding thrombomodulin) in 60 individuals with venous thrombosis and 60 controls and a functional assay showed that a common variant, c.−151G>T (rs16984852), in the 5′ UTR significantly reduced the gene expression and could cause a predisposition to venous thrombosis. Therefore, this variant was genotyped in a case-control study, and results indicated that heterozygotes had a 2.80-fold (95% confidence interval = 1.88–4.29) increased risk of venous thrombosis. The THBD c.−151G>T variant was further investigated in a family analysis involving 176 first-degree relatives from 38 index families. First-degree relatives with this variant had a 3.42-fold increased risk of venous thrombosis, and their probability of remaining thrombosis-free was significantly lower than that of relatives without the variant. In addition, five rare mutations that might be deleterious were also identified in thrombophilic individuals by sequencing. This study is the largest genetic investigation of venous thrombosis in the Chinese population. Further study on genetics of thrombosis should focus on resequencing of THBD and other hemostasis genes in different populations.
Introduction
Venous thrombosis (VT) and its sequela, postthrombotic syndrome, are major medical disorders throughout the world. VT is a common disease that has a high morbidity and mortality rate shortly after the event.1 The relevant data have shown that the incidence is approximately 1–3 per 1,000 persons per year in Western countries.2 Early epidemiological studies have suggested that the directly standardized incidence of all VT events is significantly higher among African Americans (138–141 cases per 100,000 individuals per year) than among Caucasians (80–117 cases per 100,000 individuals per year), significantly lower among Hispanic populations (55–61.5 cases per 100,000 individuals per year), and strikingly lower among Asians and Pacific Islanders (21–29 cases per 100,000 individuals per year).3,4 With the improvement of its diagnosis and the development of access to healthcare, VT in Asian populations is now believed to be common,5,6 and in particular, the absolute number is thought to be great as a result of the large population. Several genetic and environmental risk factors are involved in the pathogenesis of this complex disease.7–11 Acquired and environmental factors include advancing age, immobilization, multiple trauma, major general surgery, pregnancy, superficial vein thrombosis, oral contraceptives, hormone replacement, inflammation, and active cancer.12–14 Ethnic differences are implicated in the inherited traits of VT.15 A poor APC (plasma to activated protein C) response caused by the factor-V-Leiden mutation in the coding sequence of F5 (MIM 612309),16–18 a common prothrombin-G20210A mutation in the 3′ UTR of F2 (MIM 176930),19 and the antithrombin-Cambridge-II mutation in SERPINC1 (MIM 107300)20 are common genetic risk factors for VT in whites. However, these polymorphisms are rare in Asians, including Chinese populations. Little is known about the genetic background of VT, and no common genetic risk factors have been identified in the Chinese population until recently.21–23
However, despite the discoveries described above, the underlying molecular mechanisms of a considerable number of inherited thrombotic events remain unsolved.11 Mounting evidence indicates that genetic variation within the procoagulant, anticoagulant, and fibrinolytic pathways might be a potential risk factor for VT.24–28 Thrombomodulin (TM, encoded by THBD [MIM 188040]), a critical component of the protein C pathway, is a glycosylated type I transmembrane protein of 557 amino acids and is expressed mainly on the endothelial surface of blood vessels.29 The anticoagulant property of TM is mediated by its interaction with thrombin (the activated form of prothrombin) and protein C (PC, encoded by PROC [MIM 612283]). The high-affinity binding of thrombin to TM results in a greater than 1,000-fold amplification of the rate of thrombin-dependent PC activation. Thus, APC (the activated form of PC) inhibits the coagulation pathway by proteolysis of coagulation factor Va (the activated form of coagulation factor V) and coagulation factor VIIIa (the activated form of coagulation factor VIII, encoded by F8 [MIM 300841]).30 Akin to the cofactor function in anticoagulation, TM is also reported to be associated with fibrinolysis, embryonic development, arteriosclerosis, inflammation regulation, and tumor growth and metastasis.31–35
On the basis of the crucial antithrombotic role of TM, we investigated THBD as a candidate gene and report a common mutation for VT in the Chinese population in the present study. Then, we analyzed all these common genetic risk factors identified in the Chinese population, and the results might explain why ethnicity affects the incidence of VT.36
Subjects and Methods
Study Population
Some of the current study population was based on the cohort from the previous case-control study (1,003 VT individuals and 1,031 controls).21 To obtain fresh plasma samples and introduce more known risk factors for thrombosis, we recruited some participants from this cohort again. Meanwhile, we enrolled another cohort of 403 consecutive unselected individuals diagnosed with VT and 471 controls. In total, 1,406 Chinese individuals were diagnosed with VT and were registered at the Hubei Clinical and Research Center of Thrombosis and Hemostasis between March 1, 2008 and June 30, 2012. Among the 1,406 eligible individuals with VT, 33 died of VT or its complications before inclusion, 49 were out of contact, and 20 refused to participate because they were in the end stage of a disease, such as malignancy. Therefore, these 102 individuals did not participate in the case-control study. In addition, 168 of the 1,502 eligible healthy Chinese subjects were out of contact or refused to take part in this genetic study. Finally, a total of 1,304 cases and 1,334 controls were available.
The sample collection and the criteria for diagnosis of VT were previously described.22 The validation of VT was based on the clinical manifestations, a D-dimer assay, and compression ultrasound or venography (for deep-vein thrombosis), as well as ventilation and perfusion lung scanning, computed-tomography angiography, or pulmonary angiography (for pulmonary thromboembolism). Age- and sex-matched control participants, without an individual or family history of thrombosis (arterial thrombosis and VT), were enrolled from a community screening program during the same period. The case and control groups had similar median ages (52 years for both), 5th–95th percentile age (26–73 years versus 27–74 years), and sex distribution (49% male for both groups).
The demographic data and acquired risk factors, including age, gender, smoking, malignancy, sedentariness or immobility, pregnancy or puerperium, oral contraceptives, hormone-replacement therapy, and lupus anticoagulant, were recorded. The citrated blood sample was separated into plasma and buffy-coat cells by centrifugation. Genomic DNA was extracted from the buffy-coat cells according to standard procedures. Plasma lupus anticoagulant was detected by an activated-partial-thromboplastin-time (APTT)-based functional assay with commercial reagents (Dade Behring-Siemens Healthcare Diagnostics, Margurg, Germany) on a Sysmex CA 7000 Analyzer (Sysmex, Kobe, Japan). The two common polymorphisms (PROC c.565C>T [p.Arg189Trp] and c.574_576dup/del [p.Lys192del]) associated with VT in the Chinese population were genotyped in the study population with the PCR-RFLP (restriction-fragment-length polymorphism) methods as described.21,22
We named these series of studies by evaluating the risk of thrombophilia or VT as MAGIC (multiple assessments of genetic risk factors for thrombophilia in the Chinese population). All the procedures followed in this study were approved by the ethics committee of Union Hospital at Huazhong University of Science and Technology in accordance with the ethical standards of human experimentation and complied with the principles expressed in the Declaration of Helsinki. Written informed consent was obtained from all participants.
Resequencing of THBD
The putative promoter, 5′ UTR, coding region, and 3′ UTR of THBD were screened with the PCR and sequencing method in 60 cases randomly selected from the VT individuals who were free from antithrombin (encoded by SERPINC1 [MIM 107300]), PC, or PS deficiency, the PROC c.565C>T (p.Arg189Trp) mutation, and the PROC c.574_576del (p.Lys192del) variant. The identified variants were further detected by resequencing in 60 randomly selected controls. The primer-pair sequences used in the amplification are summarized in Table S1, available online. The resequencing was performed on an ABI PRISM 3730XL DNA sequencer (Applied Biosystems, Carlsbad, CA, USA). The RefSeq accession numbers for THBD and protein sequences reported in this paper are NG_012027.1, NM_000361.2, and NP_000352.1.
Genotyping
On the basis of the genetic screening, a SNP (c.−151G>T) in the 5′ UTR was supposed to be associated with VT and was subsequently investigated in the study population. This variant was genotyped with the PCR-RFLP assay (Figure S1). Primers 5′-GCACTTCCTTCCTTTTCCCGA-3′ and 5′-CAGAGGGGCACAGGACGC-3′ were employed in the PCR reaction. The digestion was performed by incubation of 2.5 U of Mwo I and 1 μl of 10× buffer Tango (Fermentas, Burlington, ON, Canada) with 10 μl of the PCR product for 12 hr at 37°C. The digestion fragments were separated by 2% agarose-gel electrophoresis. A total of 96 DNA samples were randomly selected and subjected to sequencing for validation of the genotyping results. The accuracy of the RFLP assay was 100%.
Measurement of Plasma TM Antigen
Plasma soluble thrombomodulin (sTM) was measured in 48 heterozygotes for the c.−151G>T variant and in 48 age- and sex-matched normal individuals from the case-control study with the use of the Human Thrombomodulin-BDCA-3 Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA).
Family Analysis of VT Risk
Family members of 38 carriers (29 individuals with VT and 9 healthy controls from the case-control study) of the c.−151G>T variant agreed to participate in the family analysis. Each family comprised at least two members (one c.−151G>T carrier and one noncarrier). For the avoidance of selection bias, the analyses were confined to the first-degree relatives of the 38 index carriers. In total, 176 first-degree relatives from 38 families were enrolled. Written inform consent was obtained, and blood samples were collected for coagulation tests (antithrombin amidolytic activity, PC amidolytic activity, and PS cofactor activity) and genotyping. The medical histories and acquired risk factors for thrombosis were also obtained. Thrombosis was also confirmed on the basis of objective techniques and documented in their medical records. For each group (affected and unaffected family relatives), we calculated the annual incidence of thrombosis by dividing the number of thrombotic events by the total number of affected individuals per year. The years of follow-up were defined as the time period between the date of birth and either the date of the first episode of VT, if any, or June 1, 2012. The relative risk for the c.−151G>T mutation was calculated with only the first thrombotic event during the follow-up.
Plasmid Construction, Cell Culture, Transfection, and Luciferase Assay
Fragments of the wild-type and mutated promoter and 5′ UTR, spanning nucleotides −419 to −64 (with respect to the ATG codon), were amplified and subcloned into the pGL3-basic vector (Promega, Madison, WI, USA) containing the firefly luciferase gene (Fluc). The genomic DNA of both an individual with the c.−151G>T mutation and a normal subject were used as templates. The primers used in the plasmid constructions are shown in Table S1. The constructs were confirmed by sequencing. Human umbilical-vein endothelial cells (HUVECs), human embryonic kidney (HEK) 293 cells, and COS-7 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) or Cellbank of the Chinese Academy of Sciences (Shanghai, China) and were cultured under the recommended conditions. Approximately 90% confluent cells in 96-well plates were transiently cotransfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) with 300 ng of the pGL3 luciferase reporter vectors and 10 ng of the pRL-SV40 inner control vector (Promega, Madison, WI, USA), which contained the Renilla luciferase gene (Rluc). Four types of pGL3 vector were separately transfected: pGL3 with the normal 5′ UTR (pGL3-WT), pGL3 with the mutant 5′ UTR (pGL3-MT), pGL3 with an efficient SV40 promoter (pGL3-promoter), and pGL3 lacking any promoter (pGL3-basic). The relative luciferase activity (the ratio of F-luciferase activity to R-luciferase activity) was determined 36 hr later with the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Six independent experiments were performed.
Statistical Analysis
The statistical analyses were performed with SPSS 13.0 software (SPSS, Chicago, IL, USA). Continuous variables were expressed as means ± SD. The comparison between groups was performed by an unpaired Student’s t test or Mann-Whitney U test, as appropriate. Categorical variables were expressed as percentages, and the differences were analyzed by a chi-square test. A Cox regression was performed for estimating the relative risk (hazard ratio) of thrombosis in the first-degree relatives with the TM defect. The thrombosis-free survival was assessed with Kaplan-Meier curves. A two-tailed p < 0.05 was considered to be statistically significant.
Results
Identification of Variants in THBD
The resequencing of THBD in 60 individuals with VT revealed ten genetic variants, including one in the promoter region, one in the 5′ UTR, five in the coding sequence, and three in the 3′ UTR (Table 1). On the basis of bioinformatics tools (PolyPhen-2 and miRDB),37,38 c.256C>A (p.Leu86Ile), c.1418C>T (p.Ala473Val), and three 3′ UTR variants might be benign or tolerated. No differences in the genotype distribution of g.4959G>A and c.1456G>T (p.Asp486Tyr) were observed between the groups. In addition, c.569C>G (p.Ser190Trp) and c.636C>A (p.Ser212∗) were located at the hydrophobic region between the lectin-like and EGF-1 domains and were highly conserved. Especially, c.636C>A (p.Ser212∗), a nonsense mutation, leads to a truncated protein lacking 346 amino acids. These two variants might have deleterious consequences. However, neither of them was observed in 60 healthy controls, indicating rare mutations. Therefore, these variants were not further studied. The remaining variant, c.−151G>T, was present in three individuals with VT and only one control. This variant was in low linkage disequilibrium with other polymorphisms and was thus detected in the whole study population.
Table 1.
Variants Identified in THBD in 60 VT Individuals and 60 Controls
| dbSNP ID | HGVS Name | Gene Region | Number of VT Cases (Hetero/Homo) | Number of Controls (Hetero/Homo) |
|---|---|---|---|---|
| rs13306848 | c.−202G>A | promoter | 8 (8/0) | 7 (7/0) |
| rs16984852 | c.−151G>T | 5′ UTR | 3 (3/0) | 1 (1/0) |
| NA | c.256C>A (p.Leu86Ile) | CDS | 8 (8/0) | 9 (9/0) |
| NA | c.569C>G (p.Ser190Trp) | CDS | 1 (1/0) | 0 (0/0) |
| NA | c.636C>A (p.Ser212∗) | CDS | 1 (1/0) | 0 (0/0) |
| rs1042579 | c.1418C>T (p.Ala473Val) | CDS | 13 (9/4) | 12 (8/4) |
| NA | c.1456G>T (p.Asp486Tyr) | CDS | 1 (1/0) | 1 (1/0) |
| rs13306852 | c.∗26C>T | 3′ UTR | 3 (3/0) | 3 (3/0) |
| NA | c.∗50A>C | 3′ UTR | 1 (1/0) | 1 (1/0) |
| rs3176134 | c.∗277G>A | 3′ UTR | 1 (1/0) | 1 (1/0) |
The following abbreviations are used: HGVS, human genome variation society; hetero, heterozygous; homo, homozygous; NA, not available; and CDS, coding sequence.
Association between the c.−151G>T Variant and VT Risk
The characteristics and demographic data of the study population are presented in Table 2. According to the genotyping results, the heterozygous THBD c.−151G>T variant was identified in 35 VT individuals and 13 controls. The homozygous genotype was absent in all the participants. Thus, carriers of the variant allele in the heterozygous state had a 2.80-fold (95% confidence interval [CI] = 1.48–5.32) risk of developing VT (Table 3). The statistical power was 0.913, and the type-I-error probability was 0.05. In addition, with respect to the two common genetic risk factors recently reported in the Chinese population, the PROC c.565C>T mutation was present in 5.21% of the VT individuals and in 0.90% of the controls. Likewise, the PROC c.574_576del polymorphism was detected in 6.52% and 2.40% of the subjects from the case and control groups, respectively.
Table 2.
Data from the Participants Enrolled in the MAGIC Study
| Variable |
Cases (n = 1,304) |
Controls (n = 1,334) |
p Value | |||
|---|---|---|---|---|---|---|
| n | % | n | % | |||
| Age (years) | 51.7 ± 13.8 | 51.3 ± 14.2 | 0.468 | |||
| Sex | male | 638 | 48.9% | 649 | 48.7% | 0.887 |
| female | 666 | 51.1% | 685 | 51.3% | ||
| Smoking | yes | 271 | 20.8% | 240 | 18.0% | 0.070 |
| no | 1,033 | 79.2% | 1,094 | 82.0% | ||
| Malignancy | yes | 81 | 6.2% | 6 | 0.4% | 1.179 × 10−16 |
| no | 1,223 | 93.8% | 1,328 | 99.6% | ||
| Immobility | yes | 30 | 2.3% | 15 | 1.2% | 0.038 |
| no | 1,274 | 97.7% | 1,319 | 98.8% | ||
| Pregnancy or puerperium | yes | 34 | 2.6% | 18 | 1.3% | 0.020 |
| no | 1,270 | 97.4% | 1,316 | 98.7% | ||
| OCT or HRP | yes | 16 | 1.2% | 7 | 0.5% | 0.052 |
| no | 1,288 | 98.8% | 1,327 | 99.5% | ||
| Lupus anticoagulant | positive | 65 | 5.0% | 12 | 0.9% | 4.61 × 10−10 |
| negative | 1,239 | 95.0% | 1,322 | 99.1% | ||
Years of age are expressed as means ± SD. The age of cases is the age at the first incident, and the age of controls is the age at enrollment. Age was further divided into three levels: under 40 years, 40–60 years, and over 60 years. Immobility and OCT reflect the 4 weeks prior to the VT incident. The following abbreviations are used: VT, venous thrombosis; OCT, oral contraceptives; and HRT, hormone-replacement therapy.
Table 3.
Association between the Common Genetic Risk Factors Identified and VT in the Chinese Population
| Genotype |
Cases |
Controls |
Risk of VT |
PAR | |
|---|---|---|---|---|---|
| n = 1,304 | n = 1,334 | OR (95% CI) | p Value | ||
| PROC c.565C>T (p.Arg189Trp) | |||||
| C/C | 1,236 (94.79%) | 1,322 (99.10%) | 1 | - | - |
| C/T | 68 (5.21%) | 12 (0.90%) | 6.06 (3.26–11.25) | 1.03 × 10−10 | 4.67% |
| PROC c.574_576dup/del (p.Lys192del) | |||||
| dup/dup | 1,219 (93.48%) | 1,302 (97.60%) | 1 | - | - |
| dup/del + del/del | 85 (6.52%) | 32 (2.40%) | 2.84 (1.88–4.29) | 2.77 × 10−7 | 4.14% |
| THBD c.−151G>T | |||||
| G/G | 1,269 (97.32%) | 1,321 (99.03%) | 1 | - | - |
| G/T | 35 (2.68%) | 13 (0.97%) | 2.80 (1.48–5.32) | 1.02 × 10−3 | 1.48% |
Hardy-Weinberg equilibrium tests for PROC c.565C>T, PROC c.574_576dup/del, and THBD c.−151G>T are p = 0.869, 0.657, and 0.858, respectively. The following abbreviations are used: VT, venous thrombosis; OR, odds ratio; CI, confidence interval; and PAR, population-attributable risk.
Association between the c.−151G>T Genotype and sTM Levels
The plasma sTM antigen was determined in 48 heterozygotes (22 males and 26 females) for this variant and in 48 subjects (22 males and 26 females) with a normal genotype. As illustrated in Figure 1, the mean values of sTM of variant carriers were significantly lower than those of noncarriers by sex group (in men: 6.18 ± 1.59 ng/ml versus 7.36 ± 1.25 ng/ml, p = 0.009; in women: 5.15 ± 1.29 ng/ml versus 6.00 ± 1.00 ng/ml, p = 0.011). Likewise, in the case group, the mean sTM level was significantly lower in THBD c.−151G>T carriers (5.74 ± 1.66 ng/ml) than in noncarriers (6.59 ± 1.38, p = 0.024). In the control group, the mean sTM level was also lower in variant carriers (5.30 ± 0.98 ng/ml) than in noncarriers (6.72 ± 1.14 ng/ml, p = 0.002).
Figure 1.
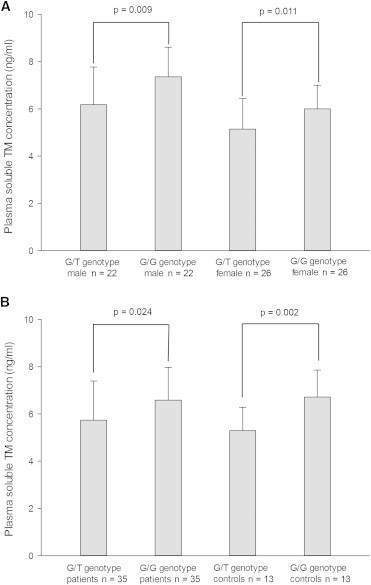
Association between the sTM Level and the c.−151G>T Genotype
(A) Sex-matched data. Error bars represent the SD.
(B) Data matched for case-control status. The sTM level was determined in 48 heterozygotes and 48 individuals with a normal genotype via the ELISA (enzyme-linked immunosorbent assay) method. Each sample was tested in duplicate. Error bars represent the SD.
Thrombotic Risk in First-Degree Relatives and Lifetime Risk of Thrombosis
Overall, 81 of the total 176 relatives were identified as carriers of the c.−151G>T variant, and one of these had the PROC c.574_576del variant. The other 95 relatives had a normal THBD genotype, and of these, one had the PROC c.574_576del variant and one had PS deficiency (PS cofactor activity: 39 U/dl). In total, 16 thrombosis events were confirmed in these relatives. Precipitating factors (pregnancy, puerperium, oral contraceptives, immobility, and malignancy) for the first episodes of thrombosis were analyzed. Thrombosis occurred spontaneously in seven, whereas predisposing factors were present in the other nine. As shown in Table 4, first-degree relatives with the c.−151G>T variant had a higher incidence of thrombosis than did those without this variant (3.41/1,000 versus 0.94/1,000 persons per year), as well as a higher relative thrombosis risk (3.42; 95% CI = 1.03–11.33). When analysis was performed for VT individuals’ relatives, first-degree relatives with the c.−151G>T variant had a higher incidence of thrombosis than did those without this variant (3.30/1,000 versus 0.95/1,000 persons per year), as well as a higher relative thrombosis risk (3.38; 95% CI = 1.02–11.14). Likewise, when analysis was performed for controls’ relatives, the incidence of thrombosis was also greater in the first-degree relatives with the c.−151G>T variant (3.83 per 1,000 persons per year) than in those without this variant (0.90 per 1,000 persons per year). Although the affected relatives had a higher relative risk (2.00; 95% CI = 0.18–22.06) of thrombosis, the analysis was not statistically significant (p = 0.571), which might be due to the limited sample size in the group of controls’ relatives. The Kaplan-Meier analysis showed that the probability of remaining thrombosis-free was significantly lower (p = 0.012 for analysis of the VT individuals’ relatives group; p = 0.029 for analysis of the combined relatives) in relatives with the c.−151G>T variant than in those without the variant (Figure 2). For the total first-degree relatives, the probability of being cumulative thrombosis-free at age 55 years was 76.6% and 97.6% for affected and unaffected relatives, respectively.
Table 4.
Family Analysis of Thrombotic Risk in c.−151G>T Carriers and Noncarriers
| Parameters |
VT Persons’ Relatives |
Controls’ Relatives |
Total Relatives |
|||
|---|---|---|---|---|---|---|
| Carriers | Noncarriers | Carriers | Noncarriers | Carriers | Noncarriers | |
| Total number of first-degree relatives | 63 | 72 | 18 | 23 | 81 | 95 |
| Median age (range) in years | 46 (12–66) | 43 (18–66) | 45 (16–69) | 46 (34–71) | 46 (12–69) | 43 (18–71) |
| Male/female | 35/28 | 41/31 | 10/8 | 16/7 | 45/36 | 57/38 |
| Number of VT events | 9 | 3 | 3 | 1 | 12 | 4 |
| Median age at first thrombosis in years | 46 | 37 | 51 | 62 | 50 | 46 |
| Years of follow-upa | 2,731 | 3,144 | 784 | 1,106 | 3,515 | 4,250 |
| Events per 1,000 persons per year | 3.30 | 0.95 | 3.83 | 0.90 | 3.41 | 0.94 |
| Relative risk for thrombosis (95% CI)b | 3.38 (1.02–11.14) | 1 | 2.00 (0.18–22.06) | 1 | 3.42 (1.03–11.33) | 1 |
The annual incidence of thrombosis was calculated by a division of the number of thrombotic events by the total number of affected individuals per year. The following abbreviations are used: VT, venous thrombosis; and CI, confidence interval.
The years of follow-up are defined as the time period between the date of birth and either the date of the first episode of VT, if any, or June 1, 2012, and are the total combined number of years of follow-up for all individuals.
Data were analyzed with a Cox regression, and the relative risk for the c.−151G>T mutation was calculated with only the first thrombotic event during the follow-up.
Figure 2.
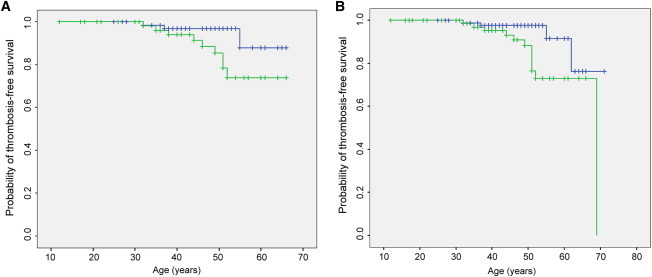
Thrombosis-free Survival in Relatives
(A) Analysis was performed for VT persons’ relatives.
(B) Analysis was performed for the total relatives. The probability of thrombosis-free survival in first-degree relatives was estimated by Kaplan-Meier analysis. The green plot shows relatives with the c.−151G>T mutation, and the blue plot shows relatives without the mutation.
Consequence of the c.−151G>T Variant on Gene Expression In Vitro
For assessment of the functional relevance of the variant, a luciferase assay was performed. In Figure 3, the gene-expression levels of different constructs are measured as relative light units (the ratio of F-luciferase activity to R-luciferase activity). The pGL3-basic vector, which did not contain any promoter elements, was used as a negative control. Compared with the wild-type allele, the c.−151G>T variant significantly reduced the reporter gene-expression level in all three kinds of cells (HUVECs: 0.88 ± 0.14 versus 0.56 ± 0.06, p = 4.62 × 10−4; HEK 293 cells: 1.94 ± 0.12 versus 1.14 ± 0.08, p = 9.13 × 10−8; and COS-7 cells: 2.56 ± 0.10 versus 1.88 ± 0.12, p = 9.21 × 10−7).
Figure 3.
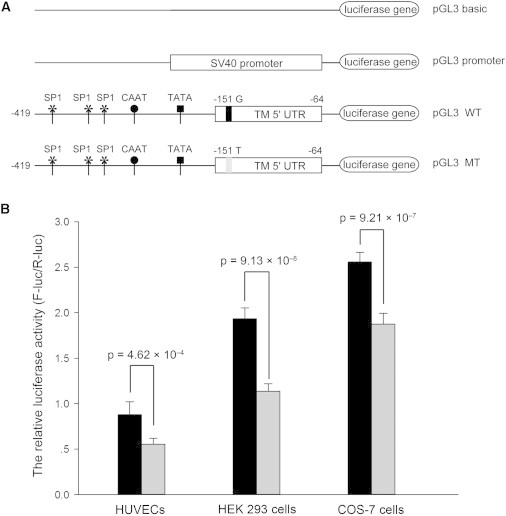
Gene-Expression Levels Determined by the Luciferase Assay
(A) Schematic of reporter gene constructs. Known regulatory elements (SP1 sites, CAAT box, and TATA box) in the promoter of THBD are schematically represented.
(B) Luciferase activity. Transfection and luciferase assay were performed in three kinds of cells (HUVECs, HEK 293 cells, and COS-7 cells). The pGL3-basic vector was used as a negative control. The luciferase activities are expressed as the ratio of firefly to Renilla luciferase units. The values represent means ± SD of six independent experiments. The black column represents the wild-type construct, and the gray column represents the mutant construct. Error bars represent the SD.
Rare Mutations
In order to verify that the prevalence of deleterious mutations in THBD among individuals with VT was relatively high in the Chinese population, we resequenced this locus in another set of thrombophilic individuals. That was, another 48 individuals enrolled in our study population were selected for THBD analysis. The selection criterion was based on an onset of age under 45 years accompanied with a recurrent thrombosis history or a family thrombosis history. Direct sequencing revealed another three variants in two severe thrombophilic individuals. A 10-year-old boy was found to be a compound heterozygote for the c.376G>T (p.Asp126Tyr) mutation and the c.∗6_∗23del variant (an 18 deoxynucleotide deletion, which was 6 deoxynucleotides downstream of the stop codon) according to the clone-sequencing analysis. A 45-year-old man was identified to be a heterozygote for the c.659T>G (p.Leu220∗) nonsense mutation. Both of the two persons suffered from at least three episodes of thrombosis, including arterial thrombosis in the lower extremity. These two persons, together with the two previously described individuals carrying either p.Ser190Trp or p.Ser212∗, were chosen for plasma sTM assay and familial analysis when family members were available. Table 5 shows that all these probands had a decreased sTM level and that mutations p.Ser190Trp and p.Ser212∗ cosegregated with a reduced sTM level in the two pedigrees. However, other members carrying these mutations had not suffered from an arterial-thrombosis or a VT event yet. With respect to the 3′ UTR mutation (c.∗6_∗23del), it occurred at the miR-3155a and miR-3155b target site (5′-GAGCCTG-3′), and it therefore might affect mRNA stability and cause suppression of protein translation. Moreover, consequences of the two nonsense mutations are probably deleterious. Therefore, the five mutations are likely to be causative. These further support the fact that THBD variants might be a relatively prevalent cause of VT.
Table 5.
Relationship between Phenotypes and Genotypes of the Five Rare Mutations
| Individual | Age (Years) | Clinical Manifestation | Genotype | sTM Level (ng/ml) |
|---|---|---|---|---|
| c.569C>G (p.Ser190Trp) | ||||
| Proband | 44 | DVT | mutation carrier | 5.19 |
| Older brother | 49 | none | mutation carrier | 4.86 |
| Son | 19 | none | noncarrier | 6.40 |
| c.636C>A (p.Ser212∗) | ||||
| Proband | 44 | DVT | mutation carrier | 4.70 |
| Younger brother | 40 | none | noncarrier | 7.61 |
| Young sister | 39 | none | noncarrier | 6.45 |
| Daughter 1 | 21 | none | noncarrier | 6.31 |
| Daughter 2 | 16 | none | mutation carrier | 3.95 |
| c.659T>G (p.Leu220∗) | ||||
| Proband | 45 | recurrent DVT, PE, andATE | mutation carrier | 4.46 |
| c.376G>T (p.Asp126Tyr) combined with c.∗6_∗23del | ||||
| Proband | 10 | recurrent DVT, PE, and ATE | mutation carrier | 3.22 |
All of these probands are Chinese males. Their family members did not experience a symptomatic thrombosis. The reference intervals of sTM in our laboratory are 5.60–9.94 ng/ml for men and 4.82–8.21 ng/ml for women. Family members of index individuals carrying the c.659T>G, c.376G>T, and c.∗6_∗23del mutations were not available. The following abbreviations are used: sTM, soluble thrombomodulin; DVT, deep-vein thrombosis; PE, pulmonary embolism; and ATE, arterial thromboembolism.
Discussion
This study investigated the relationship between possible THBD variants and the development of VT and revealed that the c.−151G>T polymorphism (rs16984852: G>T) causes a predisposition to VT. The case-control study showed that the heterozygous genotype for this variant confers an approximately 2.80-fold increased risk of VT in the Chinese population. The family study indicated that first-degree relatives bearing this variant have a 3.42-fold increased risk of VT. These findings are further supported by in vitro functional luciferase assays in which the c.−151T mutant allele reduced the total expression level to 59%–73% of the c.−151G wild-type in all three cell types.
The intronless human THBD is located in chromosomal region 20p11.21 and is 3.6 kb in length.39 The association between THBD variation and the susceptibility to VT is biologically plausible because of the essential role of TM in anticoagulation. However, to date, only a few mutations have been confirmed to be associated with arterial-thrombosis and VT events, and these mutations were all rare and only observed in some special cases or populations.40–43 As for THBD polymorphisms, several studies have mainly focused on three common polymorphisms (c.−202G>A, c.127G>A [p.Ala43Thr], and c.1418C>T [p.Ala473Val]). Sugiyama et al.44 evaluated the contribution of genetic variations in THBD to sTM level and deep-vein thrombosis in 118 Japanese VT individuals and controls. They indicated that the mean values of sTM were lower in C-allele carriers of c.∗1001A>C (in tight linkage disequilibrium with c.1418C>T [p.Ala473Val]) than in noncarriers and that the CC homozygous genotype might be a genetic risk factor for thrombosis in Japanese men. On the contrary, studies in white populations45–48 have not shown any association between the c.−202G>A, c.127G>A (p.Ala43Thr), and c.1418C>T (p.Ala473Val) variants and sTM levels or an obvious susceptibility to thrombosis, suggesting that these known polymorphisms are not likely to be a common risk factor for VT in the general population. In the present study, the c.−202G>A and c.1418C>T variants were observed in the Chinese population, and the similar genotype distribution in cases and controls also indicates that they might be no more than an extremely weak, if at all, risk factor for thrombosis. In addition, the c.−151G>T variant, which was strongly suggested to be associated with a decreased level of gene expression and an increased risk of thrombosis in this research, was not identified in those previous studies. Therefore, thrombotic disease caused by inherited TM deficiency has remained largely unexplored because of the difficulty in examining the phenotype of this endothelial transmembrane protein. Given that sTM was present in human plasma, we tested whether the plasma concentrations of sTM might reflect the endothelial expression of thrombomodulin. The ELISA (enzyme-linked immunosorbent assay) showed that the mean plasma sTM level of the c.−151G>T carriers was significantly lower than that of noncarriers. Our results are in accordance with the Atherosclerosis Risk in Communities Study, which suggested that there was a strong, graded, and inverse relation association between sTM concentration and coronary heart disease.49 Therefore, plasma concentrations of sTM might reflect endothelial expression of thrombomodulin to some degree. However, the sample size of the sTM-antigen test was rather small as a result of the relatively low penetration of this variant. Therefore, this observation should be validated by a larger study.
With the development of access to healthcare and epidemiological studies, an increase in the annual incidence of VT has been observed in the past decade, and the incidence of VT in Asian populations has been estimated to be 50–115 per 100,000 individuals per year.50–53 The difference in the incidence between Asian and Western populations is smaller than previously reported, and VT is believed to be a common disease in Asians. However, the prevalence of VT is, in fact, still lower in Asians than in whites. The ethnic variation in VT should be attributed to the genetic-background disparity among ethnicities. At present, we have identified three common genetic risk factors (PROC c.565C>T, PROC c.574_576dup/del, and THBD c.−151G>T) in the Chinese population; their respective prevalences are approximately 0.90%, 2.40%, and 0.98%, and their estimated population-attributable risk is 4.67%, 4.14%, and 1.48%, respectively (Table 3). These variants conferred a 6.06-, 2.84-, and 2.80-fold increased risk, respectively, of developing VT. Taken together, these mild to moderate thrombophilic risk factors account for ∼10% of the total VT events in the general population. It is worth noting that the prevalence of these polymorphisms is much lower than that of the most common genetic risk factor in whites, the factor V Leiden (5%–10%),24 which is associated with an approximately 5-fold higher risk of VT. This difference might partially explain the relatively lower incidence of thrombosis in Asians compared to whites.
Apart from the genetic-risk-factor findings, this study might have other important inspirations. First, we have hitherto identified three common variants in the thrombosis-pathway genes. Therefore, resequencing of the hemostasis genes might be useful for unraveling unknown genetic defects for thrombophilia and VT in future research, which is particularly helpful for Africans because these populations are reported to have the highest incidence of VT among all ethnicities; however, few common genetic risk factors have been identified in Africans so far.36 Second, the minor allele frequencies (MAFs) of the three variants are all relatively low (prevalences between 0.90% and ∼2.40%, MAF < 5%) but could confer high risk (odds ratio [OR] > 2) of VT. This is quite different from most previous genetic studies, such as genome-wide association studies54–56 in which the identified variants often had a higher frequency (MAF > 5% or 10%) but a lower risk (OR < 1.5). Hence, low-frequency polymorphisms might play a more important role in the development of VT and other complex disorders, and it might therefore be important to include low-penetrance SNPs (MAF < 5%) in future disease-association studies. Third, in addition to the common c.−151G>T variant, five other rare mutations (c.376G>T, c.569C>G, c.636C>A, c.659T>G, and c.∗6_∗23del) in THBD were also identified by sequencing in 108 individuals with VT. These mutations, especially the nonsense variants, might also be causative. This proportion (5/108) was relatively high. As a result, one might expect that more mutations in THBD would be identified in the entire VT group and that an inherited TM defect would be more common in Chinese VT individuals. We should pay more attention to THBD mutations in thrombosis-affected persons. Further systematic research on the genetics of TM defects is thus warranted. Finally, investigating the link between the THBD c.−151G>T variant and arteriosclerotic vascular disorders, such as coronary artery disease, might be valuable because it has been well established that VT and atherothrombosis share some similar physiopathological processes57,58 and that there is a strong association between TM defect and atherosclerosis.59,60
In conclusion, this study is the largest on the genetic background of VT in the Chinese population. We reported that the common THBD c.−151G>T variant is associated with both impaired 5′ UTR function and an increased risk of VT. In combination with the two PROC variants, these three common genetic risk factors could account for approximately 1/10 of the VT events in the general population. The prevalence and relative risk of VT of these variants in other populations (especially in Asians) will require further evaluation.
Acknowledgments
We are indebted to all of the participants in the MAGIC (multiple assessments of genetic risk factors for thrombophilia in the Chinese population) study and medical assistants of our hospital. Our special thanks go to the Department of Vascular Surgery (Union Hospital) and the Ministry of Education Key Lab for Environment and Health (Tongji Medical College). This work was supported by the National Basic Scientific Research Program of China (973 Program, No. 2007 CB935803), the National Natural Sciences Foundation of China (No. 30825018), the State Ministry of Health Key Clinical Construction Project (2010 No. 58), the Chinese National Science and Technology Major Project, and the Mega-Project for New Drugs Development (No. 2011ZX09302-002-01).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
References
- 1.Flinterman L.E., van Hylckama Vlieg A., Cannegieter S.C., Rosendaal F.R. Long-term survival in a large cohort of patients with venous thrombosis: Incidence and predictors. PLoS Med. 2012;9:e1001155. doi: 10.1371/journal.pmed.1001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White R.H. The epidemiology of venous thromboembolism. Circulation. 2003;107(23, Suppl 1):I4–I8. doi: 10.1161/01.CIR.0000078468.11849.66. [DOI] [PubMed] [Google Scholar]
- 3.Stein P.D., Kayali F., Olson R.E., Milford C.E. Pulmonary thromboembolism in Asians/Pacific Islanders in the United States: Analysis of data from the National Hospital Discharge Survey and the United States Bureau of the Census. Am. J. Med. 2004;116:435–442. doi: 10.1016/j.amjmed.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 4.White R.H., Zhou H., Murin S., Harvey D. Effect of ethnicity and gender on the incidence of venous thromboembolism in a diverse population in California in 1996. Thromb. Haemost. 2005;93:298–305. doi: 10.1160/TH04-08-0506. [DOI] [PubMed] [Google Scholar]
- 5.Roberts L.N., Patel R.K., Arya R. Venous thromboembolism and ethnicity. Br. J. Haematol. 2009;146:369–383. doi: 10.1111/j.1365-2141.2009.07786.x. [DOI] [PubMed] [Google Scholar]
- 6.Zakai N.A., McClure L.A. Racial differences in venous thromboembolism. J. Thromb. Haemost. 2011;9:1877–1882. doi: 10.1111/j.1538-7836.2011.04443.x. [DOI] [PubMed] [Google Scholar]
- 7.Morange P.E., Bezemer I., Saut N., Bare L., Burgos G., Brocheton J., Durand H., Biron-Andreani C., Schved J.F., Pernod G. A follow-up study of a genome-wide association scan identifies a susceptibility locus for venous thrombosis on chromosome 6p24.1. Am. J. Hum. Genet. 2010;86:592–595. doi: 10.1016/j.ajhg.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meltzer M.E., Lisman T., Doggen C.J., de Groot P.G., Rosendaal F.R. Synergistic effects of hypofibrinolysis and genetic and acquired risk factors on the risk of a first venous thrombosis. PLoS Med. 2008;5:e97. doi: 10.1371/journal.pmed.0050097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosendaal F.R. Venous thrombosis: A multicausal disease. Lancet. 1999;353:1167–1173. doi: 10.1016/s0140-6736(98)10266-0. [DOI] [PubMed] [Google Scholar]
- 10.Blom J.W., Doggen C.J., Osanto S., Rosendaal F.R. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293:715–722. doi: 10.1001/jama.293.6.715. [DOI] [PubMed] [Google Scholar]
- 11.Bezemer I.D., Bare L.A., Doggen C.J., Arellano A.R., Tong C., Rowland C.M., Catanese J., Young B.A., Reitsma P.H., Devlin J.J., Rosendaal F.R. Gene variants associated with deep vein thrombosis. JAMA. 2008;299:1306–1314. doi: 10.1001/jama.299.11.1306. [DOI] [PubMed] [Google Scholar]
- 12.Christiansen S.C., Naess I.A., Cannegieter S.C., Hammerstrøm J., Rosendaal F.R., Reitsma P.H. Inflammatory cytokines as risk factors for a first venous thrombosis: A prospective population-based study. PLoS Med. 2006;3:e334. doi: 10.1371/journal.pmed.0030334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson F.A., Jr., Spencer F.A. Risk factors for venous thromboembolism. Circulation. 2003;107(23, Suppl 1):I9–I16. doi: 10.1161/01.CIR.0000078469.07362.E6. [DOI] [PubMed] [Google Scholar]
- 14.Souto J.C., Almasy L., Borrell M., Blanco-Vaca F., Mateo J., Soria J.M., Coll I., Felices R., Stone W., Fontcuberta J., Blangero J. Genetic susceptibility to thrombosis and its relationship to physiological risk factors: The GAIT study. Genetic Analysis of Idiopathic Thrombophilia. Am. J. Hum. Genet. 2000;67:1452–1459. doi: 10.1086/316903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seligsohn U., Lubetsky A. Genetic susceptibility to venous thrombosis. N. Engl. J. Med. 2001;344:1222–1231. doi: 10.1056/NEJM200104193441607. [DOI] [PubMed] [Google Scholar]
- 16.Dahlbäck B., Carlsson M., Svensson P.J. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: Prediction of a cofactor to activated protein C. Proc. Natl. Acad. Sci. USA. 1993;90:1004–1008. doi: 10.1073/pnas.90.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koster T., Rosendaal F.R., de Ronde H., Briët E., Vandenbroucke J.P., Bertina R.M. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden Thrombophilia Study. Lancet. 1993;342:1503–1506. doi: 10.1016/s0140-6736(05)80081-9. [DOI] [PubMed] [Google Scholar]
- 18.Bertina R.M., Koeleman B.P., Koster T., Rosendaal F.R., Dirven R.J., de Ronde H., van der Velden P.A., Reitsma P.H. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–67. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 19.Poort S.R., Rosendaal F.R., Reitsma P.H., Bertina R.M. A common genetic variation in the 3′-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood. 1996;88:3698–3703. [PubMed] [Google Scholar]
- 20.Corral J., Hernandez-Espinosa D., Soria J.M., Gonzalez-Conejero R., Ordonez A., Gonzalez-Porras J.R., Perez-Ceballos E., Lecumberri R., Sanchez I., Roldan V. Antithrombin Cambridge II (A384S): an underestimated genetic risk factor for venous thrombosis. Blood. 2007;109:4258–4263. doi: 10.1182/blood-2006-08-040774. [DOI] [PubMed] [Google Scholar]
- 21.Tang L., Guo T., Yang R., Mei H., Wang H., Lu X., Yu J., Wang Q., Hu Y. Genetic background analysis of protein C deficiency demonstrates a recurrent mutation associated with venous thrombosis in Chinese population. PLoS ONE. 2012;7:e35773. doi: 10.1371/journal.pone.0035773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang L., Lu X., Yu J.M., Wang Q.Y., Yang R., Guo T., Mei H., Hu Y. PROC c.574_576del polymorphism: A common genetic risk factor for venous thrombosis in the Chinese population. J. Thromb. Haemost. 2012;10:2019–2026. doi: 10.1111/j.1538-7836.2012.04862.x. [DOI] [PubMed] [Google Scholar]
- 23.Hamasaki N. Unmasking Asian thrombophilia: Is APC dysfunction the real culprit? J. Thromb. Haemost. 2012;10:2016–2018. doi: 10.1111/j.1538-7836.2012.04893.x. [DOI] [PubMed] [Google Scholar]
- 24.Dahlbäck B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood. 2008;112:19–27. doi: 10.1182/blood-2008-01-077909. [DOI] [PubMed] [Google Scholar]
- 25.Corral J., Aznar J., Gonzalez-Conejero R., Villa P., Miñano A., Vayá A., Carrell R.W., Huntington J.A., Vicente V. Homozygous deficiency of heparin cofactor II: Relevance of P17 glutamate residue in serpins, relationship with conformational diseases, and role in thrombosis. Circulation. 2004;110:1303–1307. doi: 10.1161/01.CIR.0000140763.51679.D9. [DOI] [PubMed] [Google Scholar]
- 26.Meltzer M.E., Lisman T., de Groot P.G., Meijers J.C., le Cessie S., Doggen C.J., Rosendaal F.R. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI-1. Blood. 2010;116:113–121. doi: 10.1182/blood-2010-02-267740. [DOI] [PubMed] [Google Scholar]
- 27.Heit J.A., Cunningham J.M., Petterson T.M., Armasu S.M., Rider D.N., DE Andrade M. Genetic variation within the anticoagulant, procoagulant, fibrinolytic and innate immunity pathways as risk factors for venous thromboembolism. J. Thromb. Haemost. 2011;9:1133–1142. doi: 10.1111/j.1538-7836.2011.04272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saposnik B., Reny J.L., Gaussem P., Emmerich J., Aiach M., Gandrille S. A haplotype of the EPCR gene is associated with increased plasma levels of sEPCR and is a candidate risk factor for thrombosis. Blood. 2004;103:1311–1318. doi: 10.1182/blood-2003-07-2520. [DOI] [PubMed] [Google Scholar]
- 29.Esmon C.T., Owen W.G. Identification of an endothelial cell cofactor for thrombin-catalyzed activation of protein C. Proc. Natl. Acad. Sci. USA. 1981;78:2249–2252. doi: 10.1073/pnas.78.4.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dahlbäck B., Villoutreix B.O. Regulation of blood coagulation by the protein C anticoagulant pathway: Novel insights into structure-function relationships and molecular recognition. Arterioscler. Thromb. Vasc. Biol. 2005;25:1311–1320. doi: 10.1161/01.ATV.0000168421.13467.82. [DOI] [PubMed] [Google Scholar]
- 31.Weiler H., Isermann B.H. Thrombomodulin. J. Thromb. Haemost. 2003;1:1515–1524. doi: 10.1046/j.1538-7836.2003.00306.x. [DOI] [PubMed] [Google Scholar]
- 32.Isermann B., Sood R., Pawlinski R., Zogg M., Kalloway S., Degen J.L., Mackman N., Weiler H. The thrombomodulin-protein C system is essential for the maintenance of pregnancy. Nat. Med. 2003;9:331–337. doi: 10.1038/nm825. [DOI] [PubMed] [Google Scholar]
- 33.Delvaeye M., Noris M., De Vriese A., Esmon C.T., Esmon N.L., Ferrell G., Del-Favero J., Plaisance S., Claes B., Lambrechts D. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009;361:345–357. doi: 10.1056/NEJMoa0810739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horowitz N.A., Blevins E.A., Miller W.M., Perry A.R., Talmage K.E., Mullins E.S., Flick M.J., Queiroz K.C., Shi K., Spek C.A. Thrombomodulin is a determinant of metastasis through a mechanism linked to the thrombin binding domain but not the lectin-like domain. Blood. 2011;118:2889–2895. doi: 10.1182/blood-2011-03-341222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu K.K. TM hidden treasure: Lectin-like domain. Blood. 2012;119:1103–1104. doi: 10.1182/blood-2011-12-394544. [DOI] [PubMed] [Google Scholar]
- 36.Bounameaux H., Rosendaal F.R. Venous thromboembolism: Why does ethnicity matter? Circulation. 2011;123:2189–2191. doi: 10.1161/CIRCULATIONAHA.111.031690. [DOI] [PubMed] [Google Scholar]
- 37.Wang X., El Naqa I.M. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics. 2008;24:325–332. doi: 10.1093/bioinformatics/btm595. [DOI] [PubMed] [Google Scholar]
- 38.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jackman R.W., Beeler D.L., Fritze L., Soff G., Rosenberg R.D. Human thrombomodulin gene is intron depleted: Nucleic acid sequences of the cDNA and gene predict protein structure and suggest sites of regulatory control. Proc. Natl. Acad. Sci. USA. 1987;84:6425–6429. doi: 10.1073/pnas.84.18.6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le Flem L., Picard V., Emmerich J., Gandrille S., Fiessinger J.N., Aiach M., Alhenc-Gelas M. Mutations in promoter region of thrombomodulin and venous thromboembolic disease. Arterioscler. Thromb. Vasc. Biol. 1999;19:1098–1104. doi: 10.1161/01.atv.19.4.1098. [DOI] [PubMed] [Google Scholar]
- 41.Kunz G., Ireland H.A., Stubbs P.J., Kahan M., Coulton G.C., Lane D.A. Identification and characterization of a thrombomodulin gene mutation coding for an elongated protein with reduced expression in a kindred with myocardial infarction. Blood. 2000;95:569–576. [PubMed] [Google Scholar]
- 42.Kunz G., Ohlin A.K., Adami A., Zöller B., Svensson P., Lane D.A. Naturally occurring mutations in the thrombomodulin gene leading to impaired expression and function. Blood. 2002;99:3646–3653. doi: 10.1182/blood.v99.10.3646. [DOI] [PubMed] [Google Scholar]
- 43.Aleksic N., Folsom A.R., Cushman M., Heckbert S.R., Tsai M.Y., Wu K.K. Prospective study of the A455V polymorphism in the thrombomodulin gene, plasma thrombomodulin, and incidence of venous thromboembolism: The LITE Study. J. Thromb. Haemost. 2003;1:88–94. doi: 10.1046/j.1538-7836.2003.00029.x. [DOI] [PubMed] [Google Scholar]
- 44.Sugiyama S., Hirota H., Kimura R., Kokubo Y., Kawasaki T., Suehisa E., Okayama A., Tomoike H., Hayashi T., Nishigami K. Haplotype of thrombomodulin gene associated with plasma thrombomodulin level and deep vein thrombosis in the Japanese population. Thromb. Res. 2007;119:35–43. doi: 10.1016/j.thromres.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 45.Heit J.A., Petterson T.M., Owen W.G., Burke J.P., DE Andrade M., Melton L.J., 3rd Thrombomodulin gene polymorphisms or haplotypes as potential risk factors for venous thromboembolism: A population-based case-control study. J. Thromb. Haemost. 2005;3:710–717. doi: 10.1111/j.1538-7836.2005.01187.x. [DOI] [PubMed] [Google Scholar]
- 46.Kubisz P., Stasko J., Hollý P., Ivanková J., Pullmann R. More on: Thrombomodulin gene polymorphisms or haplotypes as potential risk factors for venous thromboembolism: A population-based case-control study. J. Thromb. Haemost. 2005;3:2825–2827. doi: 10.1111/j.1538-7836.2005.01655.x. [DOI] [PubMed] [Google Scholar]
- 47.Le Flem L., Mennen L., Aubry M.L., Aiach M., Scarabin P.Y., Emmerich J., Alhenc-Gelas M. Thrombomodulin promoter mutations, venous thrombosis, and varicose veins. Arterioscler. Thromb. Vasc. Biol. 2001;21:445–451. doi: 10.1161/01.atv.21.3.445. [DOI] [PubMed] [Google Scholar]
- 48.Reny J.L., Remones V., Fontana P., Bieche I., Desvard F., Aubry M.L., Gaussen P., Aiach M. The thrombomodulin-1208/-1209delTT gene promoter polymorphism does not affect basal or LPS-dependent monocyte TM mRNA transcription in healthy volunteers. Thromb. Haemost. 2005;94:686–687. [PubMed] [Google Scholar]
- 49.Salomaa V., Matei C., Aleksic N., Sansores-Garcia L., Folsom A.R., Juneja H., Chambless L.E., Wu K.K. Soluble thrombomodulin as a predictor of incident coronary heart disease and symptomless carotid artery atherosclerosis in the Atherosclerosis Risk in Communities (ARIC) Study: A case-cohort study. Lancet. 1999;353:1729–1734. doi: 10.1016/s0140-6736(98)09057-6. [DOI] [PubMed] [Google Scholar]
- 50.Leizorovicz A., Turpie A.G., Cohen A.T., Wong L., Yoo M.C., Dans A., SMART Study Group Epidemiology of venous thromboembolism in Asian patients undergoing major orthopedic surgery without thromboprophylaxis. The SMART study. J. Thromb. Haemost. 2005;3:28–34. doi: 10.1111/j.1538-7836.2004.01094.x. [DOI] [PubMed] [Google Scholar]
- 51.Sakuma M., Nakamura M., Yamada N., Ota S., Shirato K., Nakano T., Ito M., Kobayashi T. Venous thromboembolism: Deep vein thrombosis with pulmonary embolism, deep vein thrombosis alone, and pulmonary embolism alone. Circ. J. 2009;73:305–309. doi: 10.1253/circj.cj-08-0372. [DOI] [PubMed] [Google Scholar]
- 52.Ikeda S., Miyahara Y. Ethnicity of patients with venous thromboembolism. -The incidence of venous thromboembolism in an Asian population- Circ. J. 2011;75:1831–1832. doi: 10.1253/circj.cj-11-0623. [DOI] [PubMed] [Google Scholar]
- 53.Yang Y., Liang L., Zhai Z., He H., Xie W., Peng X., Wang C., Investigators for National Cooperative Project for Prevention and Treatment of PTE-DVT Pulmonary embolism incidence and fatality trends in chinese hospitals from 1997 to 2008: A multicenter registration study. PLoS ONE. 2011;6:e26861. doi: 10.1371/journal.pone.0026861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morange P.E., Tregouet D.A. Lessons from genome-wide association studies in venous thrombosis. J. Thromb. Haemost. 2011;9(Suppl 1):258–264. doi: 10.1111/j.1538-7836.2011.04311.x. [DOI] [PubMed] [Google Scholar]
- 55.Wason J.M., Dudbridge F. A general framework for two-stage analysis of genome-wide association studies and its application to case-control studies. Am. J. Hum. Genet. 2012;90:760–773. doi: 10.1016/j.ajhg.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manolio T.A. Genomewide association studies and assessment of the risk of disease. N. Engl. J. Med. 2010;363:166–176. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- 57.Prandoni P., Bilora F., Marchiori A., Bernardi E., Petrobelli F., Lensing A.W., Prins M.H., Girolami A. An association between atherosclerosis and venous thrombosis. N. Engl. J. Med. 2003;348:1435–1441. doi: 10.1056/NEJMoa022157. [DOI] [PubMed] [Google Scholar]
- 58.Doggen C.J., Cats V.M., Bertina R.M., Rosendaal F.R. Interaction of coagulation defects and cardiovascular risk factors: Increased risk of myocardial infarction associated with factor V Leiden or prothrombin 20210A. Circulation. 1998;97:1037–1041. doi: 10.1161/01.cir.97.11.1037. [DOI] [PubMed] [Google Scholar]
- 59.Ireland H., Kunz G., Kyriakoulis K., Stubbs P.J., Lane D.A. Thrombomodulin gene mutations associated with myocardial infarction. Circulation. 1997;96:15–18. doi: 10.1161/01.cir.96.1.15. [DOI] [PubMed] [Google Scholar]
- 60.Wu K.K., Aleksic N., Ahn C., Boerwinkle E., Folsom A.R., Juneja H., Atherosclerosis Risk in Communities Study (ARIC) Investigators Thrombomodulin Ala455Val polymorphism and risk of coronary heart disease. Circulation. 2001;103:1386–1389. doi: 10.1161/01.cir.103.10.1386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.