

Abstract
The purpose of this study was to determine the deep sequencing and analytic conditions needed to detect fetal subchromosome abnormalities across the genome from a maternal blood sample. Cell-free (cf) DNA was isolated from the plasma of 11 pregnant women carrying fetuses with subchromosomal duplications and deletions, translocations, mosaicism, and trisomy 20 diagnosed by metaphase karyotype. Massively parallel sequencing (MPS) was performed with 25-mer tags at approximately 109 tags per sample and mapped to reference human genome assembly hg19. Tags were counted and normalized to fixed genome bin sizes of 1 Mb or 100 kb to detect statistically distinct copy-number changes compared to the reference. All seven cases of microdeletions, duplications, translocations, and the trisomy 20 were detected blindly by MPS, including a microdeletion as small as 300 kb. In two of these cases in which the metaphase karyotype showed additional material of unknown origin, MPS identified both the translocation breakpoint and the chromosomal origin of the additional material. In the four mosaic cases, the subchromosomal abnormality was not demonstrated by MPS. This work shows that in nonmosaic cases, it is possible to obtain a fetal molecular karyotype by MPS of maternal plasma cfDNA that is equivalent to a chromosome microarray and in some cases is better than a metaphase karyotype. This approach combines the advantage of enhanced fetal genomic resolution with the improved safety of a noninvasive maternal blood test.
Introduction
Two major recent advances in prenatal diagnosis, chromosome microarrays (CMA) and noninvasive prenatal testing (NIPT) via sequencing of maternal plasma cell-free DNA, have already translated to clinical care and are changing widely accepted clinical paradigms.1 According to the recommendations of several expert groups, CMAs have replaced the standard metaphase karyotype in the postnatal assessment of individuals with developmental delay, intellectual disability, congenital anomalies, and autism.2 Discussion is ongoing regarding the role of CMAs in prenatal diagnosis.3,4 Recently, an NIH-sponsored clinical trial investigating the accuracy of fetal diagnosis by comparing metaphase karyotype to CMA was completed.5 The results showed an increase in the detection of clinically significant copy-number changes even when the metaphase karyotype was apparently normal at conventional levels of resolution. To use CMAs, however, an invasive procedure such as amniocentesis or chorionic villus sampling (CVS) must be performed to obtain a source of fetal genomic DNA. These procedures carry small but well-validated rates of spontaneous pregnancy loss and they can be performed only by obstetricians who have specialized training.
At the same time, major progress has been achieved in the ability to noninvasively detect whole-chromosome aneuploidy by massively parallel sequencing (MPS) of maternal plasma cell-free (cf) DNA that includes fetal cfDNA originating from the placenta.1 In two studies published in 2007, molecular counting utilizing digital PCR indicated that it was possible to determine relative over- or underrepresentation of specific chromosomes, thus inferring the presence of fetal aneuploidy from a maternal plasma sample.6,7 A year later the counting approach transitioned to MPS,8,9 rapidly followed by multiple independent clinical trials that assessed the accuracy of NIPT for the detection of trisomies 21, 18, and 13.1 The success of these trials has led to the clinical introduction of MPS as an alternative to invasive procedures for women carrying fetuses at high risk for whole-chromosome aneuploidies.10
An increasing body of literature has also documented the feasibility of detection of autosomal subchromosome fetal abnormalities by MPS of maternal plasma cfDNA. In an unblinded study, a 4.2 megabase (Mb) paternally inherited deletion of chromosome 12p11.22 to 12p12.1 was found in a maternal sample taken at 35 weeks of gestation.11 In two cases of fetuses previously diagnosed with DiGeorge syndrome, sequencing of maternal plasma demonstrated the presence of the pathognomonic 22q11 deletion.12 In blinded studies, our laboratory detected the presence of a small deletion in chromosome 11q21–2313 and a duplication of 6q.14
The success of MPS to detect both whole and subchromosome fetal copy number variation depends on the percentage of fetal DNA in the maternal blood sample, which is referred to as the relative fetal fraction (ff). Although early literature suggested that the characteristic ff averaged only 5%–6%,15 subsequent MPS studies have determined that ffs of between 10% and 15% are more typical.16,17 The higher ffs make it more likely that subchromosomal fetal abnormalities can be determined with these methods. They also provide the opportunity to confirm observed structural variations and avoid false-positive test results.
The purpose of the current study was to determine the deep sequencing and analytic conditions needed to noninvasively detect fetal subchromosome abnormalities across the genome. Our goal was to create the equivalent of a CMA that could be applied to a maternal blood sample. If successful, this would combine the advantages of the greater genomic resolution provided by CMAs with the increased safety of performing a blood test instead of an invasive procedure. Here we show that it is possible to detect fetal subchromosome abnormalities at a resolution of 100 kb across the genome, validating the concept of a noninvasive molecular karyotype of the fetus that could have clinical utility in the near future.
Material and Methods
Sample Preparation and Sequencing
Artificial Mixtures
To determine the depth of sequencing needed to detect fetal subchromosome abnormalities and to assess the effect of the relative ff of cfDNA present in the sample, we created artificial mixtures of 5% and 10% sheared genomic DNA by using paired mother and child DNAs obtained from the Coriell Institute for Medical Research (Camden, NJ). All children were males with karyotypes previously determined by metaphase cytogenetic analysis. The karyotypes of the four paired samples are shown in Table S1 available online. The children’s chromosome abnormalities were selected to represent different clinical scenarios, such as (1) whole-chromosome aneuploidy (family 2139), (2) subchromosomal deletion (family 1313), (3) mosaic subchromosomal copy-number change (family 2877, with an additional inherited deletion), and (4) a derivative chromosome that contains a subchromosomal duplication (family 1925).
The genomic DNA samples were sheared to a size of ∼200 bp via the Covaris S2 sonicator (Covaris, Woburn, MA) according to the manufacturer’s recommended protocols. DNA fragments smaller than 100 bp were removed with AmPure XP beads (Beckman Coulter Genomics, Danvers, MA). Sequencing libraries were generated with TruSeq v1 Sample Preparation kits (Illumina, San Diego, CA) from sheared DNA mixtures consisting of maternal DNA only and maternal + child DNA mixtures at 5% and 10% w/w. Samples were sequenced with single-ended 36 base pair (bp) reads on the Illumina HiSeq 2000 instrument via TruSeq v3 chemistry. Each sample was sequenced on four lanes of a flow cell, resulting in 400 × 106 to 750 × 106 sequence tags per sample.
Maternal Plasma Samples
The Maternal Blood Is Source to Accurately Diagnose Fetal Aneuploidy (MELISSA) trial was a registered clinical trial (NCT01122524) that recruited high-risk pregnant subjects and plasma samples from 60 different centers in the United States, along with the corresponding metaphase karyotype results from an invasive prenatal diagnostic procedure. Each of the medical centers conducted the study with approval by their local institutional review boards. Written informed consent was obtained from each study participant. The study was designed to prospectively determine the accuracy of MPS to detect whole-chromosome fetal aneuploidy. During this trial, all samples with any abnormal karyotype were included to emulate the real clinical scenarios in which the fetal karyotype is not known at the time of sample acquisition. The results of this study have been previously published.14 After completion of the MELISSA trial, the study database was assessed to identify ten samples that had complex karyotypes, including subchromosome abnormalities, material of unknown origin, or a marker chromosome (Table 1); we also added one MELISSA study sample with trisomy 20 as a control of performance in detection of whole-chromosome aneuploidy. The karyotypes were performed for standard clinical indications and reflected local protocols. For example, some fetal samples were analyzed with CMAs and some had metaphase analysis with or without FISH studies.
Table 1.
Karyotypes of Maternal Plasma Samples Analyzed by MPS
| Sample ID | Specimen | Procedure | Karyotype |
|---|---|---|---|
| AL1893 | chorionic villi | metaphase and 20q12 FISH | 47,XX,+20 |
| BE3096 | cultured villi | metaphase, 6q12, 6q16.3 FISH, and microarray | arr 6q12q16.3(64,075,795-101,594,105)x3, 6q16.3(102,176,578-102,827,691)x3 |
| BF3404 | chorionic villi | metaphase | 46,XY,del(7)(q36.1) |
| AK1604 | amniocytes | metaphase and 22q FISH | 46,XX,del(8)(p23.1p23.2) |
| BE3236 | chorionic villi | metaphase and Chr 15 FISH | 45,XX,-15,der(21)t(15;21)(q15;p11.2) |
| AF1019 | amniocytes | metaphase | 46,XY,add(10)(q26) |
| BC2659 | amniocytes | metaphase | 46,XX,add(X)(p22.1) |
| AL1873 | amniocytes | metaphase and FISH | 46,XY or 46,XY,add(15)(p11.2) |
| BE3129 | amniocytes | metaphase | mos 46,XY,+i(20)(q10)[8]/46,XY[17] |
| BG3701 | chorionic villi | metaphase and FISH | 47,XY,+der(14 or 22)[10]/46,XY[10] |
| AH1200 | chorionic villi | metaphase | 47,XX+mar[12]/46,XX[8] |
The last four samples have mosaic karyotypes.
In the MELISSA study, libraries were sequenced with single-end reads of 36 bp with 6 samples in a lane on an Illumina HiSeq 2000 with TruSeq v2.5 chemistry. In the present study, the previously generated MELISSA libraries were resequenced with TruSeq v3 chemistry on an Illumina HiSeq 2000 with single-end reads of 25 bp. In this study, each of the 11 maternal samples was sequenced with an entire flow cell, resulting in 600 × 106 to 1.3 × 109 sequence tags per sample. All sequencing was performed in the Verinata Health research laboratory (Redwood City, CA) by research laboratory personnel who were blinded to the fetal karyotype.
Normalization and Analysis
Sequence reads were aligned to the human genome assembly hg19 obtained from the UCSC database. Alignments were carried out utilizing the Bowtie short read aligner (version 0.12.5), allowing for up to two base mismatches during alignment. Only reads that unambiguously mapped to a single genomic location were included. Genomic sites at which reads mapped were counted as tags. Regions on the Y chromosome at which sequence tags from male and female samples mapped without any discrimination were excluded from the analysis (specifically, from base 0 to base 2 × 106; base 10 × 106 to base 13 × 106; and base 23 × 106 to the end of chromosome Y).
The genome was then further divided into contiguous 1 Mb and 100 kb bins and, for each sample, tags from both the positive and negative strand were assigned to individual bins for further analysis. The GC percentage of each bin was determined and bins were ranked by GC percentage across the entire genome. Each bin was individually normalized by calculating the ratio of tags within a bin to the sum of the number of tags in bins with the nearest GC percentages by
| (Equation 1) |
where BRVij is the bin ratio value for the jth bin of chromosome i and Tagsij is the number of tags in the jth bin of chromosome i. The sum runs over the 10 bins for the 1 Mb data and 40 bins for the 100 kb data for bins (km) with the nearest GC percentage to bin ij.
In order to detect any subchromosomal differences, we examined each of the BRVs for deviations from the median values measured across multiple samples. The medians were determined from the four maternal only DNAs (Table S1) for the artificial samples and from the 11 maternal plasma samples (Table 1) for the clinical samples and were robust to individual subchromosomal variants that might have been present in any one of the samples. Median absolute deviations (MADs) were calculated for each bin based on the medians and adjusted assuming a normal distribution for the number of tags in each bin. The MADs were adjusted (aMADs) assuming that the BRVs followed a normal distribution (i.e., MAD was multiplied by 1.4826) and were utilized to calculate a z score for each bin:
| (Equation 2) |
We expected zij to be approximately zero to ±3 for regions (i.e., ∼99.8% of values in the normal distribution) without any copy-number variants (CNVs) and significantly greater than 3 when fetal CNVs were present.
The zij values can be utilized to determine the relative ff present in the cfDNA. The value can then be compared to an independent measurement of ff to validate copy number detection, or suggest the presence of mosaicism. For a bin ratio containing a copy-number change from normal, the BRVij will increase (in the case of a duplication) or decrease (in the case of a deletion):
| (Equation 3) |
In this equation, ffn is the fetal fraction for sample n. If we define the coefficient of variation for each bin, CVij as
| (Equation 4) |
then
| (Equation 5) |
can be used to calculate ffn for sample n from zij values when a CNV is present.
Detection and Classification of CNVs
Detection of a subchromosomal abnormality was a three-step process for classifying specific regions as having a CNV present. In step 1, we identified zij values from the 1 Mb bins that exceeded ±4. The zij ± 4 thresholds are indicated in each figure by a dashed horizontal line. The ff was then calculated by applying Equation 5 and bins that had a ff of less than 4% were eliminated. For the samples from pregnant women carrying male fetuses, the ff was also calculated with all of the bins in chromosome X. This value was compared to the result obtained for putative copy number changes to validate a copy-number change or suggest a mosaic result. Finally, in cases in which only a single 1 Mb bin met the above criteria, we examined the 100 kb bins data and required that at least 2 bins within a contiguous group of 4 indicated a zij value that exceeded +4 or −4 before classifying a sample as having a CNV present. All three criteria had to be fulfilled to classify the CNV. For example, individual data points that only had a z score of greater than or less than 4 but did not meet the additional criteria were not classified as CNVs.
Results
Artificial Mixtures
Whole-Chromosome Aneuploidy of Chromosome 21
Figure S1 shows the chromosome 21 z21j values (1 Mb bins) for an artificial mixture of family 2139 with 10% of the son’s DNA (T21) mixed with the mother’s DNA. In chromosome 21, there are approximately 38 Mb (35 Mb in the q arm) that contain unique reference genome sequence in hg19. All of the chromosome 21 tags mapped to this region. With the exception of the first 4 Mb, Figure S1 shows an overrepresentation of a 34 Mb region of chromosome 21 in the 10% mixture, as would be expected with a full-chromosome aneuploidy. With Equation 5 to calculate the ff from the average z21j values of the amplified regions, we obtained ffs of 7.0% and 12.7% for the 5% and 10% mixtures, respectively. Calculating the ff average with chromosome X zXj values, we obtained ffs of 4.2% and 9.0% for the 5% and 10% mixtures, respectively.
Subchromosomal Deletion of Chromosome 7
We next tested the method on Family 1313, in which the son has a subchromosomal deletion of chromosome 7. Figure 1 shows the chromosome 7 z7j values (1 Mb bins) for the maternal sample mixed with 10% of her son’s DNA. A deletion was observed beginning at bin 38 and continuing to bin 58. This reflects the approximately 20 Mb deletion documented in the metaphase karyotype. We calculated ffs of 6.1% and 10.5% for the 5% and 10% mixtures, respectively, for this sample. Calculating the ff average with chromosome X zXj values, we obtained ffs of 5.9% and 10.4%, respectively. Interestingly, in this sample we also noted what appeared to be a duplication in the maternal sample at bin 98 of chromosome 7 (red circle in Figure 1), which did not appear in the son, i.e., was not inherited. If this duplication was maternally inherited, we would not expect the z7j value to decrease in the mixture. As seen in Figure 1, the value of z7j is lower for the 10% mixture compared to the pure maternal sample.
Figure 1.
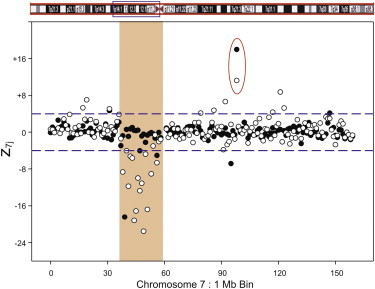
Family 1313 z7j 1 Mb Bin Results for Chr 7
The data show the 0% (solid circles) and 10% (empty circles) mixtures of the affected son’s DNA mixed with the mother’s DNA. The red circle highlights bin 98. The figure shows a 20 Mb deletion on Chr 7 in the DNA mixture, covering the region between 38 Mb and 58 Mb of the chromosome. Additionally, a potential maternal copy number increase, not shared by the son, is seen at 98 Mb.
Mosaic Duplication of Chromosome 15
In Family 2877, the maternal sample has a deletion in chromosome 11 that was inherited by the son. In addition, the son has a duplication in chromosome 15 that was not maternally inherited and is part of a mosaic karyotype in which the majority of cells are normal (Table S1). Figure 2 shows both the chromosome 11 and chromosome 15 zij values for the 1 Mb bins in the mixture with 10% of the son’s DNA. As expected, the inherited deletion in chromosome 11 from 41 Mb to 49 Mb had a consistent set of values that did not change with fetal fraction. However, the chromosome 15 duplication was clearly detected between bins 27 and 66, albeit with more noise than observed in the other artificial samples. The noise results from the reduced apparent ff for this duplication due to the mosaicism. The ffs calculated from the duplication with z15j values were 1.6% and 3.0% for the 5% and 10% mixtures, respectively. In contrast, the ffs calculated from chromosome X with zXj were 5.3% and 10.7%. The method was able to detect both the subchromosomal duplication with the low mosaic ff and to distinguish that the duplication was due to mosaicism by comparison of the ff result to an independent measurement of chromosome X.
Figure 2.
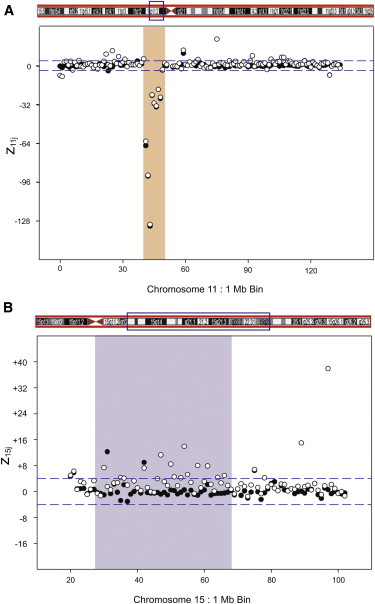
Family 2877 zij 1 Mb Bin Results
The data show results for Chr 11 (A) and Chr 15 (B) with 0% (solid circles) and 10% (empty circles) mixture of the affected son’s DNA mixed with the mother’s DNA.
(A) An 8 Mb deletion for Chr 11 in both mother-only and the mixed DNAs, covering the region from 41 Mb to 49 Mb of Chr 11; this deletion is shared by mother and son.
(B) The son-specific CNV in Chr 15 from 27 Mb to 66 Mb.
Duplications of Chromosome 22
Family 1925 consisted of a mother and two male twins, one of which had two duplications of different sizes in chromosome 22. 10% mixtures of the affected twin’s DNA and the mother were sequenced. The results indicated a 2 Mb and an 8 Mb duplication at bins 17 and 43, respectively. The ff for 10% mixture was calculated to be 11.2% from the 2 Mb duplication, 11.6% from the 8 Mb duplication, and 9.8% from chromosome X (Figure S2).
Maternal Plasma Samples
Whole-Chromosome Aneuploidy
Sample AL1893 was previously reported in the MELISSA study as detected for trisomy 20.14 The 1 Mb bin deep sequencing results for this sample contain ∼960 million tags across the genome. The extra copy of chromosome 20 was clearly detected and the ff calculated from the 1 Mb bin data is 4.4%.
Duplications and Deletions
Sample BE3096 (Table 1) had a complex fetal karyotype that involved the long arm of chromosome 6 (6q) and two duplications, one of which was ∼38 Mb in size. The second duplication was reported as approximately 650 kb from the chromosome microarray analysis of cultured villi. By using MPS we previously reported that this sample showed an increased whole-chromosome normalized chromosome value (NCV) in chromosome 6 (NCV = 3.6).14 This value was insufficient to classify this sample as having a full-chromosome aneuploidy, but it was consistent with the presence of a large duplication. Figure 3 shows the 1 Mb bin results for this sample. All the chromosomes other than chromosome 6 showed z values that clustered around 0. By focusing only on chromosome 6 (Figure 3), the exact region of the 38 Mb duplication was identified. This 38 Mb corresponded to the large duplication seen in the microarray karyotype, and the ff calculated from this duplication was 11.9%. The second duplication in the microarray karyotype would not have been detected by our three-step criteria. Improved analytic methodology and/or deeper sequencing should allow this duplication to be detected. Finally, a 300 kb gain in chromosome 7 at 7q22.1 was also identified by MPS in agreement with the microarray results (Table 2).
Figure 3.
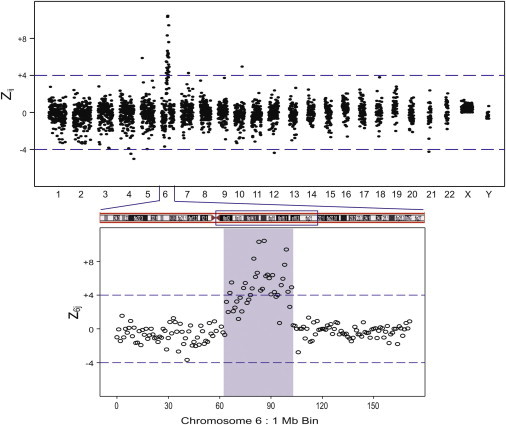
Maternal Plasma Sample BE3096 zij 1 Mb Bin Results with a Fetal Karyotype with a Duplication in Chromosome 6
Expanded regions show z6j 1 Mb bin results. The figure shows a 38 Mb duplication, covering the region between 64 Mb and 102 Mb of Chr 6.
Table 2.
MPS Results on Maternal Plasma Samples that Are Concordant with the Clinically Reported Karyotype
| Sample ID | Affected Chr | Gain/Loss | Start Bin | End Bin | Size (Mb) | Chromosome Region |
|---|---|---|---|---|---|---|
| BE3096 | 6 | gain | 64 | 102 | 38 | 6q12–6q16.3 |
| 7 | gain | 98.1 | 98.3 | 0.3 | 7q22.1 | |
| BF3404 | 7 | loss | 150.3 | 150.6 | 0.3 | 7q36.1 |
| AK1604 | 8 | loss | 6 | 12 | 6 | 8p23.2–8p23.2 |
| BE3236 | 15 | loss | 22 | 39 | 17 | 15q11.2–15q14 |
| AF1019 | 17 | gain | 62 | 81 | 19 | 17q23.3–17q25.3 |
| 10 | loss | 134 | 135 | 2 | 10q26.3 | |
| BC2659 | 3 | gain | 158 | 198 | 40 | 3q25.32–3q29 |
| X | loss | 1 | 10 | 9 | Xp22.33–Xp22.31 |
Sample BF3404 came from a pregnant woman carrying a fetus with a 7q36.1 deletion detected by metaphase karyotype analysis of chorionic villi. Figure 4A shows the 1 Mb bin results for this sample. Only chromosomes 7 and 8 showed 1 Mb bins with z values that met our criteria for classification. Chromosome 7 showed a single 1 Mb bin with a significant decrease in the z value at 7q36.1 (denoted by blue circle in Figure 4A). An examination of the data at higher resolution (100 kb bins) (Figure 4B) showed a deletion of approximately 300 kb, which was consistent with the karyotype report (Table 2). In this sample we also observed an approximately 1 Mb deletion in both the 1 Mb and 100 kb bin data close to the centromere of chromosome 8 (Table 3 and denoted by the red circle in Figure 4). The chromosome 8 deletion was not reported in the karyotype obtained from chorionic villi (Table 1). The ffs calculated from the chromosome 7 and 8 deletions were 18.4% and 68.5%, respectively. The ff calculated from chromosome X was 2.8%. In this case, the high ff value for chromosome 8 indicated that this deletion, which was not reported in the fetal metaphase karyotype, was maternal in origin. In addition, the discordant value of the chromosome 7 compared to chromosome X ff values suggests that part of the signal could be due to the mother. The karyotype report indicated that the chromosome 7 “abnormality is most likely a derivative from a carrier parent,” which is consistent with the MPS data.
Figure 4.
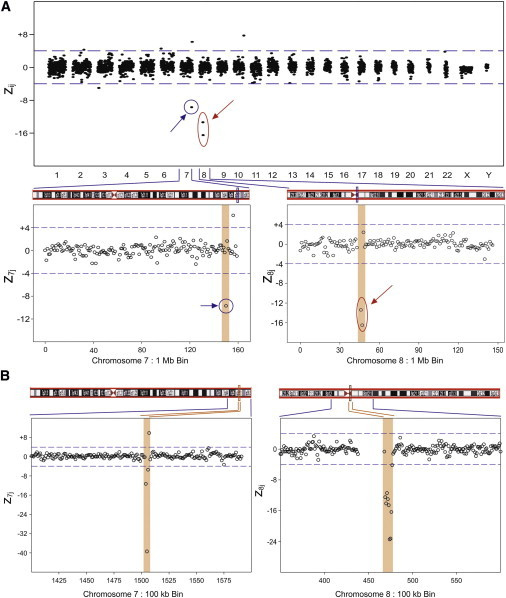
Maternal Plasma Sample BF3404 zij 1 Mb Bin Results across the Genome with a Fetal Karyotype with a Deletion in Chromosome 7
This clinical sample has a karyotype with a small deletion in chromosome 7 (blue circle). Another small deletion is detected in chromosome 8 (red circle). Expanded regions show z7j and z8j 1 Mb and 100 kb bin data. The figure shows a 1 Mb deletion at bin number 150 Mb on Chr 7 (A). At higher resolution (B), this deletion is found to be 300 kb long, in the region from 150.3 Mb to 150.6 Mb of Chr 7. Note: the putative copy-number gain seen in Chr 7 at bin number 156 in the 1 Mb data is not seen in the same region in the 100 kb data. The figure also shows a 2 Mb deletion on Chr 8 (A) covering bins 46 Mb and 47 Mb. At higher resolution (B), this resolves into a 900 kb deletion, covering the region from 46.9 Mb to 47.7 Mb of Chr 8.
Table 3.
Copy-Number Variants Detected by MPS that Were Not Reported in the Clinical Karyotypes
| Sample ID | Affected Chr | Gain or Loss | Start Bin | End Bin | Size (Mb) | Chromosome Region |
|---|---|---|---|---|---|---|
| AL1893 | 2 | gain | 87.3 | 87.9 | 0.6 | 2p11.2 |
| 2 | loss | 89.8 | 90.2 | 0.5 | 2p11.2 | |
| BF3404 | 8 | loss | 46.9 | 47.7 | 0.9 | 8q11.1 |
| AF1019 | 7 | gain | 158.7 | 158.9 | 0.3 | 7q36.3 |
| BC2659 | 3 | loss | 114 | 114.5 | 0.6 | 3q13.31 |
| 11 | loss | 55.3 | 55.4 | 0.2 | 11q11 | |
| 17 | gain | 81 | 81.1 | 0.2 | 17q25.3 | |
| AL1873 | 1 | loss | 12.8 | 13 | 0.3 | 1p36.21 |
| BE3129 | 7 | loss | 39.3 | 40 | 0.8 | 7p14.1 |
| 14 | loss | 58 | 58.1 | 0.2 | 14q23.1 | |
| BG3701 | 9 | gain | 40.7 | 41 | 0.4 | 9p31.1 |
| AH1200 | 6 | loss | 151.4 | 151.5 | 0.2 | 6q25.1 |
| 22 | gain | 25.6 | 25.9 | 0.4 | 22q11.23 |
Sample AK1604 had a partial deletion of the short arm of chromosome 8. The 1 Mb bin results (Figure S3) indicated an approximately 6 Mb deletion in the p arm of chromosome 8 in agreement with the karyotype (Table 2). The fetal fraction calculated from this chromosome deletion was 8.4%.
Translocations
The fetal metaphase karyotype for sample BE3236 showed an unbalanced translocation consisting of 45,XX,−15,der(21)t(15;21)(q15;p11.2). The 1 Mb bin results for this sample are shown in Figure 5. There was a clear 17 Mb deletion in chromosome 15 in agreement with the karyotype (Table 2). The ff calculated from the chromosome 15 deletion was 11.3%. No subchromosomal abnormalities were detected in the chromosome 21 data to indicate the translocation breakpoint.
Figure 5.
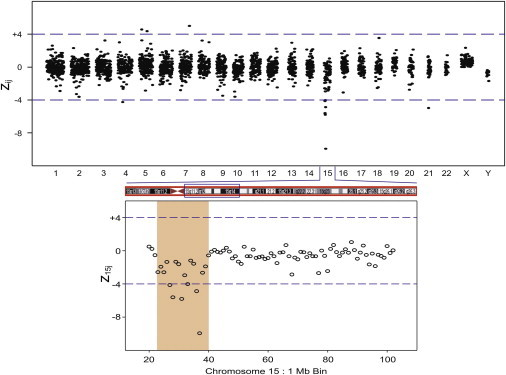
Maternal Plasma Sample BE3236 zij 1 Mb Bin Results across the Genome for Clinical Sample with a Fetal Karyotype with an Unbalanced Translocation Involving Chromosome 15
Expanded region shows z15j 1 Mb bin data. The figure shows a 17 Mb deletion in the region between 22 Mb and 39 Mb of Chr 15.
Identification of Additional Material Not Identified by Karyotype
Two maternal samples had fetal karyotypes with added material of unknown origin at specific chromosomes. The 1 Mb bin results for sample AF1019 are shown in Figure 6. From the MPS data, the additional material of unknown origin on the long arm of chromosome 10 appeared to be derived from an approximately 19 Mb duplication at the q terminus of chromosome 17. There was also an approximately 2 Mb deletion at the q terminus of chromosome 10 that was confirmed by the 100 kb bin data. The ffs calculated from the chromosome 17 duplication and chromosome X (male fetus) were 12.5% and 9.4%, respectively. The 2 Mb deletion on chromosome 10 had a calculated ff of 19.4%. Finally, the MPS results for this sample indicated a small (300 kb) duplication in chromosome 7 that was not reported in the metaphase karyotype (Table 3).
Figure 6.
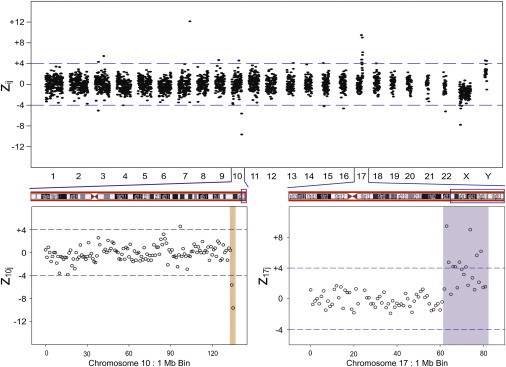
Maternal Plasma Sample AF1019 zij 1 Mb Bin Results across the Genome with Additional Unspecified Material in the Fetal Karyotype
Expanded regions show z10j and z17j 1 Mb bin data. The figures show a 19 Mb duplication of the region from 62 Mb to 81 Mb on Chr 17 and a 2 Mb deletion on Chr 10 from 134 Mb to 135 Mb.
The 1 Mb bin results for sample BC2659 are shown in Figure S4. The karyotype for this sample indicated additional chromosomal material on the short arm of one of the X chromosomes. The additional material of unknown origin appeared to originate from a 40 Mb duplication at the q terminus of chromosome 3. There was also an approximately 9 Mb deletion on the p arm of chromosome X (Figure S4) although the signal from this deletion did not meet our criteria for classifying it as a CNV. The ffs calculated from the chromosome 3 duplication and chromosome X deletion were 9.5% and 6.7%, respectively. The MPS results for this sample also indicated three small subchromosomal changes that were not reported in the metaphase karyotype (Table 3).
Mosaic Karyotypes
Four of the samples listed in Table 1 (AL1873, BE3129, BG3701, AH1200) had mosaic karyotypes with subchromosomal abnormalities. Unfortunately, for three of the samples (AL1873, BG3701, AH1200) the putative subchromosomal abnormality originates in regions of the genome for which information is either unavailable in the genome build or highly repetitive and not accessible for analysis. Thus, we were unable to determine the subchromosomal abnormalities reported in these three samples. The zij values for these samples were all close to and centered around zero. Sample BE3129 had a mosaic karyotype with isochromosome 20q, an abnormality that is reported to be associated with an event secondary to postzygotic error.18 Because cfDNA primarily originates from placental cytotrophoblasts, it is not expected that this abnormality would be detected with MPS. There were 1–2 small subchromosomal changes detected in these samples by MPS that were not reported in the karyotypes (Table 3).
Discussion
This study demonstrates that in nonmosaic cases, it is possible to obtain a fetal molecular karyotype that is equivalent to CMA by MPS of maternal plasma cfDNA. In some cases the MPS results provided better resolution than a metaphase karyotype obtained from chorionic villi or amniocytes. Such a noninvasive test could have clinical utility in the near future, particularly for women who either have a medical contraindication or lack of access to an invasive procedure.
With 25-mer tags at ∼109 tags/sample, the results shown here indicate that sufficient precision can be obtained between sequencing runs to reliably achieve 100 kb resolution across the genome. Even greater resolution can be achieved with deeper sequencing. The improvements in the Illumina v3 sequencing chemistry allowed for the use of 25-mer tags, compared to the 36-mers used in our previous work.14 These short tags mapped with high efficiency across the genome, and the quantitative behavior demonstrated with the artificial mixture analyses validates the methodology. Today, the current cost of sequencing a human genome with 30–60× coverage via paired-end sequencing of 75–100 bp from each end is ∼$5,000. However, with the single-ended 25-mer reads utilized here, one billion tags can be obtained for less than $1,000 per sample. This is comparable to the cost of a CMA but employs a risk-free blood draw rather than an invasive procedure. Deeper sequencing would allow for even finer resolution at an additional cost. Thus, this type of analysis could be implemented as a reflex test when other clinical factors are present (such as sonographically detected anomalies that are not typical of whole-chromosome aneuploidy) and the pregnant woman declines an invasive procedure or prefers a blood test.
The lack of results on the mosaic samples (except for the artificial mixture) highlights the current limitations of both the microarray and MPS approaches. Subchromosomal abnormalities that originate in regions of the genome for which information is either unavailable in the genome build or highly repetitive will not be accessible for analysis. Such inaccessible genome regions are typically focused in the telomeres and centromeres of different chromosomes and in the short arms of acrocentric chromosomes. Also, balanced translocations are a challenge for both the CMA and MPS methods although MPS may be able to detect translocation junctions with deeper sequencing. Finally, the mosaic portion of a sample may be more challenging for detection because of its lower effective ff. This may require even deeper sequencing for effective classification.
Metaphase cytogenetic analysis from cell cultures, although considered “standard,” has some limitations that need to be considered. For example, the ability to detect subchromosomal abnormalities is typically limited to sizes of 5 Mb or greater. This constraint is what led to the recent recommendation of using CMAs as a first tier test in clinical practice. Cell culture is biased toward the detection of more stable chromosomal configurations over significant structural alterations. In the case of fluorescence in situ hybridization (FISH), only the regions of the genome that are addressed by design of the FISH probes can be analyzed. Finally, as shown here, in actual clinical practice metaphase karyotypes can be reported to contain “chromosomal material of unknown origin.” The MPS methodology of measuring copy-number variation introduced in this work overcomes these limitations of karyotyping.
Importantly, our results showed that MPS was able to identify the potential source of the material of unknown origin for clinical samples AF1019 and BC2659. In addition, the MPS data showed small deletions in the termini of the chromosomes that the metaphase karyotype indicated were the breakpoints for the unknown chromosomal material in each of these samples. Such deletions at the breakpoints of translocations have been reported repeatedly in the literature.19 Based on these results, MPS may have the capabilities to both identify the presence of a subchromosomal duplication and suggest a translocation position based on small deletions (or duplications) elsewhere in the genome. More data on translocations will need to be collected to further validate translocation classifications.
Two recent papers have utilized even deeper sequencing than used here to identify fetal single-nucleotide polymorphisms and haplotypes from maternal plasma samples.20,21 Although this work suggests an exciting future path toward routine noninvasive detection of the entire fetal genome, most clinicians are not yet ready to interpret the massive amounts of information that will come from the entire sequence. They are, however, already familiar with CMAs, so our work can potentially be translated to clinical care more expeditiously and eventually provide a rationale for whole-genome sequencing of the fetus.
The methodologies developed here also have applications beyond the determination of fetal subchromosomal abnormalities from cfDNA in maternal plasma. Ultimately, MPS can be applied to any mixed biological sample in which one wishes to determine the subchromosomal abnormalities in the minor component, even when the minor component represents only a few percent of the total DNA in the specimen. In prenatal diagnostics, samples obtained from chorionic villi could be analyzed for mosaic karyotypes or maternal contamination. Outside of prenatal diagnosis, many different cancers have been associated with copy-number changes that could potentially be detected from cfDNA in the blood of an individual or a solid tumor sample that contains both normal and cancer cells. As the cost of MPS continues to drop, we expect that its application for detecting subchromosomal abnormalities in mixed samples will find broad clinical utility.
In summary, determination of fetal subchromosome abnormalities via deep sequencing of maternal plasma allows for a molecular karyotype of the fetus to be determined noninvasively. Such a test could be available in the near future at a cost comparable to an invasive procedure but without the associated procedural risks.
Acknowledgments
The authors wish to acknowledge the help of the coinvestigators in the MELISSA trial in acquiring samples. A.S., H.H., A.J.S., and R.P.R. are employees of Verinata Health, Inc. D.W.B. is the chair of the Clinical Advisory Board of Verinata Health, and for this position she receives an honorarium and equity options. She also has a sponsored research grant from Verinata Health that is administered through Tufts Medical Center.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
UCSC Genome Bioinformatics, Feb. 2009 assembly of the human genome, http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Bianchi D.W. From prenatal genomic diagnosis to fetal personalized medicine: progress and challenges. Nat. Med. 2012;18:1041–1051. doi: 10.1038/nm.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller D.T., Adam M.P., Aradhya S., Biesecker L.G., Brothman A.R., Carter N.P., Church D.M., Crolla J.A., Eichler E.E., Epstein C.J. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vetro A., Bouman K., Hastings R., McMullan D.J., Vermeesch J.R., Miller K., Sikkema-Raddatz B., Ledbetter D.H., Zuffardi O., van Ravenswaaij-Arts C.M. The introduction of arrays in prenatal diagnosis: a special challenge. Hum. Mutat. 2012;33:923–929. doi: 10.1002/humu.22050. [DOI] [PubMed] [Google Scholar]
- 4.Novelli A., Grati F.R., Ballarati L., Bernardini L., Bizzoco D., Camurri L., Casalone R., Cardarelli L., Cavalli P., Ciccone R. Microarray application in prenatal diagnosis: a position statement from the cytogenetics working group of the Italian Society of Human Genetics (SIGU), November 2011. Ultrasound Obstet. Gynecol. 2012;39:384–388. doi: 10.1002/uog.11092. [DOI] [PubMed] [Google Scholar]
- 5.Wapner R.J., Martin C.L., Levy B., Ballif B.C., Eng C.M., Zachary J.M., Savage M., Platt L.D., Saltzman D., Grobman W.A. Chromosomal microarray versus karyotyping for prenatal diagnosis. N. Engl. J. Med. 2012;367:2175–2184. doi: 10.1056/NEJMoa1203382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan H.C., Quake S.R. Detection of aneuploidy with digital polymerase chain reaction. Anal. Chem. 2007;79:7576–7579. doi: 10.1021/ac0709394. [DOI] [PubMed] [Google Scholar]
- 7.Lo Y.M., Lun F.M., Chan K.C., Tsui N.B., Chong K.C., Lau T.K., Leung T.Y., Zee B.C., Cantor C.R., Chiu R.W. Digital PCR for the molecular detection of fetal chromosomal aneuploidy. Proc. Natl. Acad. Sci. USA. 2007;104:13116–13121. doi: 10.1073/pnas.0705765104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan H.C., Blumenfeld Y.J., Chitkara U., Hudgins L., Quake S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA. 2008;105:16266–16271. doi: 10.1073/pnas.0808319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu R.W., Chan K.C., Gao Y., Lau V.Y., Zheng W., Leung T.Y., Foo C.H., Xie B., Tsui N.B., Lun F.M. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc. Natl. Acad. Sci. USA. 2008;105:20458–20463. doi: 10.1073/pnas.0810641105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.American College of Obstetricians and Gynecologists Committee opinion no. 545: Noninvasive prenatal testing for fetal aneuploidy. Obstet. Gynecol. 2012;120:1532–1534. doi: 10.1097/01.AOG.0000423819.85283.f4. [DOI] [PubMed] [Google Scholar]
- 11.Peters D., Chu T., Yatsenko S.A., Hendrix N., Hogge W.A., Surti U., Bunce K., Dunkel M., Shaw P., Rajkovic A. Noninvasive prenatal diagnosis of a fetal microdeletion syndrome. N. Engl. J. Med. 2011;365:1847–1848. doi: 10.1056/NEJMc1106975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jensen T.J., Dzakula Z., Deciu C., van den Boom D., Ehrich M. Detection of microdeletion 22q11.2 in a fetus by next-generation sequencing of maternal plasma. Clin. Chem. 2012;58:1148–1151. doi: 10.1373/clinchem.2011.180794. [DOI] [PubMed] [Google Scholar]
- 13.Sehnert A.J., Rhees B., Comstock D., de Feo E., Heilek G., Burke J., Rava R.P. Optimal detection of fetal chromosomal abnormalities by massively parallel DNA sequencing of cell-free fetal DNA from maternal blood. Clin. Chem. 2011;57:1042–1049. doi: 10.1373/clinchem.2011.165910. [DOI] [PubMed] [Google Scholar]
- 14.Bianchi D.W., Platt L.D., Goldberg J.D., Abuhamad A.Z., Sehnert A.J., Rava R.P., MatErnal BLood IS Source to Accurately diagnose fetal aneuploidy (MELISSA) Study Group Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet. Gynecol. 2012;119:890–901. doi: 10.1097/AOG.0b013e31824fb482. [DOI] [PubMed] [Google Scholar]
- 15.Lo Y.M., Tein M.S., Lau T.K., Haines C.J., Leung T.N., Poon P.M., Wainscoat J.S., Johnson P.J., Chang A.M., Hjelm N.M. Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am. J. Hum. Genet. 1998;62:768–775. doi: 10.1086/301800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan H.C., Blumenfeld Y.J., Chitkara U., Hudgins L., Quake S.R. Analysis of the size distributions of fetal and maternal cell-free DNA by paired-end sequencing. Clin. Chem. 2010;56:1279–1286. doi: 10.1373/clinchem.2010.144188. [DOI] [PubMed] [Google Scholar]
- 17.Lo Y.M., Chan K.C., Sun H., Chen E.Z., Jiang P., Lun F.M., Zheng Y.W., Leung T.Y., Lau T.K., Cantor C.R., Chiu R.W. Maternal plasma sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Trans. Med. 2010;2 doi: 10.1126/scitranslmed.3001720. 61ra91. [DOI] [PubMed] [Google Scholar]
- 18.Chen C.-P. Detection of mosaic isochromosome 20q in amniotic fluid in a pregnancy with fetal arthrogryposis multiplex congenita and normal karyotype in fetal blood and postnatal samples of placenta, skin, and liver. Prenat. Diagn. 2003;23:85–87. doi: 10.1002/pd.524. [DOI] [PubMed] [Google Scholar]
- 19.Howarth K.D., Pole J.C.M., Beavis J.C., Batty E.M., Newman S., Bignell G.R., Edwards P.A.W. Large duplications at reciprocal translocation breakpoints that might be the counterpart of large deletions and could arise from stalled replication bubbles. Genome Res. 2011;21:525–534. doi: 10.1101/gr.114116.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan H.C., Gu W., Wang J., Blumenfeld Y.J., El-Sayed Y.Y., Quake S.R. Non-invasive prenatal measurement of the fetal genome. Nature. 2012;487:320–324. doi: 10.1038/nature11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitzman J.O., Snyder M.W., Venture M., Lewis A.P., Qiu R., Simmons L.E., Gammill H.S., Rubens C.E., Santillan D.A., Murray J.C. Noninvasive whole-genome sequencing of a human fetus. Sci. Trans. Med. 2012;4 doi: 10.1126/scitranslmed.3004323. 137ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.