

Abstract
Urofacial syndrome (UFS) (or Ochoa syndrome) is an autosomal-recessive disease characterized by congenital urinary bladder dysfunction, associated with a significant risk of kidney failure, and an abnormal facial expression upon smiling, laughing, and crying. We report that a subset of UFS-affected individuals have biallelic mutations in LRIG2, encoding leucine-rich repeats and immunoglobulin-like domains 2, a protein implicated in neural cell signaling and tumorigenesis. Importantly, we have demonstrated that rare variants in LRIG2 might be relevant to nonsyndromic bladder disease. We have previously shown that UFS is also caused by mutations in HPSE2, encoding heparanase-2. LRIG2 and heparanase-2 were immunodetected in nerve fascicles growing between muscle bundles within the human fetal bladder, directly implicating both molecules in neural development in the lower urinary tract.
Main Text
Lower-urinary-tract (LUT) and/or kidney malformations occur in 1–2 out of 1,000 pregnancies and are common causes of childhood renal failure.1 Advances have been made regarding genetic causes of kidney malformations,2 but less is known about LUT malformations despite the fact that some, including nonsyndromic vesicoureteric reflux (VUR), are common and familial.3 Autonomic nerve activity controls the bladder’s ability to act as a low-pressure reservoir, which intermittently and completely expels its contents per urethra.4 Several congenital disorders feature dysfunctional bladders. Bladder muscle is weak in prune belly syndrome (PBS [MIM 100100]), a condition sometimes associated with mutation of CHRM3 (MIM 118494), encoding the M3 receptor that mediates detrusor contraction.5 In urofacial syndrome (UFS [MIM 236730]), or Ochoa syndrome, detrusor muscle is overactive yet fails to fully expel urine because of concomitant internal sphincter contraction.6 Individuals with PBS and UFS can experience lifelong urinary incontinence, recurrent urosepsis, VUR, and kidney failure.5–7 In addition, some individuals with UFS have severe constipation, indicating a generalized elimination defect.
In 2010, biallelic, loss-of-function mutations in HPSE2 (MIM 613469) were identified as causing UFS.8,9 HPSE2 encodes heparanase-2, which binds heparan sulfate and inhibits heparanase-1 activity.10 HPSE2 mutations were not detected in all UFS cases,8 consistent with genetic heterogeneity. Accordingly, after institutional ethical review and approval (University of Manchester [06138] and National Health Service ethics committees [06/Q1406/52 and 11/NW/0021]) and written informed consent, copy-number analysis and autozygosity mapping (in consanguineous families only) were performed with the Affymetrix SNP 6.0 array, as previously described,8 and genotype calling was carried out with AutoSNPa.11 We performed whole-exome capture (Agilent SureSelect 38 Mb) followed by massively parallel sequencing (Illumina HiSeq2000) on two such unrelated individuals (IV:2 from family 1 and II:1 from family 2). Over 3 Gb of sequence was generated for each subject. Reads were aligned with the human reference genome version GRCh37/Hg19, and >73% of the targeted exome was represented by at least 10-fold coverage. Single-nucleotide substitutions and small insertion and/or deletion variants were identified with our in-house variant-calling pipeline (Table S1, available online), and exome variant profiles were analyzed with a model of a rare autosomal-recessive disorder. In the same gene in both individuals, we prioritized rare putative loss-of-function variants that were absent from variome databases, including dbSNP build 137 and the National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server (EVS, ESP6500). Putative pathogenic variants were confirmed by Sanger sequencing. PCR amplification of genomic DNA was performed with primers designed to cover the exon and intron-exon boundaries with NCBI Primer BLAST (Table S2). Direct sequencing was performed with the ABI BigDye Terminator v.3.1 cycle sequencer system (Applied Biosystems) on an ABI 3730 sequencer.
In family 1 (Figure 1A), IV:2 is an 8-year-old Turkish girl with facial features of UFS (Figure S1), and she is the second child of consanguineous parents. At the age of 5 years, she presented with urosepsis and constipation. Investigations revealed a low-capacity, overactive bladder, as well as bilateral VUR, hydronephrosis, and mild renal impairment. She was treated with intermittent catheterization, antimuscarinic drugs, and surgical bladder augmentation; however, despite these treatments, renal failure progressed, and she is about to start peritoneal dialysis. Autozygosity mapping, undertaken with Affymetrix v.6.0 SNP arrays as previously described,8 revealed in 1p13.2 a 52 Mb homozygous region containing 577 coding genes; she shares this region with her younger brother (IV:3, Figure S1). At 5 years old, he was recognized to have facial features of UFS but had no urinary-tract symptoms. His urinary tract was normal as assessed by ultrasound and uroflowmetry. Invasive micturating cystourethrogram and urodynamic studies were not undertaken because he remains asymptomatic. Such phenotypic variability has been reported previously in families affected by UFS12 such that some affected individuals had only the facial or urinary tract phenotype. The varied expression might be accounted for by genetic modifiers or environmental factors, either intrauterine or postnatal, that remain to be identified.
Figure 1.

Identification of Mutations in LRIG2 in Three Families Affected by UFS
Pedigrees and LRIG2 mutation analysis in families 1 (A), 2 (B), and 3 (C).
(A) In family 1, a homozygous frameshift (c.1230delA [p.Glu410Aspfs∗6]) in exon 10 was identified.
(B) In family 2, affected child II-I is shown at the age of 6 months. Her voiding cystourethrogram shows a trabeculated bladder and severe left-sided VUR (Bi). A compound-heterozygous frameshift (c.2088delC [p.Ser697Hisfs∗11]) (Bii) and a compound-heterozygous AluYa5-like insertion (c.1980_1981ins371, GenBank JX891452) (Biii) were identified. Gel electrophoresis demonstrated a larger band present in I:2, II:1, and II:2, and sequencing of cDNA confirmed the heterozygous skipping of exon 14 in the same individuals.
(C) In family 3, a homozygous nonsense mutation in exon 15 (c.2125C>T [p.Arg709∗]) was identified. All individuals genotyped for LRIG2 variants are indicated by an asterisk. The mutations segregated in the three families consistently with the phenotype.
Within the 1p13.2 chromosomal region, exome sequencing identified a homozygous single-base-pair deletion resulting in a frameshift in exon 10 of LRIG2 (MIM 608869): c.1230delA (p.Glu410Aspfs∗6) (RefSeq accession number NM_014813.1). This deletion was confirmed by Sanger sequencing (Figure 1A) and segregated with the disease. The mutation was absent from variome databases, 116 local exomes, and 94 healthy Turkish controls.
Exome sequencing was also performed on a 5-year-old Spanish girl (II:1 from family 2; Figure 1B) of nonconsanguineous parents. She is one of a pair of dizygotic twins conceived after intracytoplasmic sperm injection. Megacystis was visualized on ultrasonography in late gestation. An abnormal smile was noted at 6 months of age, when investigations revealed an overactive, trabeculated bladder and left-sided VUR. Her course was complicated by urosepsis and constipation, and she was treated with intermittent catheterization and anticholinergic drugs. In this child, exome and Sanger sequencing identified a heterozygous frameshift mutation, c.2088delC (p.Ser697Hisfs∗11), in exon 15 and a heterozygous insertion, c.1980_1981ins371 (GenBank JX891452), in exon 14 of LRIG2 (Figure 1B). The insertion was defined as the antisense insertion of an Alu element with homology to AluYa5. Sequencing of lymphocyte cDNA determined that the insertion resulted in a transcript that skips exon 14 and that is not subject to nonsense-mediated decay (NMD) (Figure 1B). Lymphocyte RNA from family members was extracted with the PAXgene blood RNA system (QIAGEN). Reverse transcription and PCR amplification of LRIG2 cDNA were performed with the SuperScript III one-step RT-PCR system (Life Technologies) and specific primers (Table S2).
A second consanguineous Turkish family (family 3; Figure 1C) has two siblings affected by UFS. II:1 is a 9-year-old girl with facial features of UFS (Figure S1) and constipation. At 4 years old, she presented with urosepsis and enuresis when a low-capacity, overactive bladder and bilateral VUR were detected. At the age of 6 years, she underwent a left nephrectomy for Wilms tumor.13 At the age of 2 years, her younger sister, II:2, presented with enuresis and a low-capacity, overactive bladder and VUR and subsequently underwent bladder augmentation. Autozygosity mapping identified in chromosomal region 1p13.2 a 8.5 Mb segment overlapping that in family 1, and Sanger sequencing of LRIG2 identified homozygous nonsense mutation c.2125C>T (p.Arg709∗) in exon 15. This mutation segregated with the disease in the family and was absent from variome databases, 116 local exomes, and 94 healthy Turkish controls. Previous studies have implicated the loss of a 1 Mb locus in chromosomal region 1p13 in Wilms tumor,14 but LRIG2 lies outside the critical region.
The identified nonsense and frameshift LRIG2 mutations were predicted to result in loss of function via NMD of transcripts. cDNA analysis showed that the large insertion within exon 14 of family 2 results in an in-frame skipping of this exon from the transcript; this is likely due to disruption of exon splice enhancers, consistent with the effects of previously reported exonic insertions of AluYa5 elements.15 The transcript was not subject to NMD but was predicted to result in loss of most of the second immunoglobulin (Ig)-like domain and most conserved part of the gene within the LRIG family (Figure S2).16 Despite somewhat variable LUT phenotypes, there were no consistent clinical differences between these UFS individuals with LRIG2 mutations and those previously reported to have HPSE2 mutations.8 At present, it is difficult to draw a conclusion about the relative contribution of each gene to UFS. To date, of the 14 families affected by classical UFS, we identified nine (64.3%) with mutations in HPSE2, three (21.4%) with mutations in LRIG2, and two (14.3%) with mutations in neither gene. Other groups have only reported individuals with mutations in HPSE2.9
Bladder dysfunction in UFS is similar to that found in Hinman syndrome, or “non-neurogenic neurogenic bladder.”17,18 Accordingly, we screened LRIG2 in 23 individuals (13 multiethnic British and 10 previously reported Turkish cases) with Hinman syndrome.19 Of these, variants in LRIG2 were found in a single individual, a white British male who, at the age of 10 years, presented with a 2 year history of secondary enuresis. He was hypertensive and had mild renal impairment. Investigations revealed a trabeculated bladder with a thickened wall and bilateral severe hydronephrosis. A micturating cystourethrogram showed no obstruction, and urodynamic analysis revealed a poorly compliant bladder with detrusor overactivity and detrusor sphincter dyssynergia. Renal failure progressed despite bladder augmentation. He lacks the facial features of UFS. He has compound heterozygous missense variants c.1648C>T (p.Arg550Cys) (exon 13) and c.2554A>T (p.Ile852Phe) (exon 16, Figure 2). c.1648C>T (p.Arg550Cys) is reported in the EVS as a rare variant (2 in 8,600 European American alleles), whereas c.2554A>T (p.Ile852Phe) is not present in the EVS but is reported in dbSNP (minor allele frequency = 0.003). Both the 550 and 852 residue positions are conserved to Tetraodon (Figure S2). The in silico tool PolyPhen-2 predicted that both variants are damaging (0.99), and SIFT predicted that both are most likely deleterious. LRIG proteins share a domain structure with 15 leucine-rich repeats, three Ig-like domains, a transmembrane domain, and a cytoplasmic tail.16,20 The crystal structure of LRIG2 is not determined, but Arg550 resides in the first Ig-like domain of the extracellular milieu and is orthologous to an experimentally determined structure from murine LRIG3; it has >50% sequence identity to LRIG2 in this region (Figure S3). Probably, this substitution disrupts oligomer formation of the Ig domains. The Ile852 residue is located in the cytoplasmic exposed region, close to the predicted transmembrane helix (amino acids 808–830). Its function, as well as its mutation consequence, is less predictable as a result of a lack of structural information. Screening of LRIG2 in further individuals with nonsyndromic voiding dysfunction will be required for establishing the contribution of this gene to this broader phenotype.
Figure 2.
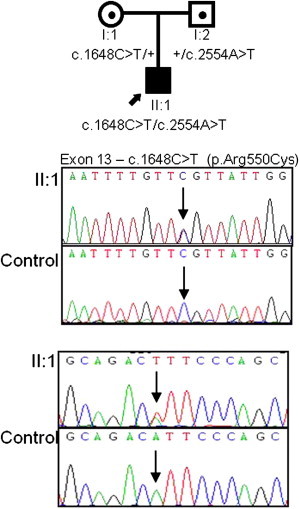
Identification of Mutations in LRIG2 in an Individual with Nonsyndromic Dysfunctional Voiding
Pedigree and LRIG2 mutation analysis in this family revealed compound heterozygous missense mutations, c.1648C>T [p.Arg550Cys] (exon 13) and c.2554A>T [p.Ile852Phe] (exon 16), in affected individual II:1.
LRIG2 modulates cell turnover and adhesion in neural support cells via altered growth-factor signaling,21 and altered LRIG2 localization occurs in various types of neural tumors.22–24 Detection of LUT abnormalities antenatally and abnormal grimacing in infancy implies that pathogenic mechanisms underlying UFS initiate during development. Indeed, in embryonic mice, VII nerve ganglia express Lrig2.16 The human bladder initiates at 7 weeks of gestation.5,25 At this stage, we detected by immunohistochemistry neither LRIG2 nor heparanase-2 (data not shown). Later in the first trimester, detrusor muscle differentiates25 and is invaded by autonomic nerves.26 At this stage, both LRIG2 and heparanase-2 were immunodetected within nerve fascicles located between muscle bundles (Figures 3A–3D). LRIG2 was also weakly immunolocalized in smooth-muscle bundles themselves (Figure 3A). Coimmunostaining with β3-tubulin, a neuronal axonal cytoskeletal protein, demonstrated that LRIG2 appears to be localized in cells adjacent to neurons (Figures 3E–3G). These might be Schwann-like cells within preganglionic parasympathetic nerves, the neurons of which synapse with second-order neurons within intramural ganglia.4 Furthermore, we detected LRIG2 transcripts in 12-week-postconception human fetal bladders and ureters, but not in kidney tissue (Figure S4), whereas the three transcripts of HPSE227 were detected in the bladder, but not in ureter or kidney tissue. Heparanase-2 immunoreactivity partially overlapped with β3-tubulin (Figures 3H–3J), consistent with its being present in neurons themselves. Evidence already implicates heparanase-1 in neural biology,28–30 and its enzymatic activity is inhibited by heparanase-2.10
Figure 3.
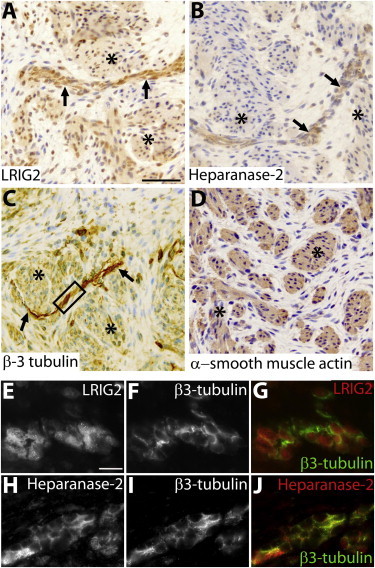
Immunohistochemistry of a Normal Urinary Bladder at 12 Weeks of Gestation
(A–D) Bright-field images counterstained with hematoxylin (blue nuclei).
(A) Immunodetection of LRIG2 (brown). Note the prominent signal in a linear structure (arrows) between muscle bundles (asterisks), which themselves show weaker LRIG2 immunoreactivity. The scale bar represents 50 μm.
(B) Heparanase-2 immunodetected in a nerve-like structure between muscle bundles.
(C) The linear structure contains components with β3-tubulin immunoreactivity, an axonal protein, thus confirming that it is a nerve.
(D) Detrusor muscle bundles express α-smooth muscle actin.
(E–J) High-power dark-field images of an area of nerve similar to that in the outlined box in (C). LRIG2 and β3-tubulin immunoreactivity (white) are depicted in (E) and (F), respectively. The color merged image (G) shows that the signal for LRIG2 (red) is generally discrete from the signal (green) for β3-tubulin; given that the latter is an axonal marker, LRIG2 might be mainly localized in neural support cells, the exact nature of which remains to be established. Heparanse-2 and β3-tubulin immunoreactivity (white) are depicted in (H) and (I), respectively. The merged image (J) shows that the signal for heparanase-2 partially overlaps with that of β3-tubulin to generate a yellow color. Histology, incorporating 5 μm paraffin sections of paraformaldehyde-fixed tissues, was undertaken with human fetal tissue collected with ethical approval under the Codes of Practice of the UK Human Tissue Authority. Endogenous peroxidise was quenched by incubation with hydrogen peroxide, and antigen retrieval was undertaken by heating at 95°C for 5 minutes in sodium citrate (pH 6). The primary antibodies used were goat anti-α-smooth muscle actin (Sigma-Aldrich SAB2500963), chicken anti-β3-tubulin (Millipore AB9354), rabbit anti-LRIG2 (Abgent AP13821b), and rabbit anti-heparanase-2 (Generon). The latter was generated against a unique epitope (NH2-QLDPSIIHDGWLDC-CONH2). After application of appropriate secondary antibodies, a streptavidin-horseradish-peroxidase-DAB system was used. For coimmunostaining immunofluorescence studies, the quenching step was omitted and species-specific secondary antibodies, each conjugated to a different fluorophores with nonoverlapping emission spectra, were used for detecting the proteins under study. The scale bar in (E) represents 10 μm.
Collectively, the evidence indicates that both LRIG2 and heparanase-2 are required for normal LUT innervation and/or neural function. It is curious why loss-of-function mutations in these two genes, expressed more widely in the peripheral nervous system, should result in a specific phenotype confined to elimination and facial expression. Mild degrees of dysfunctional bladder voiding, including overactivity, are common in otherwise healthy children after the age when conscious bladder control has normally been acquired.18 Nonsyndromic VUR, which affects 1% of children, is commonly familial.3 We speculate that the biological pathways mediated by LRIG2 and the heparanases might be implicated in these common disorders of elimination.
Acknowledgments
H.M.S. is funded through a Wellcome Trust Clinical Training Fellowship. N.A.H. is a Wellcome Trust Senior Fellow in Clinical Science. The study was also funded by project grants from Kidney Research UK and Kidneys for Life and was also supported from the Manchester Biomedical Research Centre.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/genbank/
NHLBI Exome Variant Server Exome Sequencing Project, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Primer-BLAST, http://www.ncbi.nlm.nih.gov/tools/primer-blast/
SIFT, http://sift.jcvi.org/
Accession Numbers
The GenBank accession number for the LRIG2 Alu insertion reported in this paper is JX891452.
References
- 1.Kerecuk L., Schreuder M.F., Woolf A.S. Renal tract malformations: Perspectives for nephrologists. Nat. Clin. Pract. Nephrol. 2008;4:312–325. doi: 10.1038/ncpneph0807. [DOI] [PubMed] [Google Scholar]
- 2.Adalat S., Bockenhauer D., Ledermann S.E., Hennekam R.C., Woolf A.S. Renal malformations associated with mutations of developmental genes: Messages from the clinic. Pediatr. Nephrol. 2010;25:2247–2255. doi: 10.1007/s00467-010-1578-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lambert H.J., Stewart A., Gullett A.M., Cordell H.J., Malcolm S., Feather S.A., Goodship J.A., Goodship T.H., Woolf A.S., UK VUR Study Group Primary, nonsyndromic vesicoureteric reflux and nephropathy in sibling pairs: A United Kingdom cohort for a DNA bank. Clin. J. Am. Soc. Nephrol. 2011;6:760–766. doi: 10.2215/CJN.04580510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benarroch E.E. Neural control of the bladder: Recent advances and neurologic implications. Neurology. 2010;75:1839–1846. doi: 10.1212/WNL.0b013e3181fdabba. [DOI] [PubMed] [Google Scholar]
- 5.Weber S., Thiele H., Mir S., Toliat M.R., Sozeri B., Reutter H., Draaken M., Ludwig M., Altmüller J., Frommolt P. Muscarinic acetylcholine receptor M3 mutation causes urinary bladder disease and a prune-belly-like syndrome. Am. J. Hum. Genet. 2011;89:668–674. doi: 10.1016/j.ajhg.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ochoa B. Can a congenital dysfunctional bladder be diagnosed from a smile? The Ochoa syndrome updated. Pediatr. Nephrol. 2004;19:6–12. doi: 10.1007/s00467-003-1291-1. [DOI] [PubMed] [Google Scholar]
- 7.Ochoa B., Gorlin R.J. Urofacial (ochoa) syndrome. Am. J. Med. Genet. 1987;27:661–667. doi: 10.1002/ajmg.1320270320. [DOI] [PubMed] [Google Scholar]
- 8.Daly S.B., Urquhart J.E., Hilton E., McKenzie E.A., Kammerer R.A., Lewis M., Kerr B., Stuart H., Donnai D., Long D.A. Mutations in HPSE2 cause urofacial syndrome. Am. J. Hum. Genet. 2010;86:963–969. doi: 10.1016/j.ajhg.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pang J., Zhang S., Yang P., Hawkins-Lee B., Zhong J., Zhang Y., Ochoa B., Agundez J.A., Voelckel M.A., Fisher R.B. Loss-of-function mutations in HPSE2 cause the autosomal recessive urofacial syndrome. Am. J. Hum. Genet. 2010;86:957–962. doi: 10.1016/j.ajhg.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy-Adam F., Feld S., Cohen-Kaplan V., Shteingauz A., Gross M., Arvatz G., Naroditsky I., Ilan N., Doweck I., Vlodavsky I. Heparanase 2 interacts with heparan sulfate with high affinity and inhibits heparanase activity. J. Biol. Chem. 2010;285:28010–28019. doi: 10.1074/jbc.M110.116384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 12.Aydogdu O., Burgu B., Demirel F., Soygur T., Ozcakar Z.B., Yalcinkaya F., Tekgul S. Ochoa syndrome: A spectrum of urofacial syndrome. Eur. J. Pediatr. 2010;169:431–435. doi: 10.1007/s00431-009-1042-9. [DOI] [PubMed] [Google Scholar]
- 13.Emir S., Kan R., Demir H.A., Cakar N., Güler M. Occurrence of Wilms tumor in a child with urofacial (OCHOA) syndrome. Pediatr. Hematol. Oncol. 2011;28:616–618. doi: 10.3109/08880018.2011.600803. [DOI] [PubMed] [Google Scholar]
- 14.Natrajan R., Williams R.D., Grigoriadis A., Mackay A., Fenwick K., Ashworth A., Dome J.S., Grundy P.E., Pritchard-Jones K., Jones C. Delineation of a 1Mb breakpoint region at 1p13 in Wilms tumors by fine-tiling oligonucleotide array CGH. Genes Chromosomes Cancer. 2007;46:607–615. doi: 10.1002/gcc.20446. [DOI] [PubMed] [Google Scholar]
- 15.Claverie-Martín F., Flores C., Antón-Gamero M., González-Acosta H., García-Nieto V. The Alu insertion in the CLCN5 gene of a patient with Dent’s disease leads to exon 11 skipping. J. Hum. Genet. 2005;50:370–374. doi: 10.1007/s10038-005-0265-5. [DOI] [PubMed] [Google Scholar]
- 16.Homma S., Shimada T., Hikake T., Yaginuma H. Expression pattern of LRR and Ig domain-containing protein (LRRIG protein) in the early mouse embryo. Gene Expr. Patterns. 2009;9:1–26. doi: 10.1016/j.gep.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Vidal I., Héloury Y., Ravasse P., Lenormand L., Leclair M.D. Severe bladder dysfunction revealed prenatally or during infancy. J. Pediatr. Urol. 2009;5:3–7. doi: 10.1016/j.jpurol.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Leclair M.D., Héloury Y. Non-neurogenic elimination disorders in children. J. Pediatr. Urol. 2010;6:338–345. doi: 10.1016/j.jpurol.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Silay M.S., Tanriverdi O., Karatag T., Ozcelik G., Horasanli K., Miroglu C. Twelve-year experience with Hinman-Allen syndrome at a single center. Urology. 2011;78:1397–1401. doi: 10.1016/j.urology.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 20.Guo D., Holmlund C., Henriksson R., Hedman H. The LRIG gene family has three vertebrate paralogs widely expressed in human and mouse tissues and a homolog in Ascidiacea. Genomics. 2004;84:157–165. doi: 10.1016/j.ygeno.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 21.Wang B., Han L., Chen R., Cai M., Han F., Lei T., Guo D. Downregulation of LRIG2 expression by RNA interference inhibits glioblastoma cell (GL15) growth, causes cell cycle redistribution, increases cell apoptosis and enhances cell adhesion and invasion in vitro. Cancer Biol. Ther. 2009;8:1018–1023. doi: 10.4161/cbt.8.11.8375. [DOI] [PubMed] [Google Scholar]
- 22.Hedman H., Henriksson R. LRIG inhibitors of growth factor signalling - Double-edged swords in human cancer? Eur. J. Cancer. 2007;43:676–682. doi: 10.1016/j.ejca.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 23.Ghasimi S., Haapasalo H., Eray M., Korhonen K., Brännström T., Hedman H., Andersson U. Immunohistochemical analysis of LRIG proteins in meningiomas: Correlation between estrogen receptor status and LRIG expression. J. Neurooncol. 2012;108:435–441. doi: 10.1007/s11060-012-0856-x. [DOI] [PubMed] [Google Scholar]
- 24.Holmlund C., Haapasalo H., Yi W., Raheem O., Brännström T., Bragge H., Henriksson R., Hedman H. Cytoplasmic LRIG2 expression is associated with poor oligodendroglioma patient survival. Neuropathology. 2009;29:242–247. doi: 10.1111/j.1440-1789.2008.00970.x. [DOI] [PubMed] [Google Scholar]
- 25.Jenkins D., Winyard P.J.D., Woolf A.S. Immunohistochemical analysis of Sonic hedgehog signalling in normal human urinary tract development. J. Anat. 2007;211:620–629. doi: 10.1111/j.1469-7580.2007.00808.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gilpin S.A., Gosling J.A. Smooth muscle in the wall of the developing human urinary bladder and urethra. J. Anat. 1983;137:503–512. [PMC free article] [PubMed] [Google Scholar]
- 27.McKenzie E., Tyson K., Stamps A., Smith P., Turner P., Barry R., Hircock M., Patel S., Barry E., Stubberfield C. Cloning and expression profiling of Hpa2, a novel mammalian heparanase family member. Biochem. Biophys. Res. Commun. 2000;276:1170–1177. doi: 10.1006/bbrc.2000.3586. [DOI] [PubMed] [Google Scholar]
- 28.Cui H., Shao C., Liu Q., Yu W., Fang J., Yu W., Ali A., Ding K. Heparanase enhances nerve-growth-factor-induced PC12 cell neuritogenesis via the p38 MAPK pathway. Biochem. J. 2011;440:273–282. doi: 10.1042/BJ20110167. [DOI] [PubMed] [Google Scholar]
- 29.Navarro F.P., Fares R.P., Sanchez P.E., Nadam J., Georges B., Moulin C., Morales A., Pequignot J.M., Bezin L. Brain heparanase expression is up-regulated during postnatal development and hypoxia-induced neovascularization in adult rats. J. Neurochem. 2008;105:34–45. doi: 10.1111/j.1471-4159.2007.05116.x. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y., Yeung M.N., Liu J., Chau C.H., Chan Y.S., Shum D.K. Mapping heparanase expression in the spinal cord of adult rats. J. Comp. Neurol. 2006;494:345–357. doi: 10.1002/cne.20811. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.