

Abstract
Rare copy-number variants (CNVs) have been implicated in autism and intellectual disability. These variants are large and affect many genes but lack clear specificity toward autism as opposed to developmental-delay phenotypes. We exploited the repeat architecture of the genome to target segmental duplication-mediated rearrangement hotspots (n = 120, median size 1.78 Mbp, range 240 kbp to 13 Mbp) and smaller hotspots flanked by repetitive sequence (n = 1,247, median size 79 kbp, range 3–96 kbp) in 2,588 autistic individuals from simplex and multiplex families and in 580 controls. Our analysis identified several recurrent large hotspot events, including association with 1q21 duplications, which are more likely to be identified in individuals with autism than in those with developmental delay (p = 0.01; OR = 2.7). Within larger hotspots, we also identified smaller atypical CNVs that implicated CHD1L and ACACA for the 1q21 and 17q12 deletions, respectively. Our analysis, however, suggested no overall increase in the burden of smaller hotspots in autistic individuals as compared to controls. By focusing on gene-disruptive events, we identified recurrent CNVs, including DPP10, PLCB1, TRPM1, NRXN1, FHIT, and HYDIN, that are enriched in autism. We found that as the size of deletions increases, nonverbal IQ significantly decreases, but there is no impact on autism severity; and as the size of duplications increases, autism severity significantly increases but nonverbal IQ is not affected. The absence of an increased burden of smaller CNVs in individuals with autism and the failure of most large hotspots to refine to single genes is consistent with a model where imbalance of multiple genes contributes to a disease state.
Introduction
Repeat architecture of the human genome predisposes certain regions to nonallelic homologous recombination (NAHR), resulting in copy-number variants (CNVs).1,2 Rare (<0.1% frequency) CNVs created by NAHR between large (>10 kbp) segmental duplications (SDs), termed “genomic hotspots,” have been implicated in a range of neurodevelopmental disorders, including intellectual disability, autism (MIM 209850), epilepsy, and schizophrenia (SCZD [MIM 181500]).3 Recent studies have suggested that a large CNV burden is, in fact, associated with lower measures for cognitive ability.4,5 However, the specificity of CNVs to autism susceptibility as opposed to a broader developmental-delay phenotype is not clear.6 Although rare CNVs account for ∼15% of pediatric neurodevelopmental disease, more than 30% of these cases correspond to genomic hotspots—i.e., segments flanked by high-identity SDs.7 In fact, most CNVs categorized as “unequivocally” pathogenic map to these genomic hotspots. Their predisposition to recurrent rearrangement corresponds to a higher mutation rate, requiring fewer cases and controls to be screened in order for statistical significance to be reached. Most recurrent pathogenic CNVs are large (>400 kbp), and the regions recurrently deleted typically involve several to dozens of genes; therefore, the genes responsible for neurodevelopmental disease have not yet been discovered. Atypical pathogenic CNVs, which are not mediated by SDs, will be much rarer and require the detection of smaller events from exceedingly larger populations.8–10 Despite advances in exome sequencing,11–14 the discovery and genotyping of smaller (<50 kbp) CNVs associated with disease have been challenging, especially in repeat-rich regions. Furthermore, the discovery of extremely rare variants has precluded replication and enrichment analysis for further genotype-phenotype correlations.
One approach is to identify genomic regions that are prone to recurrent rearrangements and under strong selection in the human population. Although other high-identity repeat sequences that are smaller than SDs have been documented,15,16 their role in disease rearrangements under the NAHR model has not been systematically explored, especially for cases of sporadic and familial autism. On the basis of the original model of NAHR and the repeat architecture of the human genome, we identified 1,367 gene-rich regions capable, in principle, of recurrent copy-number variation. The sites ranged in size from 5 kbp to 5 Mbp, and most traversed or intersected genes. We designed a customized targeted, high-density (one probe every 50 bp to 1 kbp) microarray to screen individuals with autism (from the Simons Simplex Collection [SSC] and Autism Genetic Resource Exchange [AGRE]) for four purposes: (1) to assess genomic instability of 1,367 hotspot regions, including 120 SD-mediated hotspots, 253 minihotspots (flanked by smaller SD blocks), 410 microhotspots (flanked by identical pairs of >100 bp sequences), and 584 Alu-mediated hotspots; (2) to identify CNVs that are specific to autism as opposed to developmental delay; (3) to determine whether smaller atypical pathogenic CNVs could be identified within 17 genomic regions previously associated with developmental delay and autism; and (4) to characterize candidate genes disrupted more often in autism cases than in controls.
Subjects and Methods
Samples from Affected Individuals
Families with children affected with idiopathic autism (n = 2,478) were identified through the SSC.17 The SSC includes families with no more than one child with autism. The families were ascertained through 12 data-collection sites across North America. The criteria for inclusion require that a child meet the autism spectrum disorder (ASD) diagnosis on the Autism Diagnostic Observation Schedule (ADOS)18 and on the Autism Diagnostic Interview, Revised (ADI-R)19 and that the child passes an expert clinical evaluation. Standardized head-circumference scores were calculated on the basis of norms that Roche and colleagues20 established to account for age and gender. A nonverbal IQ (NVIQ) greater than 35 is also required. Exclusion criteria consist of significant hearing, vision, or motor problems, significant birth complications, or a diagnosis of an ASD-related disorder, such as fragile X syndrome. Individuals with up to a third-degree relative with ASD or a sibling with features of ASD were also excluded. Diagnostic evaluations, cognitive assessment, and phenotypic characterization were conducted at each site, and data collection, entry, and validation methods were standardized across sites to ensure reliability of sample collection. In addition, affected probands (n = 719) from multiplex families with autism were identified through AGRE.21 All family members were tested with ADOS and ADI-R, and cytogenetic analyses were performed so that gross chromosomal abnormalities could be ruled out. All individuals from this study were recruited after appropriate approval and informed consent were obtained from all participants.
Controls
Control populations were obtained from two cohorts. The National Institute of Mental Health (NIMH) control cohort22 consisted of 207 DNA samples obtained from the Rutgers University Cell and DNA Repository. These individuals were ascertained through an online self-report based on the Composite International Diagnostic Instrument short-form (CIDI-SF).23 Those who did not meet DSM-IV criteria for major depression, denied a history of bipolar disorder or psychosis, and reported exclusively European origins were included.24,25 In addition, 373 individuals from ClinSeq, designed to study a spectrum of atherosclerotic disease, were included.26 The frequency of the identified CNVs was also assessed from an expanded set of control CNV calls derived from 8,329 individuals assessed via SNP microarrays for the 120 SD-mediated hotspots8 and from a subset of this cohort (n = 2,090 individuals), characterized through the Wellcome Trust Case-Control Consortium (WTCCC) by the high-density Illumina 1.2M SNP microarray for smaller hotspot regions and candidate-gene events.27 No cognitive assessments were available for these controls, who were deemed normal, healthy volunteers. As such, they do not represent disease-matched controls but rather population controls.
Hotspot Definition and Microarray Design
On the basis of the original model of NAHR, where the presence of high-identity repeat sequences can promote unequal crossover and lead to deletions or duplications of the intervening unique sequence of DNA, we identified 1,367 regions, including (1) regular nonredundant hotspots, defined as regions flanked by SDs (≥95%, ≥10 kbp), whose size ranged from 50 kbp to 5 Mbp and which were mapped within 120 genomic sites (Figures S1–S3 available online);28,29 (2) minihotspots, defined here to correspond to 1–100 kbp intervals of unique sequence flanked by smaller (1–10 kbp) SDs with ≥90% sequence identity; (3) microhotspots, defined here as 1–50 kbp regions that contain at least one gene and are flanked by nearly perfect-identity repeats (one mismatch) of at least 100 bp in length; and (4) Alu hotspots, defined as events identified within 50 kbp nonoverlapping genomic windows containing at least 15 kbp of Alu elements (specifically AluY) and one or more RefSeq genes. We also surveyed CNVs in regions (n = 250) that were enriched for highly conserved bases (on the basis of Genomic Evolutionary Rate Profiling scores) and contained 15 kbp or more of Alu repeats within a 50 kbp window.30 In total, our customized microarray (Agilent 2 × 400K) targeted 1,367 sites totaling ∼346 Mbp of genomic sequence. Probe density varied from one probe every 640 bp to 5 kbp depending on size (Table 1, Figure S2). For smaller hotspots, we included an additional 5–20 kbp of flanking sequence to improve sensitivity and boundary definition. We interrogated 3,883 genes for dosage sensitivity and 920 genes for gene breakage as a consequence of CNVs within the 1,367 hotspots (Table S1). In addition, a backbone of probes (including probes used by the International Standards for Cytogenomic Arrays [ISCA] Consortium microarray design31), at a density of one probe every 14.5 kbp, were distributed across the genome.
Table 1.
Definition of Targeted Genomic Hotspots
| Regions | Total Sites |
Size in Base Pairs |
Probe Spacing |
Number of Genes Completely Contained within the CNV | Number of Genes Disrupted by CNV Breakpoints | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total Size | Median | Max | Min | Median | Max | Min | ||||
| SD hotspot | 120 | 270,139,571 | 1,777,180 | 12,991,283 | 241,831 | 5,000 | 20,000 | 5,000 | 2,971 | 67 |
| Microhotspot | 410 | 12,565,293 | 32,068 | 55,726 | 7,018 | 641 | 1,115 | 140 | 143 | 145 |
| Minihotspot | 253 | 11,342,110 | 41,701 | 96,127 | 3,176 | 1,043 | 2,403 | 79 | 77 | 105 |
| AluY hotspot | 584 | 52,560,000 | 90,000 | 90,000 | 90,000 | 2,500 | 2,500 | 2,500 | 692 | 603 |
| 1,367 | 346,606,974 | 3,883 | 920 | |||||||
CNV Discovery and Genotyping
Array comparative genomic hybridization (CGH), washing, and analysis were performed according to the manufacturer's instructions. We used sex-matched reference samples with NA12878 (CEU [Utah residents with ancestry from northern and western Europe from the CEPH collection] female) as the reference for females and NA18507 (YRI [Yoruba in Ibadan, Nigeria] male) as the reference for males (from Coriell). Microarray hybridization data for each probe was computed with Agilent Feature Extraction software, converted to log2 ratio values, and uploaded to an in-house structured SQL database. We applied a first-pass quality-control (QC) filter of >0.23 derivative log-ratio threshold per sample (per the manufacturer's instructions). CNV calling was then performed by targeted genotyping and/or Agilent’s Aberration Detection Method 2 (ADM-2) algorithm,32 built within the Agilent Genomic Workbench software, requiring an absolute log2 ratio cutoff of 0.5 with at least three probes. In brief, we performed targeted genotyping of hotspots by calculating the average log2 intensity ratios of a region (Figures S3 and S4). By using chromosome-specific means and standard deviations, we generated a normalized log2 intensity ratio of each hotspot for each sample (z score).8 We plotted and manually visualized samples with an absolute z score ≥ 1.5 for each targeted region to remove false-positive calls. Events were considered to be mediated by smaller repeats if the identified CNV had a reciprocal overlap of 50% of its length with the targeted micro- or minihotspot sites (Figures S3 and S4). Events with one or both breakpoints mapping within flanking repeat sequences were categorized as “hotspot associated” or “hotspot mediated,” respectively (Figure S3). Detection of CNVs outside of hotspots, in the genomic backbone, was performed with the ADM-2 algorithm. We applied additional filters (<50% SD content, <0.3 standard deviation of log2 ratios across chromosomes for a given sample, <1% frequency in the controls) to remove false-positive calls and copy-number polymorphisms (Figure S5). Because most of these CNVs are not recurrent and do not share common breakpoints, we have limited power to derive statistically significant associations. Furthermore, smaller atypical events within disease-associated hotspots and CNVs involving autism candidate genes, compiled by the Simons Foundation Autism Research Initiative (SFARI) gene database and from recent exome sequencing projects, were identified by both targeted genotyping and the ADM-2 algorithm. We manually curated all identified CNVs by loading the log-transformed signal intensities on a custom UCSC Genome Browser. CNVs within the population control cohort were characterized in a previous study.33 Validation experiments were performed on a NimbleGen array with 135K probes as described previously.4
Results
We initially screened 2,478 individuals ascertained for ASD from the SSC, 719 individuals from the AGRE collection, and 580 controls from the ClinSeq and NIMH collections. All analyses of copy-number variation were performed by a custom microarray with a high density of probes (640 bp to 5 kbp) targeted to 1,367 regions with a susceptible genomic architecture and a median probe spacing of 14.5 kbp in the genomic backbone. After QC measures, a total of 2,588 autism and 580 control samples were included for an initial analysis (Table S2). We determined the performance of our array by assessing inheritance for a subset (n = 550) of CNVs and performed validation experiments by a second customized NimbleGen array. We estimated a high sensitivity and specificity (>99%) of the array to detect events >10 kbp in the targeted regions and >250 kbp in the genomic backbone. In addition, to assess the frequency of variants as well as to exclude copy-number polymorphisms, we genotyped candidate sites on an expanded set of 8,329 population controls for SD-mediated genomic hotspots8 and a subset of these controls for smaller CNVs (n = 2,090 WTCCC controls) genotyped by SNP microarray data (see Subjects and Methods).
Rare Recurrent Rearrangements within SD-Mediated Genomic Hotspots
We initially focused on rare recurrent CNVs within genomic hotspot regions; these corresponded to 120 regions in the human genome and were flanked by large, highly identical SDs.28 As expected, the burden of these large CNVs was higher in cases than in controls (2.8%, 72/2,588 autism cases versus 1.3%, 108/8,329 controls; p = 7.20 × 10−7, OR = 2.18); there was a slight bias toward duplications (42/72) versus deletions (30/72). Inheritance data from available parental samples showed that 52% (36/69) of these CNVs were de novo variants and that the remainder were inherited (16 maternal, 17 paternal) (Table S3). These CNVs corresponded to 29/120 (24%) genomic hotspots. Mutations in 17 of these, including 16p11.2, 1q21.1, 15q13.3, 17q12, and 17p12 chromosomal regions, were previously known to be associated with disease (Figure 1; Table S3).
Figure 1.
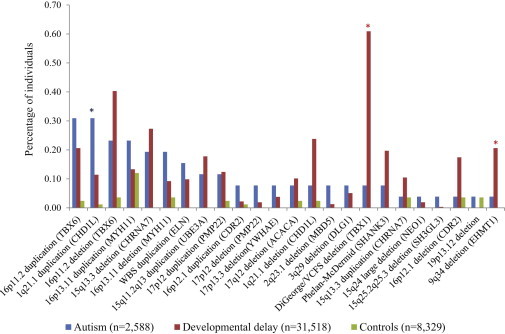
Recurrent Large CNVs in Individuals with Autism versus Developmental Delay
Data from individuals with developmental delay (n = 31,518) were derived from recently published reports.8,34 CNVs (>400 kbp) enriched among children with autism (blue asterisk) as opposed to developmental delay (red asterisk) and vice versa.
We compared the prevalence of various genomic disorders in this set of clinically defined individuals with autism to the frequency observed in a more broadly defined set of children with developmental delay (n = 31,518) and a large population control set (n = 8,329).8,34 In most cases the frequencies of specific genomic disorders were comparable, although some differences were noted. We found a significant enrichment of 1q21.1 duplications (MIM 612475) in autism compared to developmental delay (Fisher’s exact test, p = 0.01; OR = 2.7; 95% CI, 1.1–5.9). No specific clinical features served as a hallmark of these duplications, ruling out any autism-specific syndromes. Furthermore, we observed duplications of the Williams-Beuren syndrome (WBS [MIM 194050]) region in the autism cohort (4/2,588, 0.15%), as previously reported,35 that were not significantly enriched in autism compared to developmental delay (31/31,518, 0.10%; p = 0.27; OR = 1.5). Other recurrent CNVs were more commonly observed in children diagnosed with developmental delay as opposed to autism. For example, 22q11.2 deletions (Velocardiofacial DiGeorge syndromes [MIM 188400]) (p = 3.11 × 10−11, OR = 7.9, 95% CI, 2.6–66) and nonrecurrent 9q34 deletions (Kleefstra syndrome [MIM [610253]) (p = 0.04, OR = 5.34, 95% CI, 1.1–Inf) were significantly associated with developmental delay as opposed to autism (Figure 1). Trends toward association with developmental delay were observed for the 1q21.1 deletion (p = 0.07, OR = 3.04, 95% CI, 0.9–Inf) and the 16p12.1 deletion (p = 0.07, OR = 4.52, 95% CI, 0.92–Inf).
Interestingly, we identified 12 regions harboring SD-mediated (e.g., 10q11.23, 11q14.3, 5p15.33, 12p12.1, 7q11.23, and 2q11.2) or SD-associated (e.g., 2q12.1, 9p24, and 16q22) events not previously reported in autism. In a subset of these regions, CNVs were also identified in children with developmental delay (out of 15,767 cases)8 but not in controls (out of 8,329 controls), although this result was not statistically significant (Figure S6). Interesting regions include a 1.6 Mbp recurrent 10q11.23 duplication (paternally inherited in SSC proband 13465.p1), which was previously reported in a case study of children with developmental delay and intellectual disability;36 a 940 kbp recurrent deletion (two paternally inherited in SSC probands 12550.p1 and 14043.p1) of the 2q11.2 locus; 2 Mbp deletions of the region distal to the WBS region in autism (a de novo event in SSC proband 14484.p1) and developmental delay cases not observed in controls; and SD-associated events at the 16q22.2 locus, spanning axonemal central pair apparatus protein (HYDIN), in three children with autism but not in children with developmental delay or controls.
Phenotypic Trends: Duplications versus Deletions
In individuals with autism, we observed an apparent enrichment of reciprocal duplications (e.g., 1q21.1 and WBS) corresponding to microdeletion hotspots associated with developmental delay (Figure 1). With the phenotypic data collected for the SSC samples,11–13,17,37 we investigated the phenotypic features of affected individuals with large CNVs and, more specifically, contrasted those carrying deletions with those carrying duplications. A significant difference was observed between individuals with large CNVs and individuals with smaller events in the restricted/repetitive interests/behaviors domain (multivariate ANOVA, F(3,1887) = 4.7, p = 0.003, η2 = 0.007), but no differences in the social or communication domains were observed (Figure S7). Furthermore, post hoc comparisons of the ADI-R data showed significantly higher mean scores (p = 0.007), indicating more impairment with respect to restricted or repetitive interests in children with duplications than in those with deletions. Notably, individuals with deletions had lower mean scores in this domain, indicating that they had less impairment (p = 0.016) than individuals without any identified mutations.
Head circumference was also contrasted in individuals with deletions and duplications (Table S4). Children with 16p11.2 deletions (mean z score = 2.3, SD = 1.6) had significantly larger heads than children with 16p11.2 duplications (mean z score = −0.2, SD = 0.9; t(11) = 3.56, p = 0.004), consistent with previous reports.38 Even though the mean head circumference of the ASD individuals examined in this study was 0.69 standard deviations larger than that of the normative population, the individuals with 16p11.2 deletions (MIM 611913) were, on average, 1.22 standard deviations above the mean of the ASD sample set. Likewise, the head circumference of individuals with 16p11.2 duplications (MIM 614671) was, on average, 0.72 standard deviations below the ASD mean.
To further untangle the relationship between duplications and deletions in relation to autism or developmental delay phenotypes, we calculated correlations between CNV size across all genomic regions and NVIQ, as a measure of developmental delay and autism severity (as measured by the calibrated severity scale39). A significant negative correlation between the size of the event and NVIQ was observed for the deletions (Pearson correlation, R(222) = −0.24, p < 0.0001) but not for the duplications (R(317) = −0.06, p = 0.257). In contrast, a significant correlation was observed between event size and autism severity for the duplications (Spearman correlation, rs (301) = 0.13, p = 0.02) but not for the deletions (rs (212) = −0.02, p = 0.86) (Figure 2). It is possible that the observed relationships between CNV duplication size and severity and between CNV deletion size and NVIQ are driven by a few key regions but, given the limited sample sizes in each region, it is difficult to draw definitive conclusions (Table S5).
Figure 2.
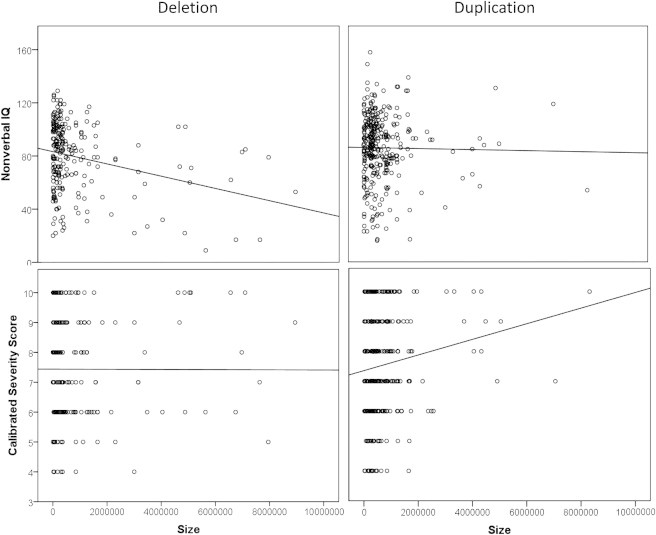
Phenotypic Features of Deletions and Duplications as a Function of Size
The scatter plot demonstrates that as the size of the deletion increases, nonverbal IQ significantly decreases, whereas as the size of duplication increases, autism severity significantly increases.
With specific regard to regions showing enrichment of duplications in autism versus developmental delay, the median and standard deviations for NVIQ were 94 and 31.9 for 1q21.1 duplications and 86 and 31.9 for WBS duplications (MIM 609757), respectively. Interestingly, the majority of probands carrying these duplications scored above the average for NVIQ. These results suggest that increased dosage of genes within certain genomic hotspots might enhance the severity of the autism phenotype but that haploinsufficiency might lead to reduced NVIQ associated with developmental delay.
Putative Smaller Hotspots
We next focused on 1,247 genomic regions that are potentially predisposed to unequal crossover events because they are flanked by smaller repeat elements (see Subjects and Methods; Figure S1). The flanking sequence of these hotspots included smaller (1–10 kbp) SDs (minihotspots), perfectly identical sequences at least 100 bp in size (microhotspots), or regions enriched for Alu repeats. These smaller hotspots potentially affect 1,765 genes—of which 853 would be disrupted if the CNVs occurred (Table S1). We performed targeted CNV discovery within 1,106 of these smaller hotspots, which could be accurately genotyped on our microarray, on the same sample set (see Subjects and Methods). We identified CNVs in 22% (83/378) of microhotspots, 6% (13/227) of minihotspots, and 4% (21/501) of AluY hotspots (Tables S6–S9). After excluding variants observed in controls by using the same microarray platform (n = 580) or >0.1% of the expanded set of 2,090 WTCCC controls, we found that 3% (76/2,588) of the cases carried smaller hotspot-mediated or hotspot-associated events (Table S10). We assessed the inheritance status of these events and found that the majority of the tested events were transmitted from a carrier parent (60/70, 86%). All ten de novo events were greater than 100 kbp in size. We found no significant enrichment or global increase in burden among the smaller hotspots in cases compared to controls (p = 0.13, OR = 1.2) (Figure 3A; Table S9). Although none of these genes have been reported to be associated with autism and no single locus reaches statistical significance in this study, functional analysis suggests coexpression and interactions in metabolic pathways among several of these genes (Figure S8).
Figure 3.
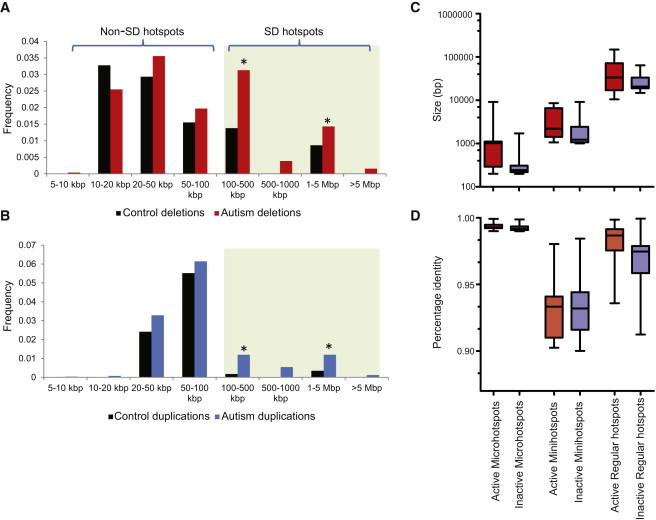
Properties of CNV Hotspots
(A and B) Frequency of deletions (A) and duplications (B) at different size ranges is shown for SD-mediated hotspots and smaller (micro, mini, and AluY) hotspots. Although no enrichment for smaller non-SD hotspots is observed among autistic children, a significant enrichment for larger, SD-mediated CNVs is observed. An asterisk denotes a significant difference (p < 0.05) in cases compared to controls via a Mann-Whitney t test.
(C and D) Sequence properties of genomic hotspots are shown. A comparison of size (C) and sequence identity (D) of repeats flanking “active” hotspots versus “inactive” genomic hotspots. The error bars indicate the minimum and maximum range of the data points.
We compared the structure and sequence properties of repeat sequences flanking active and inactive hotspots as defined by this study. We categorized hotspots as predisposing and nonpredisposing for NAHR-mediated rearrangements on the basis of the orientation of the SDs within the human reference genome.7 Although all microhotspots were chosen to contain the predisposing structure, we found that the repeat sequences flanking active microhotspots were larger (median size 284 bp; p = 0.0001, Mann Whitney U test) and had higher sequence identity (median sequence identity 99.29%; p = 0.0001, Mann-Whitney U test) than those flanking inactive hotspots (median size and sequence identity 244 bp; 99.18%) (Figure 3B). We note that the high correlation between the size of the repeats and sequence identity is due to ascertainment of these hotspots (see Subjects and Methods). We curated all events mapping within the 120 regular hotspots by assessing the 2,588 autism samples analyzed in this study and the previously reported 15,767 cases with developmental delay.8 As observed previously,8 we found both the size and identity of SDs flanking active hotspots to be greater than those of SDs flanking inactive hotspots. However, when minihotspots were analyzed for sequence properties, we found that the activity within minihotspot sites was dependent upon the size (hotspot median size and sequence identity 2,198 bp; 93.3%) of the flanking repeat (p = 0.021, Mann-Whitney U test) and not on the extent of sequence identity (p = 0.55, Mann-Whitney U test); this is in contrast to results for inactive sites (coldspot median size and sequence identity 1,459 bp; 93.4%) (Figure 3B). No events were identified in minihotspot regions that had a nonpredisposing or protective genomic architecture, suggesting no alternative SD structure within these regions.40 Overall, our results suggest that, apart from predisposing orientation, the size of the repeats and high sequence identity are major determinants of activity of NAHR hotspots.
Atypical CNVs within Candidate Regions
In an effort to detect smaller events within known pathogenic regions, we targeted 17 larger, disease-associated hotspot CNVs with a probe density of one probe every 750 bp. We screened individuals by array CGH in order to refine these loci to one or a few candidate genes. Within the autism cohort (1.4% of cases), we discovered and validated 36 smaller deletions (n = 23) and duplications (n = 13) that were not detected in our control cohort (n = 580) and were at <0.1% frequency among the 2,090 WTCCC controls (requiring ≥10 probe coverage) (Table 2). Interesting candidates specific to the autism cohort include a 323 kbp deletion encompassing chromodomain helicase DNA binding protein 1-like (CHD1L [MIM 613039]) within the chromosome 1q21.1 deletion region (MIM 612474) in SSC proband 12719.p1 (Figure 4A); a 132 kbp tandem duplication of neuregulin-3 (NRG3 [MIM 605533]) exon 4 at 10q23.1 in SSC proband 13889.p1; and a 304 kbp deletion of acetyl-CoA carboxylase alpha (ACACA [MIM 200350]) within the 17q12 microdeletion region in SSC proband 11234.p1 (Figure 4B). All atypical events detected were inherited from an apparently unaffected parent. In addition, we also identified two individuals (SSC proband 12132.p1 and AGRE proband AU084503) with inherited in-frame deletions of exons 3 and 4 of BBS4 (MIM 600374), a gene implicated in the autosomal-recessive Bardet-Biedl syndrome (MIM 209900), although the size of the event was too small to assess in the WTCCC controls. At the chromosome 15q13.3 locus, we detected an enrichment (5/2,588) for deletions of TRPM141,42 (MIM 603576) in individuals with autism (SSC proband 13686.p1 and AGRE probands AU0316301, AU2275301, AU079904, and AU1006301) compared to controls (0/2,670 controls; Fisher’s exact test p = 0.029) (Figure 5A). Interestingly, all TRPM1 deletions were inherited, and one individual inherited the deletion from both parents and thus had a homozygous deletion.
Table 2.
Atypical Exon-Disrupting Events of Disease-Associated Hotspot Regions in Children with Autism
| Region | Genomic Coordinates (hg18) | Sample | Size (bp) | Genes | Event | Inheritance | Case Probes | Control Probes | Control Eventsa | p Value |
|---|---|---|---|---|---|---|---|---|---|---|
| 1q21.1 | chr1: 145,182,393–145,442,505 | 12719.p1 | 260,112 | CHD1L, LINC00624 | del | maternal | 323 | 118 | 0 | n/a |
| 1q21.1 | chr1: 145,302,783–145,354,227 | 12345.p1 | 51,444 | LINC00624 | del | maternal | 59 | 4 | n/a | n/a |
| 1q21.1 | chr1: 145,302,783–145,354,227 | 11712.p1 | 51,444 | LINC00624 | del | maternal/paternal | 59 | 4 | n/a | n/a |
| 3q29 | chr3: 197,762,959–197,767,300 | 14045.p1 | 4,341 | WDR53 | del | paternal | 7 | 3 | n/a | n/a |
| 3q29 | chr3: 198,022,697–198,054,675 | 13144.p1 | 31,978 | PAK2 | dup | paternal | 47 | 44 | 0 | n/a |
| 3q29 | chr3: 198,682,714–198,836,982 | AU1087301 | 154,268 | BDH1 | dup | paternal | 221 | 75 | 0 | 0.242 |
| 3q29 | chr3: 198,682,714–198,836,982 | 13746.p1 | 154,268 | BDH1 | dup | maternal | 221 | 75 | 0 | 0.242 |
| 10q23 | chr10: 81,575,201–81,959,837 | 13214.p1 | 384,636 | MBL1P1, SFTPD, LOC219347, C10orf57, PLAC9, ANXA11 | del | maternal | 534 | 247 | 1 | 0.301 |
| 10q23 | chr10: 81,809,415–81,833,598 | 13582.p1 | 24,183 | LOC219347, C10orf57 | del | paternal | 35 | 6 | n/a | n/a |
| 10q23 | chr10: 81,824,603–81,834,112 | 12969.p1 | 9,509 | LOC219347, C10orf57 | del | paternal | 16 | 3 | n/a | n/a |
| 10q23 | chr10: 82,084,686–82,106,599 | 12378.p1 | 21,913 | DYDC1 | dup | maternal | 34 | 9 | n/a | n/a |
| 10q23 | chr10: 82,084,686–82,106,599 | 13634.p1 | 21,913 | DYDC1 | dup | paternal | 34 | 9 | n/a | n/a |
| 10q23 | chr10: 82,084,686–82,106,599 | 14055.p1 | 21,913 | DYDC1 | dup | maternal | 34 | 9 | n/a | n/a |
| 10q23 | chr10: 82,084,686–82,106,599 | 14343.p1 | 21,913 | DYDC1 | dup | maternal | 34 | 9 | n/a | n/a |
| 10q23 | chr10: 82,084,686–82,106,599 | 11545.p1 | 21,913 | DYDC1 | dup | maternal | 34 | 9 | n/a | n/a |
| 10q23 | chr10: 82,084,686–82,106,599 | 13611.p1 | 21,913 | DYDC1 | dup | maternal | 34 | 9 | n/a | n/a |
| 10q23 | chr10: 82,084,686–82,106,599 | AU1763301 | 21,913 | DYDC1 | dup | maternal | 34 | 9 | n/a | n/a |
| 10q23 | chr10: 82,359,933–82,366,203 | 14348.p1 | 6,270 | SH2D48 | del | paternal | 10 | 5 | n/a | n/a |
| 10q23 | chr10: 84,593,836–84,680,305 | 13889.p1 | 86,469 | NRG3 | dup | maternal | 132 | 40 | 0 | n/a |
| 15q13.3 | chr15: 29,142,646–29,178,949 | 13686.p1 | 36,303 | TRPM1 | del | maternal | 54 | 18 | 0 | 0.029 |
| 15q13.3 | chr15: 29,142,646–29,178,949 | AU0316301 | 36,303 | TRPM1 | del | paternal | 54 | 18 | 0 | 0.029 |
| 15q13.3 | chr15: 29,142,646–29,178,949 | AU2275301 | 36,303 | TRPM1 | del | paternal | 54 | 18 | 0 | 0.029 |
| 15q13.3 | chr15: 29,142,646–29,178,949 | AU079904 | 36,303 | TRPM1 | del | maternal/paternal | 54 | 18 | 0 | 0.029 |
| 15q13.3 | chr15: 29,176,938–29,188,508 | AU1006301 | 11,570 | TRPM1 | del | maternal | 17 | 6 | n/a | n/a |
| 15q24.1 | chr15: 70,788,521–70,793,560 | 12132.p1 | 5,039 | BBS4 | del | maternal | 5 | 1 | n/a | 0.242 |
| 15q24.1 | chr15: 70,788,521–70,793,560 | AU084503 | 5,039 | BBS4 | del | paternal | 5 | 1 | n/a | 0.242 |
| 15q24.2 | chr15: 74,294,283–74,297,672 | AU1228303 | 3,389 | ETFA | del | paternal | 15 | 2 | n/a | n/a |
| 16p13.11 | chr16: 15,476,243–15,515,874 | 13215.p1 | 39,631 | C16orf45 | del | paternal | 61 | 19 | 0 | n/a |
| 16p11.2 | chr16: 29,961,147-29974677 | 14004.p1 | 13,530 | ALDOA | del | maternal | 16 | 6 | n/a | n/a |
| 17q12 | chr17: 32560142-32732763 | 11234.p1 | 172,621 | ACACA | del | maternal | 304 | 92 | 0 | n/a |
| 17q12 | chr17: 33,179,017–33,351,760 | 11002.p1 | 172,743 | HNF1B (TCF2), LOC284100 | dup | maternal | 282 | 68 | 0 | n/a |
| 22q11.21 | chr22: 17,413,909–17,429,856 | 12686.p1 | 15,947 | DGCR2 | del | paternal | 26 | 6 | n/a | n/a |
| 22q11.21 | chr22: 17,703,043–17,719,750 | 11599.p1 | 16,707 | HIRA | del | maternal | 17 | 6 | n/a | n/a |
| 22q11.21 | chr22: 19,455,582–19,462,562 | 12878.p1 | 6,980 | SERPIND1 | del | paternal | 11 | 7 | n/a | n/a |
| 22q13.31 | chr22: 46,964,885–47,079,942 | 13698.p1 | 115,058 | MIR3201 | dup | paternal | 163 | 95 | 0 | 0.242 |
| 22q13.32 | chr22: 47,131,579–47,224,607 | AU1678301 | 93,028 | MIR3201 | dup | maternal | 134 | 75 | 0 | 0.242 |
| 22q13.32 | chr22: 48,026,222–48,302,803 | 13722.p1 | 276,581 | C22orf34 | del | maternal | 406 | 284 | 1 | n/a |
None of the above events were observed in controls assayed with our custom microarray (n = 580).
Control events, defined here as atypical CNVs that overlap with the reported event, were identified from population controls ran on Illumina 1M SNP array platform (n = 2,090). Control CNVs were assessed only if coverage exceeded ten probes on the SNP array.
Figure 4.
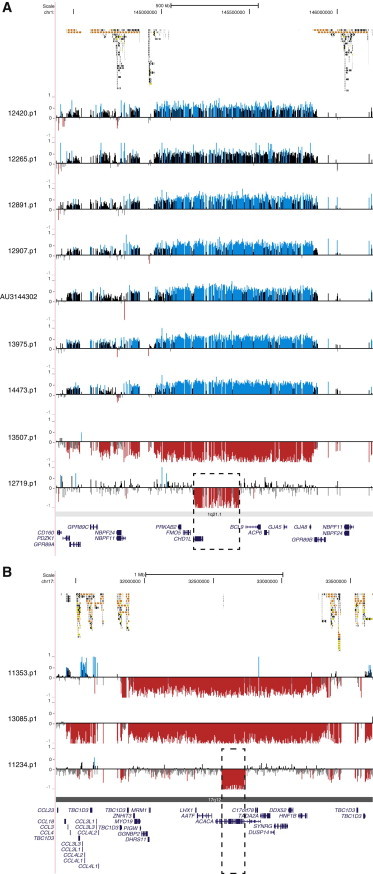
Atypical Copy-Number Variants within Disease-Associated Regions
Large recurrent CNVs were identified within the 1q21.1 (A) and 17q12 (B) microdeletion loci in autism cases. Private atypical deletion events reveal potential candidate genes CHD1L (A) and ACACA (B), highlighted by the dashed box. Blue (duplication) and red (deletion) histograms depict log2 relative hybridization signals. SDs flanking the larger recurrent events are mapped at the top.
Figure 5.
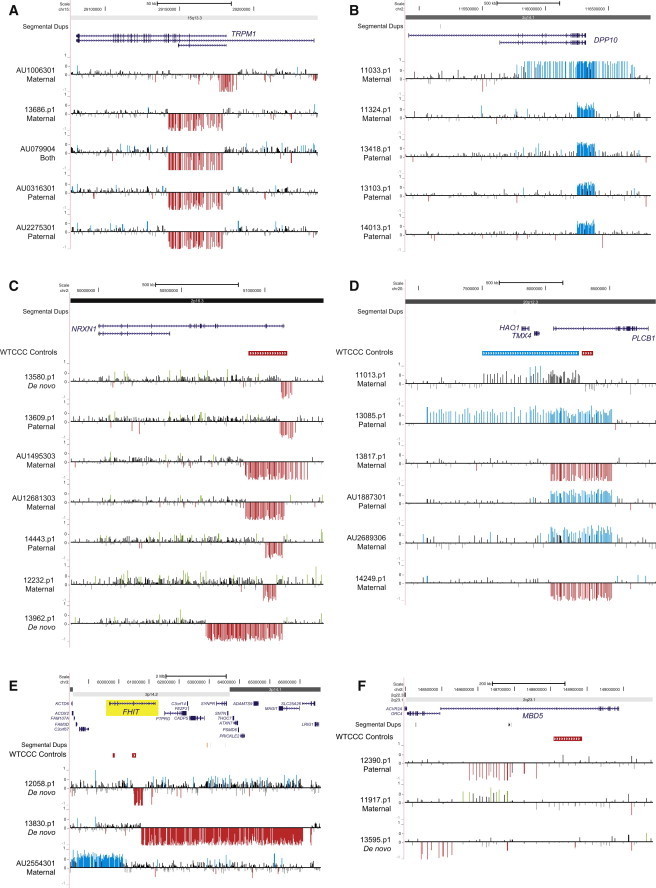
Autism Candidate Genes Enriched for Copy-Number Variants in Cases versus Controls
Atypical deletions of TRPM1 (A) were observed in cases but not in controls within the 15q13.3 microdeletion locus. Targeted genotyping of autism candidate genes DPP10 (B), NRXN1 (C), PLCB1 (D), FHIT (E), and MBD5 (F) revealed multiple deletions (red) and duplications (blue) of genes observed in cases at a higher frequency than in controls. Blue (duplication) and red (deletion) histograms depict log2 relative hybridization signals.
Targeted Discovery of CNVs in “Autism" Genes
Because our custom microarray also carried a suitable (14 kbp density) coverage of probes across the genomic backbone, we performed targeted discovery of CNVs affecting known autism candidate genes (n = 430 genes). The set included genes reported in recent exome sequencing studies11–13 as well as those curated in autism databases. Of the 253 candidate genes that we could genotype with high specificity, 76 showed evidence of copy-number variation; 60 of these genes were enriched in or exclusive to the autism cohort as opposed to controls (Table 3, Figure 5; Table S11). Of these autism-enriched candidate genes, 55 were directly “broken” or disrupted by a CNV, which in some cases was characterized as a hotspot-mediated or -associated event. Although we observed no significant difference in overall CNV burden of these autism-candidate genes between individuals with autism (8.5%; 221/2,588) and controls (6.9%; 40/580), several genes stood out as being enriched for CNVs in cases versus our complete set of controls (n = 2,670; 580 in this study and 2,090 WTCCC controls) (Figures 5 and S9). Some examples include dipeptidyl peptidase 10 (DPP10 [MIM 608209]) (p = 0.029, Fisher’s exact test) (Figure 5B), a gene that is involved in synaptogenesis and which was previously reported in a CNV study of autism;43 neurexin 1 (NRXN1 [MIM 600565]) (p = 0.032) (Figure 5C), a gene encoding a cell-adhesion molecule previously implicated in numerous neurocognitive disorders, including autism;44 phospholipase C, beta 1 (PLCB1 [MIM 607120]) (Figure 5D), a gene implicated in epilepsy45and schizophrenia;46 fragile-histidine triad (FHIT [MIM 601153]) (Figure 5E), implicated in a genome-wide association study for anxiety;47 methyl-CpG binding domain protein 5 (MBD5 [MIM 611472]) (Figure 5F), which lies within chromosomal region 2p23.1 and was previously implicated in autism, epilepsy, and developmental delay;48,49 and Parkinson protein 2 (PARK2 [MIM 602544]) (p = 0.009), a gene associated with Parkinson disease and previously implicated in autism.50 Interestingly, we also observed three large SD-associated CNVs (one deletion and two duplications) that include HYDIN (MIM 610812) on chromosome 16q22 (Figure 6; described above). Of the events affecting HYDIN, two were de novo, and one was inherited in a multiplex autism family where the CNV segregated with disease. A majority (88.2%; 149/169) of the autism-enriched gene-specific events identified were inherited from unaffected parents. The 20 de novo CNVs were all greater than 50 kbp in size and were observed in 17 genes, including NRXN1, FHIT, and HYDIN, all three of which had recurrent events.
Table 3.
Autism Candidate Genes Enriched for Exon-Disrupting CNVs in Cases versus Controls
| Candidate Gene(s) |
Autism Cases (n = 2,588) |
Controls (n = 580) |
WTCCC Controls (n = 2,090) |
Enriched Event | p Valuea | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Deletions | Duplications | Total | Deletions | Duplications | Total | Deletions | Duplications | Total | |||
| A2BP1 | 6 | 0 | 6 | 0 | 0 | 0 | 6 | 0 | 6 | del | 0.591 |
| ANO5 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| CACNA2D3 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | del | n/a |
| CADPS2 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| CNR1 | 0 | 7 | 7 | 0 | 1 | 1 | 0 | 0 | 0 | dup | 0.032 |
| CNTNAP2 | 1 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | del/dup | 0.242 |
| CTNNA3b | 21 | 1 | 22 | 4 | 0 | 4 | 8 | 0 | 8 | del/dup | 0.050 |
| CTNND2b | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | del | n/a |
| DIAPH3 | 1 | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 2 | del | 0.869 |
| DISC1 | 2 | 5 | 7 | 0 | 3 | 0 | 0 | 0 | 0 | del | 0.242 |
| DNAH5 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| DOCK1b | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | del | 0.242 |
| DPP10 | 0 | 5 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | dup | 0.029 |
| DPP6 | 1 | 4 | 5 | 0 | 1 | 1 | 0 | 0 | 0 | del/dup | 0.102 |
| EML1 | 0 | 4 | 4 | 0 | 0 | 0 | 0 | 1 | 1 | dup | 0.178 |
| EPHA6 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| ERBB4 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | del | n/a |
| F13A1b | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| FHITc | 2 | 1 | 3 | 0 | 0 | 0 | 2 | 0 | 2 | del/dup | 0.485 |
| FOXP1 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | del | 0.242 |
| GABRB3b | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| GALNT13 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | del | 0.742 |
| GIMAP8 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | del | n/a |
| GPC6 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | del | n/a |
| GRID2b | 1 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | del/dup | 0.242 |
| GRIN2A | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| HYDINc | 1 | 2 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | del/dup | 0.119 |
| IARSb | 0 | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | dup | 0.242 |
| ICA1, NXPH1 | 0 | 3 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | dup | 0.119 |
| IQGAP2 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | del | n/a |
| KANK1 | 2 | 6 | 8 | 0 | 1 | 1 | 0 | 2 | 2 | del/dup | 0.103 |
| KHDRBS2 | 0 | 3 | 3 | 0 | 0 | 0 | 1 | 2 | 3 | dup | 0.641 |
| KIAA1586 | 3 | 5 | 8 | 1 | 0 | 1 | 1 | 0 | 1 | del/dup | 0.049 |
| MBD5b | 2 | 1 | 3 | 0 | 0 | 0 | 1 | 0 | 1 | del | 0.301 |
| MCC | 2 | 0 | 2 | 0 | 1 | 0 | 1 | 1 | 1 | del | 0.488 |
| MCPH1 | 2 | 2 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | del/dup | 0.059 |
| MCPH1, DLGAP2 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | del | n/a |
| MET | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| MKL2 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| MLL3b | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | del | n/a |
| MLL5 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| MPHOSPH8b | 0 | 3 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | dup | 0.119 |
| NLGN1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| NRXN1c | 7 | 0 | 7 | 0 | 0 | 0 | 1 | 0 | 1 | del | 0.032 |
| NRXN3 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | del | n/a |
| NTRK3 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | del | n/a |
| NXPH1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | del | n/a |
| PARK2b | 12 | 12 | 24 | 1 | 2 | 3 | 4 | 3 | 7 | del/dup | 0.009 |
| PCDH9 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| PDE4A | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| PLCB1 | 2 | 4 | 6 | 0 | 0 | 0 | 0 | 2 | 2 | del/dup | 0.134 |
| POGZ | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| RGS7 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | dup | 0.742 |
| ROBO1b | 1 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | del/dup | 0.242 |
| RSRC1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | dup | n/a |
| SEMA5A | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 2 | del | 0.869 |
| SLC1A1 | 0 | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | dup | 0.242 |
| TBC1D4 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | del | n/a |
| THSD7A | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 2 | 2 | dup | 0.869 |
| ZMYND11b | 0 | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | dup | 0.242 |
p value determined by a Fisher’s exact test.
Candidate genes with private de novo events.
Candidate genes with recurrent de novo events. See Table S11 for a complete list of CNVs.
Figure 6.
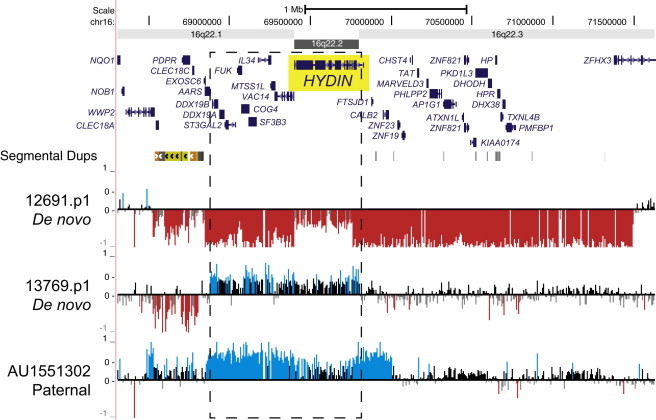
SD-Associated Copy-Number Variants of HYDIN
A de novo deletion and duplication, as well as an inherited duplication, were identified in children with autism. The child with the inherited duplication shares the event with his affected identical twin, but not with his unaffected sibling. Though these events are large and encompass numerous genes, the de novo duplication directly breaks HYDIN. Blue (duplication) and red (deletion) histograms depict log2 relative hybridization signals.
Discussion
In this study, we systematically assessed regions prone to recurrent copy-number variation in 2,588 children with ASD—this is one of the largest studies to date for clinically defined autism. Many of the smaller regions targeted in this study would have been previously unassayable35 because of the paucity of markers on standard commercial SNP microarrays. As expected, we observed a significantly more CNVs (total = 72 events) among the larger 120 hotspot regions that were flanked by large SDs than we did in controls (Figure 1; Table S3). Almost half of these larger events were de novo (Figure S10). Although 10.6% (117/1,106) of our smaller regions showed evidence of copy-number variation consistent with unequal crossing over of flanking repetitive DNA, we observed no enrichment of smaller CNVs when we compared individuals with autism to controls (Table S9). In contrast to the larger hotspots, in these smaller regions the majority (86%) of the CNVs were transmitted from unaffected parents. One possibility is that the 1,866 genes within these smaller hotspot regions are simply irrelevant to the etiology of autism. We note, for example, a slight functional enrichment of genes associated with metabolism within these smaller hotspot regions (Figure S8). Alternatively, it might be that larger events, because of their potential to affect multiple genes, are more likely to have a larger impact during neurodevelopment, consistent with an oligogenic model.6,51–53 Smaller CNVs disrupting a single gene might not be sufficiently deleterious and might, therefore, be more likely to be transmitted within the human population without adverse effect.
The large number of autistic individuals screened in this study allowed us to make some novel insights with respect to larger recurrent CNVs. We found that the chromosome 1q21.1 duplication, 16p11.2 deletion, and 16p11.2 duplication (Table S3) are the most prevalent CNVs significantly enriched in individuals with autism versus controls. Despite the fact that less than 25% of the autistic individuals screened in this study would qualify as having intellectual disability,17 the pattern and frequency of various genomic disorders is remarkably similar to recent large-scale screens of children with developmental delay (Figure 1).8,34 Nevertheless, some differences in frequency were noted. We found significant enrichment of 1q21.1 duplications (a novel association for autism) and a trend for WBS duplications (as reported in Sanders et al.35) in autistic children compared to those classified as developmentally delayed. No distinguishable additional phenotypes for these variants in cases could be deduced from the available clinical descriptions. Of note, most of the probands carrying these duplications have average to above-average NVIQ. Conversely, 22q11.2 and 9q34 deletions were significantly associated with developmental delay but not with autism. Although it appears there are no recurrent CNVs specific to autism, there are biases in frequency.
Our analysis also suggests that the type of CNV (i.e., duplication or deletion) has an important influence on phenotypic outcome among individuals with autism. As shown previously,4,35 increasing size of deletions correlates with a significant decrease in NVIQ; the same effect, however, is not observed for increasing size of CNV duplications (Figure 2). In contrast, we find that as the size of duplications increases, autism severity increases as measured by the calibrated autism severity score (p = 0.02) and repetitive or stereotypic behavior (ADI criteria) (p = 0.007). The basis for this difference is unclear, but it might be that an increase in gene dosage perturbs neurodevelopmental processes, whereas haploinsufficiency disrupts it more severely and leads to reduced NVIQ and developmental delay. These findings are also consistent with the reciprocal nature of some CNVs, such as those in the WBS and 1q21.1 regions, where duplications are enriched in autism cases but deletions are found primarily among children with developmental delay.
In this study, we also targeted 17 disease-associated regions at high-probe density (1 probe per 650 bp) in an attempt to identify smaller atypical events that might help refine a smaller region of overlap and thereby pinpoint specific genes. Within the 1q21.1 deletion region, we identified a 323 kbp deletion involving CHD1L (Figure 4A). This is a compelling candidate gene because it is related to CHD8 (MIM 610528), one of the genes with recurrent mutations identified from exome screens of idiopathic autism.11–14 Within 10q23, we identified a smaller duplication within NRG3, a schizophrenia-associated signaling molecule that binds and activates ErbB4, a receptor implicated in cellular proliferation, migration, and differentiation.54,55 If the duplicated exon were transcribed, it would create an in-frame duplication of ten amino acids within the ErbB4-binding domain. Additionally, although CHRNA7 (MIM 118511) has been previously implicated as contributing to neurological defects at the 15q13.3 locus,56 in our cohort we identified no deletions of this gene; instead, we identified five cases with deletions of a different gene, TRPM1, at this locus; these included a homozygous deletion not observed in any controls. Although appealing candidates, all of the atypical CNVs discovered within these 17 regions were inherited from unaffected parents, in contrast to the typical larger hotspot events that were de novo more than 50% of the time. If multiple genes within a critical region are required for an autism phenotype, it might be that some of these genomic hotspots will never be refined by smaller atypical CNVs. This is consistent with recent experimental data that suggest that synergistic combinations of downregulated genes in cis are required for recapitulation of some phenotypes of the 16p11.2 deletion,38 one of the most frequent CNVs associated with autism.
Additional smaller recurrent CNVs were also identified in this study outside of typical genomic hotspot regions as a result of our backbone of genomic probes. These included, for example, two recurrent de novo deletions involving Neurexin1 (NRXN1)—a gene with a longstanding association with autism—as well as single de novo mutations in GABRB3 (MIM 137192) and DOCK1 (MIM 601403). Recurrent inherited mutations and de novo mutations were observed in other strong autism candidates, such as PARK2, CNR1, MCPH1, and DPP10, and reached nominal significance in comparison to controls. The finding of three disruptive SD-associated CNVs of HYDIN is particularly interesting because the gene has been reported to cause hydrocephalus and impaired ciliary motility in mice 57 but has never been associated with autism. In our study, the two de novo events are found in probands from simplex autism families, whereas the inherited event segregates with the disease in a multiplex family.
In summary, we have comprehensively characterized recurrent CNVs for both large and putative smaller hotspots across the human genome. Irrespective of the size of the hotspot, our analysis shows that both sequence identity and the size of flanking repetitive sequences are key determinants in their mutability. Although smaller hotspots contribute to CNVs, we do not observe in aggregate an increased CNV burden for the mini- and microhotspot regions. This is in contrast to larger hotspot regions, for which there is a clear increase in CNV burden in children with ASD. Moreover, no recurrent larger hotspots appear to be specific to autism as opposed to intellectual disability, although some, such as the 1q21 duplication, appear to be significantly enriched. Although some hotspots have been refined to a single or few candidate genes by atypical events, we note that most of these smaller regions of overlap correspond to genomic disorders associated with recognizable syndromic diseases.7–10 Failure of the current study as well as previous reports to refine most testable candidate gene(s) in autism suggests that a genomic imbalance of multiple genes could be necessary for the manifestation of autism phenotypes.35,58 Consistent with this model, we present quantitative evidence for a phenotypic dependence on the size of the CNV. An increasing size of deletions correlates with a significant decrease in NVIQ, but the same effect is not observed for an increasing size of CNV duplications, although the latter might correspond with increased behavioral deficits (other than IQ) in children with autism. We predict that such recurrent CNVs will contribute to disease risk and that the identification of such de facto hotspots will be important in proving their pathogenic relevance. Because many of the individuals characterized here for CNVs are now part of ongoing exome11–14 and genome sequencing studies, it will be critical to integrate these CNV data with other disruptive inherited and de novo substitutions and indel mutations59 in order to provide a more complete picture of the genetic etiology of autism.
Acknowledgments
We thank Tonia Brown, Emre Karakoc, Nik Krumm, Arthur Ko, Daryl Dhanraj, Jason Lu, John Huddleston, and Ben Nelson for valuable discussions and technical assistance. We also thank the Autism Genetic Resource Exchange (AGRE) for providing DNA samples. We are grateful to all of the families at the participating Simons Simplex Collection (SSC) sites, as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, and E. Wijsman). We appreciate obtaining access to phenotypic data on SFARI Base. Approved researchers can obtain the SSC population dataset described in this study by applying at Simons Simplex Collection online (see Web Resources). M.Y.D. is supported by a US National Institutes of Health (NIH) Ruth L. Kirchstein National Research Service Award (NRSA) Fellowship through Eunice Kennedy Shriver National Institute of Child Health and Human Development (F32HD071698). C.D.C. was supported by NIH National Human Genome Research Institute NRSA (F32HG006070). This work was supported by Simons Foundation Autism Research Initiative award SFARI 137578 (E.E.E.) and NIH grant HD065285 (E.E.E.). E.E.E. is an investigator with the Howard Hughes Medical Institute.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Autism Genetic Resource Exchange (AGRE), http://agre.autismspeaks.org/site/c.lwLZKnN1LtH/b.5332889/k.9412/Agre_Splash_Page.htm
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Rutgers University Cell and DNA Repository, http://www.rucdr.org
Simons Foundation for Autism Research Initiative (SFARI), Simons Simplex Collection, http://sfari.org/
Accession Numbers
The raw microarray data reported in this paper have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus under accession number GSE39655.
References
- 1.Lupski J.R. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 2.Sharp A.J., Cheng Z., Eichler E.E. Structural variation of the human genome. Annu. Rev. Genomics Hum. Genet. 2006;7:407–442. doi: 10.1146/annurev.genom.7.080505.115618. [DOI] [PubMed] [Google Scholar]
- 3.Mefford H.C., Eichler E.E. Duplication hotspots, rare genomic disorders, and common disease. Curr. Opin. Genet. Dev. 2009;19:196–204. doi: 10.1016/j.gde.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Girirajan S., Brkanac Z., Coe B.P., Baker C., Vives L., Vu T.H., Shafer N., Bernier R., Ferrero G.B., Silengo M. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 2011;7:e1002334. doi: 10.1371/journal.pgen.1002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coe B.P., Girirajan S., Eichler E.E. A genetic model for neurodevelopmental disease. Curr. Opin. Neurobiol. 2012;22:829–836. doi: 10.1016/j.conb.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Girirajan S., Eichler E.E. Phenotypic variability and genetic susceptibility to genomic disorders. Hum. Mol. Genet. 2010;19(R2):R176–R187. doi: 10.1093/hmg/ddq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zollino M., Orteschi D., Murdolo M., Lattante S., Battaglia D., Stefanini C., Mercuri E., Chiurazzi P., Neri G., Marangi G. Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype. Nat. Genet. 2012;44:636–638. doi: 10.1038/ng.2257. [DOI] [PubMed] [Google Scholar]
- 10.Koolen D.A., Kramer J.M., Neveling K., Nillesen W.M., Moore-Barton H.L., Elmslie F.V., Toutain A., Amiel J., Malan V., Tsai A.C. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat. Genet. 2012;44:639–641. doi: 10.1038/ng.2262. [DOI] [PubMed] [Google Scholar]
- 11.O’Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neale B.M., Kou Y., Liu L., Ma’ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deininger P.L., Batzer M.A. Alu repeats and human disease. Mol. Genet. Metab. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- 16.Kidd J.M., Graves T., Newman T.L., Fulton R., Hayden H.S., Malig M., Kallicki J., Kaul R., Wilson R.K., Eichler E.E. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell. 2010;143:837–847. doi: 10.1016/j.cell.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischbach G.D., Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–195. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Lord C., Risi S., Lambrecht L., Cook E.H., Jr., Leventhal B.L., DiLavore P.C., Pickles A., Rutter M. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 2000;30:205–223. [PubMed] [Google Scholar]
- 19.Lord C., Rutter M., Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 20.Roche A.F., Mukherjee D., Guo S.M., Moore W.M. Head circumference reference data: birth to 18 years. Pediatrics. 1987;79:706–712. [PubMed] [Google Scholar]
- 21.Geschwind D.H., Sowinski J., Lord C., Iversen P., Shestack J., Jones P., Ducat L., Spence S.J., AGRE Steering Committee The autism genetic resource exchange: a resource for the study of autism and related neuropsychiatric conditions. Am. J. Hum. Genet. 2001;69:463–466. doi: 10.1086/321292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moldin S.O. NIMH Human Genetics Initiative: 2003 update. Am. J. Psychiatry. 2003;160:621–622. doi: 10.1176/appi.ajp.160.4.621. [DOI] [PubMed] [Google Scholar]
- 23.Kessler R.C., Ustün T.B. The World Mental Health (WMH) Survey Initiative Version of the World Health Organization (WHO) Composite International Diagnostic Interview (CIDI) Int. J. Methods Psychiatr. Res. 2004;13:93–121. doi: 10.1002/mpr.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Talati A., Fyer A.J., Weissman M.M. A comparison between screened NIMH and clinically interviewed control samples on neuroticism and extraversion. Mol. Psychiatry. 2008;13:122–130. doi: 10.1038/sj.mp.4002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baum A.E., Akula N., Cabanero M., Cardona I., Corona W., Klemens B., Schulze T.G., Cichon S., Rietschel M., Nöthen M.M. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol. Psychiatry. 2008;13:197–207. doi: 10.1038/sj.mp.4002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biesecker L.G., Mullikin J.C., Facio F.M., Turner C., Cherukuri P.F., Blakesley R.W., Bouffard G.G., Chines P.S., Cruz P., Hansen N.F., NISC Comparative Sequencing Program The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19:1665–1674. doi: 10.1101/gr.092841.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Craddock N., Hurles M.E., Cardin N., Pearson R.D., Plagnol V., Robson S., Vukcevic D., Barnes C., Conrad D.F., Giannoulatou E., Wellcome Trust Case Control Consortium Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 29.Sharp A.J., Hansen S., Selzer R.R., Cheng Z., Regan R., Hurst J.A., Stewart H., Price S.M., Blair E., Hennekam R.C. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 30.Cooper G.M., Brown C.D. Qualifying the relationship between sequence conservation and molecular function. Genome Res. 2008;18:201–205. doi: 10.1101/gr.7205808. [DOI] [PubMed] [Google Scholar]
- 31.Baldwin E.L., Lee J.Y., Blake D.M., Bunke B.P., Alexander C.R., Kogan A.L., Ledbetter D.H., Martin C.L. Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genet. Med. 2008;10:415–429. doi: 10.1097/GIM.0b013e318177015c. [DOI] [PubMed] [Google Scholar]
- 32.Lipson D., Aumann Y., Ben-Dor A., Linial N., Yakhini Z. Efficient calculation of interval scores for DNA copy number data analysis. J. Comput. Biol. 2006;13:215–228. doi: 10.1089/cmb.2006.13.215. [DOI] [PubMed] [Google Scholar]
- 33.Christian F., Szaszák M., Friedl S., Drewianka S., Lorenz D., Goncalves A., Furkert J., Vargas C., Schmieder P., Götz F. Small molecule AKAP-protein kinase A (PKA) interaction disruptors that activate PKA interfere with compartmentalized cAMP signaling in cardiac myocytes. J. Biol. Chem. 2011;286:9079–9096. doi: 10.1074/jbc.M110.160614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaminsky E.B., Kaul V., Paschall J., Church D.M., Bunke B., Kunig D., Moreno-De-Luca D., Moreno-De-Luca A., Mulle J.G., Warren S.T. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet. Med. 2011;13:777–784. doi: 10.1097/GIM.0b013e31822c79f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stankiewicz P., Kulkarni S., Dharmadhikari A.V., Sampath S., Bhatt S.S., Shaikh T.H., Xia Z., Pursley A.N., Cooper M.L., Shinawi M. Recurrent deletions and reciprocal duplications of 10q11.21q11.23 including CHAT and SLC18A3 are likely mediated by complex low-copy repeats. Hum. Mutat. 2012;33:165–179. doi: 10.1002/humu.21614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lord C., Petkova E., Hus V., Gan W., Lu F., Martin D.M., Ousley O., Guy L., Bernier R., Gerdts J. A multisite study of the clinical diagnosis of different autism spectrum disorders. Arch. Gen. Psychiatry. 2012;69:306–313. doi: 10.1001/archgenpsychiatry.2011.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golzio C., Willer J., Talkowski M.E., Oh E.C., Taniguchi Y., Jacquemont S., Reymond A., Sun M., Sawa A., Gusella J.F. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gotham K., Pickles A., Lord C. Standardizing ADOS scores for a measure of severity in autism spectrum disorders. J. Autism Dev. Disord. 2009;39:693–705. doi: 10.1007/s10803-008-0674-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antonacci F., Kidd J.M., Marques-Bonet T., Teague B., Ventura M., Girirajan S., Alkan C., Campbell C.D., Vives L., Malig M. A large and complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat. Genet. 2010;42:745–750. doi: 10.1038/ng.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Audo I., Kohl S., Leroy B.P., Munier F.L., Guillonneau X., Mohand-Saïd S., Bujakowska K., Nandrot E.F., Lorenz B., Preising M. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2009;85:720–729. doi: 10.1016/j.ajhg.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Genderen M.M., Bijveld M.M., Claassen Y.B., Florijn R.J., Pearring J.N., Meire F.M., McCall M.A., Riemslag F.C., Gregg R.G., Bergen A.A., Kamermans M. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am. J. Hum. Genet. 2009;85:730–736. doi: 10.1016/j.ajhg.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim H.G., Kishikawa S., Higgins A.W., Seong I.S., Donovan D.J., Shen Y., Lally E., Weiss L.A., Najm J., Kutsche K. Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 2008;82:199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poduri A., Chopra S.S., Neilan E.G., Elhosary P.C., Kurian M.A., Meyer E., Barry B.J., Khwaja O.S., Salih M.A., Stödberg T. Homozygous PLCB1 deletion associated with malignant migrating partial seizures in infancy. Epilepsia. 2012;53:e146–e150. doi: 10.1111/j.1528-1167.2012.03538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lo Vasco V.R., Cardinale G., Polonia P. Deletion of PLCB1 gene in schizophrenia-affected patients. J. Cell. Mol. Med. 2012;16:844–851. doi: 10.1111/j.1582-4934.2011.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luciano M., Huffman J.E., Arias-Vásquez A., Vinkhuyzen A.A., Middeldorp C.M., Giegling I., Payton A., Davies G., Zgaga L., Janzing J. Genome-wide association uncovers shared genetic effects among personality traits and mood states. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2012;159B:684–695. doi: 10.1002/ajmg.b.32072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Talkowski M.E., Mullegama S.V., Rosenfeld J.A., van Bon B.W., Shen Y., Repnikova E.A., Gastier-Foster J., Thrush D.L., Kathiresan S., Ruderfer D.M. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am. J. Hum. Genet. 2011;89:551–563. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams S.R., Mullegama S.V., Rosenfeld J.A., Dagli A.I., Hatchwell E., Allen W.P., Williams C.A., Elsea S.H. Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur. J. Hum. Genet. 2010;18:436–441. doi: 10.1038/ejhg.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Glessner J.T., Wang K., Cai G., Korvatska O., Kim C.E., Wood S., Zhang H., Estes A., Brune C.W., Bradfield J.P. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Girirajan S., Rosenfeld J.A., Coe B.P., Parikh S., Friedman N., Goldstein A., Filipink R.A., McConnell J.S., Angle B., Meschino W.S. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med. 2012;367:1321–1331. doi: 10.1056/NEJMoa1200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schaaf C.P., Sabo A., Sakai Y., Crosby J., Muzny D., Hawes A., Lewis L., Akbar H., Varghese R., Boerwinkle E. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum. Mol. Genet. 2011;20:3366–3375. doi: 10.1093/hmg/ddr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.State M.W., Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nat. Neurosci. 2011;14:1499–1506. doi: 10.1038/nn.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang D., Sliwkowski M.X., Mark M., Frantz G., Akita R., Sun Y., Hillan K., Crowley C., Brush J., Godowski P.J. Neuregulin-3 (NRG3): a novel neural tissue-enriched protein that binds and activates ErbB4. Proc. Natl. Acad. Sci. USA. 1997;94:9562–9567. doi: 10.1073/pnas.94.18.9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen P.L., Avramopoulos D., Lasseter V.K., McGrath J.A., Fallin M.D., Liang K.Y., Nestadt G., Feng N., Steel G., Cutting A.S. Fine mapping on chromosome 10q22-q23 implicates Neuregulin 3 in schizophrenia. Am. J. Hum. Genet. 2009;84:21–34. doi: 10.1016/j.ajhg.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Helbig I., Mefford H.C., Sharp A.J., Guipponi M., Fichera M., Franke A., Muhle H., de Kovel C., Baker C., von Spiczak S. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat. Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davy B.E., Robinson M.L. Congenital hydrocephalus in hy3 mice is caused by a frameshift mutation in Hydin, a large novel gene. Hum. Mol. Genet. 2003;12:1163–1170. doi: 10.1093/hmg/ddg122. [DOI] [PubMed] [Google Scholar]
- 58.Moreno-De-Luca D., Sanders S.J., Willsey A.J., Mulle J.G., Lowe J.K., Geschwind D.H., State M.W., Martin C.L., Ledbetter D.H. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol. Psychiatry. 2012 doi: 10.1038/mp.2012.138. Published online October 9, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karakoc E., Alkan C., O’Roak B.J., Dennis M.Y., Vives L., Mark K., Rieder M.J., Nickerson D.A., Eichler E.E. Detection of structural variants and indels within exome data. Nat. Methods. 2012;9:176–178. doi: 10.1038/nmeth.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.