

Abstract
Spastic paraplegia 46 refers to a locus mapped to chromosome 9 that accounts for a complicated autosomal-recessive form of hereditary spastic paraplegia (HSP). With next-generation sequencing in three independent families, we identified four different mutations in GBA2 (three truncating variants and one missense variant), which were found to cosegregate with the disease and were absent in controls. GBA2 encodes a microsomal nonlysosomal glucosylceramidase that catalyzes the conversion of glucosylceramide to free glucose and ceramide and the hydrolysis of bile acid 3-O-glucosides. The missense variant was also found at the homozygous state in a simplex subject in whom no residual glucocerebrosidase activity of GBA2 could be evidenced in blood cells, opening the way to a possible measurement of this enzyme activity in clinical practice. The overall phenotype was a complex HSP with mental impairment, cataract, and hypogonadism in males associated with various degrees of corpus callosum and cerebellar atrophy on brain imaging. Antisense morpholino oligonucleotides targeting the zebrafish GBA2 orthologous gene led to abnormal motor behavior and axonal shortening/branching of motoneurons that were rescued by the human wild-type mRNA but not by applying the same mRNA containing the missense mutation. This study highlights the role of ceramide metabolism in HSP pathology.
Main Text
Hereditary spastic paraplegias (HSPs) are heterogeneous inherited neurodegenerative disorders. Affected individuals suffer from pyramidal motor neuron dysfunction such as spasticity, brisk reflexes, and pyramidal weakness of the lower limbs primarily caused by dysfunction or degeneration of upper motor neurons.1,2 These clinical symptoms are sometimes associated with additional neurological or extraneurological signs and structural abnormalities of the central nervous system (CNS) observed on brain magnetic resonance imaging (MRI), such as thin corpus callosum (TCC), cortical atrophy, and cerebellar atrophy. This clinical heterogeneity partially reflects the large genetic heterogeneity of this group of disorders with ∼50 loci mapped to date and accounting for all modes of inheritance.3
We mapped SPG46 to chromosome 9 (MIM 614409) in a consanguineous Tunisian family (TUN35) (Figure 1A) with autosomal-recessive (AR) HSP, mental impairment, cataract, and TCC.4 We excluded the involvement of large genomic rearrangements in affected individual V.33 by comparative genomic hybridization (NimbleGen chromosome 9-specific 385K array, data not shown). We then sequenced all coding exons of the SPG46 candidate interval (n = 1,727 exons and their flanking splice sites in 183 genes, including 1 kb of their UTRs) by targeted next-generation sequencing (custom NimbleGen capture and sequencing in a Roche 454-FLX) in affected subjects V.32 and V.33 (Table S1 available online). After exclusion of variants reported as validated polymorphisms in public (1000 Genomes, dbSNP) and local databases, a total of 52 homozygous variants remained common to both individuals, including 48 synonymous or intronic changes or variants in pseudogenes. In the absence of nonsense or intronic/exonic splice-site mutations, we focused our analysis on the four missense variants. Three of them cosegregated with the disease, were predicted to be damaging by various online software packages (PolyPhen-2, Mutation Tasting), and affected conserved amino acids. Two were excluded as the cause of the disease either because of the frequency in controls (c.88C>T [p.Arg30Trp], rs75679360, in FAM166B [RefSeq accession number NM_001164310.1], found in 8/182 chromosomes of North African controls, including at the homozygous state in one subject) or because the corresponding gene had previously been involved, when mutated, in another autosomal-recessive disease not overlapping with HSP (c.2909T>C [p.Val970Ala] in NPR2 [MIM 108961, RefSeq NM_003995.3]). Indeed, we did not detect segregating variants in NPR2 in 68 AR-HSP index subjects and 44 simplex individuals subjected to exome sequencing. The remaining variant, c.1888C>T (p.Arg630Trp), found in exon 12 of GBA2 (MIM 609471, RefSeq NM_020944.2), segregated with the disease in the family (Figures 1A and S1) and was detected neither in 1,038 control chromosomes of 519 French, West Indian, Brazilian, and North African healthy subjects nor among 6,500 exomes on the Exome Variant Server. The variant affected an amino acid conserved from Caenorhabditis elegans and located in the six-hairpin-glucosidase-like domain of the enzyme (Figure 1E).
Figure 1.
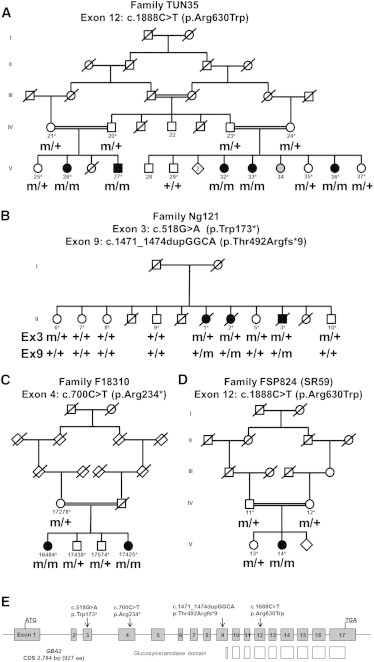
Mutations in GBA2
(A–D) Family trees and segregation analysis of the mutations identified in individuals of families TUN35 (A), Ng121 (B), F18310 (C), and FSP824 (D). The following symbols are used: squares for males, circles for females; filled symbols for affected individuals; gray symbols for subjects not clinically assessed; double line for consanguinity; m, mutation; +, wild-type; ∗, sampled individuals. Sequencing chromatograms of mutations c.518G>A (p.Trp173∗), c.700C>T (p.Arg234∗), c.1888C>T (p.Arg630Trp), and c.1471_1474dupGGCA (p.Thr492Argfs∗9) are in Figure S1.
(E) Graphic representation of the exon organization of GBA2 on chromosome 9 (gray boxes) and the location of the mutations (black arrows). The GBA2 transcript is 3,611 bp long (CDS 2,784 bp) and is composed of 17 exons that encode a 927 amino acid protein. White boxes show the exons encoding the glucosylceramidase domain.
In parallel, exome capture via the SureSelect Human All Exon 50Mb kit (Agilent) followed by sequencing on a Hiseq 2000 sequencer (Illumina) was performed on the genomic DNA of two index HSP individuals of Belgian and Turkish ancestry, Ng121-II.2 and F18310-V.16484, respectively (Figures 1B and 1C). The Burrows-Wheeler algorithm5 was used to align 100 bp length paired-end reads to the hg19 version of the human genome (Ensembl); variants were called with the Genome Analysis Toolkit (GATK) software package.6,7 Data were then imported into the Genomes Management Application (GEM-app) database and its associated toolset for further analysis. On average, 110.4 million reads were produced per sample, 95% of which could be aligned to the targeted sequence. Mean coverage of the targeted sequence was 97-fold. On average, 87,591 single-nucleotide variations (SNV) and 8,876 indels were called per sample. Variants were filtered for occurrence in the normal population (minor allele frequency < 1% in dbSNP135 and at the Exome Variant Server), conservation (Genomic Evolutionary Rate Profiling [GERP] score > 3.5 or PhastCons score > 0.7), and quality (GATK QUAL score > 100, GATK GQ score > 75). Variants occurring and segregating in more than two unrelated families in GEM-app were removed. Compound heterozygous variants in eight genes, in the absence of homozygous variants, were identified in the index subject of family Ng121. In family F18310, 23 homozygous changes and compound heterozygous variants in three genes were identified in the sequenced subject. Among all variants detected in both families, three were truncating mutations and cosegregated with the disease (Figures 1B, 1C, and S1), all located in GBA2: c.700C>T (p.Arg234∗) at the homozygous state in exon 4 of individual F18310-V.16484, and the heterozygous variants c.518G>A (p.Trp173∗) and c.1471_1474dupGGCA (p.Thr492Argfs∗9) in exons 3 and 9 of subject Ng121-II.2.
Direct Sanger sequencing (Table S2) of the 17 coding exons of GBA2 in 95 index HSP subjects with an AR compatible inheritance (study approved by the Paris-Necker ethics committee; approval No. 03-12-07 granted to A.B. and A.D.), identified one simplex subject of Portuguese ancestry (FSP824-V.14) harboring the same missense mutation (c.1888C>T [p.Arg630Trp]) as in family TUN35, although with a different associated haplotype (Figures 1D and S1). The mutation, which was homozygous, was detected at the heterozygous state in her parents.
The GBA2 protein encoded by GBA2 is an enzyme of sphingolipid metabolism that is the source of a variety of mediators of cell signaling responses and of structural components of the plasma membrane involved in its dynamics.8 GBA2 was initially identified as a microsomal bile acid β-glucosidase or β-glucosidase 2,9,10 but it is also a nonlysosomal glucosylceramidase, a ubiquitous enzyme that catalyzes the conversion of glucosylceramide to free glucose and ceramide as well as the reverse reaction consisting in the transfer of glucose to different lipid substrates.11,12 The primary catabolic pathway for glucosylceramide involves the lysosomal enzyme glucocerebrosidase GBA1 (MIM 606463), which is defective in persons with Gaucher disease, the most common inherited lysosomal storage disorder.13 GBA1 is located in the lysosomal compartment and ceramides generated by this enzyme are degraded into sphingosine and fatty acids.11,14 In contrast, GBA2 has been localized in the endoplasmic reticulum (ER)12 and at the cell surface,11 and ceramides formed by this enzyme are rapidly converted into sphingomyelin.11,12,15 With a published protocol,16,17 we assessed the GBA2 glucosylceramidase enzymatic activity in lymphoblasts and leukocytes of affected persons harboring the missense c.1888C>T (p.Arg630Trp) mutation in presence of specific inhibitors of the GBA1 enzyme, which is responsible for the vast majority of the glucosylceramidase activity.18 No detectable GBA2 activity could be observed in the cells of subject FSP824-V.14 compared to healthy controls (p = 0.001, Figure S2), indicating a complete loss of function by the mutation. Complete loss of function was also evidenced in nontransformed leukocytes of individuals TUN35-V.27 and V.33 (data not shown).
The neurological phenotype of the 11 affected individuals was similar (Table S3). Onset occurred in infancy or childhood (range 1–16 years) with disturbances that progressed slowly to the need for a cane at 22–32 years and the need for a wheelchair at the age of 54–60. During the course of the disease, cerebellar ataxia and cataract were observed in all individuals. All affected subjects also had mild to moderate mental impairment; two explored during their 50s showed an IQ level equivalent to that of 6-year-old. In the oldest affected subjects, disease course was complicated by a memory deficit in their 60s and they died in a demented state aged 61, 63, and 72 years. The initial phenotypic description4 was enriched by other frequently observed signs, such as hearing loss (n = 3) and axonal neuropathy evidenced at electroneuromyography (ENMG, n = 5) and sural nerve biopsy (n = 3). Brain MRI showed cerebral, cerebellar, and corpus callosum atrophy in all seven subjects tested, which seemed to worsen with disease duration (Figure 2). Interestingly, the two affected men presented a bilateral testicular hypotrophy in the absence of hormonal dysfunctions. Semen analysis of subject TUN35-V.27 revealed extremely severe spermatozoid head abnormalities with necrospermia and severe reduction in velocity (Table S4). Hormonal and genital exploration of affected woman FSP824-V.14 did not reveal abnormalities but woman Ng121-II.2 was reported to have few pubic hairs. The infertility highlighted in SPG46 males is reminiscent of the consequences of the pharmacological inhibition of glucosylceramidases in mice19,20 or observed in the Gba2 knockout (KO) mice in which an abnormal glucosylceramide accumulation in Sertoli cells is observed.12 This infertility together with the neurological symptoms in our SPG46 subjects points to the critical role of this enzyme in multiple tissues, including testis and brain. Indeed, even if no overt neurological signs, liver dysfunction, or reduced viability were observed in the Gba2 KO mice at 4 months of age, an accumulation of glycolipid species in the brain, liver, and testis was observed by mass spectrometry.12 Longer times may be required in mouse as shown in other HSP mouse models in which signs are very subtle during the first months of life.21,22 The neurological phenotype may also be obscured by the different structure of corticospinal tracts in mice or by compensation by other GBA enzymes early during development. Such compensation in GBA1 mutated individuals by GBA2 activity has been shown and may explain the absence of notable glucosylceramide accumulation in some Gaucher disease subjects.17 The reverse situation was not true because, in our study, no difference in GBA1 activity could be observed between lymphoblasts of the Portuguese subject and five controls (data not shown). This compensation may, however, differ between tissues and between different species.20
Figure 2.
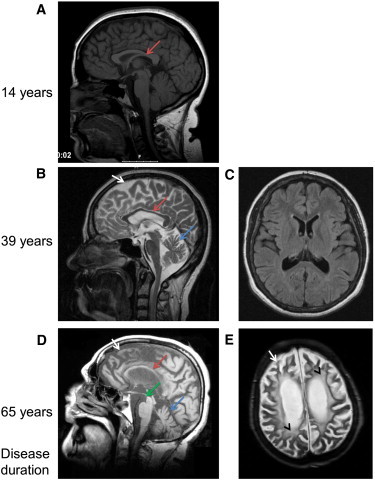
Brain Imaging in SPG46-Affected Individuals
(A) Sagittal brain magnetic resonance imaging (MRI) in subject FSP824-V.14 after 14 years of disease duration, showing a very slight thinning of the corpus callosum (red arrow).
(B and C) Sagittal and transversal flair MRI of individual F18310-V.16484 after 39 years of disease duration. The atrophy of the corpus callosum (B, red arrow) and of the cerebellar (B, blue arrow) and cerebral (B, white arrow) cortex are evident in the absence of relevant white matter disease (C).
(D and E) Sagittal T1- and transversal axial T2-weighted MRI images of subject Ng121-II.2 after 65 years of disease duration, showing severe atrophy of her cerebellum (D, blue arrow) and cerebrum (white arrows) with mesencephalon atrophy (D, hummingbird/colibri sign, green arrow) and pronounced white matter lesions (E, black arrowhead).
To validate the functional phenotype of the GBA2 mutations in vivo, and in particular in the CNS, we used the zebrafish model and antisense morpholino oligonucleotide (AMO) technology to knock down the unique GBA2 ortholog (zGba2; RefSeq NM_ENSDARG00000061472) by using classical methodologies.23 To inactivate zGba2, we used an AMO targeting the initiation codon of the mRNA (zGba2aug). As a control for our results, we designed a mismatch zGba2aug AMO, mmzGba2aug, comprising five mismatched bases, whose injection did not significantly lead to the appearance of phenotypes in injected embryos compared to uninjected ones (p > 0.05). We determined that microinjection of 0.1 pmol of zGba2aug did not cause significant lethality of injected embryos compared to uninjected (p > 0.05) or mmzGba2aug-injected (p > 0.05) embryos. This AMO dose induced a curly tail phenotype in 12.5% of morphants (Figures 3D and 3E) although 73.8% had no visible phenotype (Figure 3C). To further characterize the phenotype of normal-looking embryos (Figure 3C), we assessed their motor behavior by performing a touch-response test on 2 day postfertilization morphants (Figure S3). We clearly observed that a substantial proportion of the normal-looking zGba2aug-injected morphants showed significant motility defects (24%), characterized by slower movements and shorter touch-induced escape distances, when compared to control embryos injected with mmzGba2aug (5.6%, p < 0.001) (Figure 3E). We next investigated the conservation of the GBA2 protein functions between human and zebrafish by coinjecting zGba2aug with the wild-type (WT) human GBA2 mRNA. When double injected, the number of normal-looking morphants with no locomotor impairment increased significantly (68.4% versus 49.8%) compared to zGba2aug-injected larvae (Figure 3E, p < 0.001), suggesting that, in zebrafish, human GBA2 mRNA can compensate, at least partially, for the loss of endogenous zebrafish mRNA. Indeed, the orthologous genes have a 65.8% identity and an 86.5% similarity. In contrast, no significant difference (p > 0.05) was observed after the coinjection of the zGba2aug AMO with the human GBA2 mRNA containing the c.1888C>T missense mutation (Figures 3E and 3S), meaning that the mutated mRNA failed to compensate the zebrafish mRNA decrease. Injection of WT or mutant mRNA alone, without morpholinos, did not lead to a curly-tail or a locomotor phenotype and did not influence lethality in embryos compared to mmzGba2aug-injected control embryos (p > 0.05). Histochemical analyses were then performed on zGba2aug-injected morphant embryos showing a motor phenotype without visible anomalies. The Znp-1 antibody, which labels the synaptic protein synaptotagmin 2 in motor neuron axons,24 showed abnormal development of the spinal motoneurons, observed at mid-distance between head and tail (Figure 3G). We observed and evaluated the morphology of motor neuron axons in embryos injected with zGba2aug, which appeared to be shorter and presented with more terminal branches in contrast to the proper axonal outgrowth observed in embryos injected with the mmzGba2aug control (Table S5, p < 0.00001). This abnormal outgrowth was totally rescued when zGba2aug was coinjected with the WT human mRNA (Figure 3H and Table S5, p = 0.87), supporting the specificity of our model. In contrast, we did not observe any rescue when the coinjection was performed with the mutated human c.1888C>T mRNA (Figure 3I and Table S5, p < 0.00001). These observations suggest that, in SPG46, as in other HSPs,23,25–29 the neuronal tract formation is defective and they confirm the role of GBA2 in the CNS development.
Figure 3.
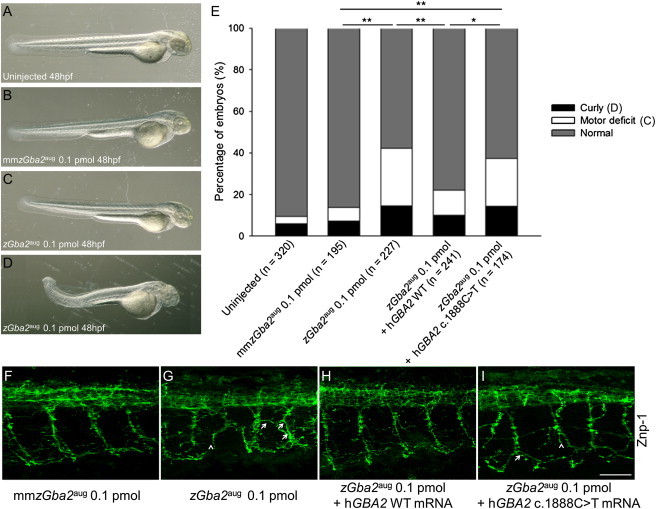
zGba2 Inactivation Induces Morphological Phenotypes and Abnormal Motoneuronal Outgrowth in Zebrafish Larvae
(A–D) Whereas 48 hpf embryos injected with 0.1 pmol of the control AMO mmzGba2aug (B) were phenotypically indistinguishable from noninjected controls (A), injection of 0.1 pmol zGba2aug (D) impaired embryonic development of the caudal region, with some morphants showing shortened and/or twisted tails. Most of the injected morphants were normal looking even when they showed abnormal locomotion (C). Magnification ×44.
(E) Classification of morphant phenotypes observed after injection of AMOs with or without mRNA. The numbers of noninjected controls and different zGba2 morphants are indicated in parentheses. ∗p < 0.002, ∗∗p < 0.001.
(F–I) Noninjected and zGba2 morphants were analyzed for Znp-1 staining in spinal neurons. In contrast to noninjected larvae and to morphants injected with mmzGba2 (F) or zGba2aug + hGBA2 WT mRNA (H), morphants injected with the zGba2aug AMO in presence (I) or absence (G) of the mutated c.1888C>T RNA showed dramatically impaired axonal outgrowth leading to truncated axons (arrowheads). Axon trajectories are disturbed; ectopic and aberrant motor axon branches can be seen (arrows). Scale bar represents 50 μm.
Could treatments in use for Gaucher disease, due to GBA1 mutations, be useful in GBA2 mutated individuals? Enzyme replacement with imiglucerase (Cerezyme, Genzyme; Velaglucerase alpha, Shire; etc.) will not be effective because it does not cross the blood-brain barrier and has been optimized to target lysosomes, which is not the localization of GBA2. Another therapy, called substrate reduction therapy, uses the iminosugar-based inhibitor N-butyldeoxynojirimycin (miglustat or Zarvesta, Actelion) or a structural analog (Gen 529448, Genzyme) that decrease glucosylceramide biosynthesis in vitro and have been reported to increase survival and motor functions of mice models of Niemann Pick type C1 (MIM 257220) and Sandhoff (MIM 268800) diseases.20,30–32 However, these inhibitors also acted on GBA2 enzymatic activity,32 making them unsuitable for SPG46 subjects. Moreover, a pharmacological chaperone strategy, such as isofagomine used in Gaucher disease, may be an interesting therapeutic option in SPG46 subjects due to missense mutations, if adapted to the GBA2 protein, because it was shown to increase GBA1 activity in GBA1 mutant fibroblasts, reduce glucosylceramide and glucosylsphingosine accumulations in mouse models, and improve neurological symptoms in subjects with type III Gaucher disease (MIM 231000).33,34
In conclusion, we have identified mutations in GBA2 leading to a specific phenotype that connects glucosylceramide metabolism and HSP. Lipid metabolism defects have already been implicated in other forms of HSP,35 including SPG35 (MIM 612319), SPG28 (MIM 609340), or SPG49 caused by mutations in FA2H (MIM 611026), DDHD1 (MIM 614603), and CYP2U1 (MIM 610670), respectively.36,37 Interestingly, measurement of specific activities of enzymes involved in HSP could be useful to facilitate diagnosis in clinical practice and to monitor drug response in future therapeutic trials, even when done in the peripheral blood cells as in SPG5A (MIM 270800)38 or in SPG46 subjects in our study. Measurement of GBA1 activity is already in use in Gaucher disease assessment.
Acknowledgments
We are grateful to the family members for their participation. We would like to thank S. Ciura, M. Nawara, J. Gomez, P. Coutinho, I. Alonso, N. Jezequel, P. Touraine, G. Gyapay, V. Meyer, S. Rivaud-Péchoux, S. Forlani, and the DNA and Cell Bank of the Centre de Recherche de l’Institut du Cerveau et de la Moelle épinière for their contribution to this study. We also thank F. Mochel, F. Darios, K.H. El-Hachimi, and R. Schiffmann for their advice and the clinicians and collaborators of the Spastic paraplegia and ataxia (SPATAX) network who referred to us some of the affected subjects. This work was supported by the Association Strumpell-Lorrain (to SPATAX), the Agence Nationale de la Recherche (ANR) (“SPAX” to A.D. and “LIGENAX” and “SPG11” to G.S.), the Association Française contre les Myopathies (“LIGENAX” to G.S.), the European Union and ANR through the Eranet E-Rare program (“EUROSPA” to A.B. and L.S.), the Deutsches Zentrum für Neurodegenerative Erkrankungen (to L.S.), the Interdisziplinären Zentrums für Klinische Forschung University of Tübingen (grant 1970-0-0 to R.S.), and the Verum foundation (to A.B.). This study also benefited from funding from the program “Investissements d’avenir” ANR-10-IAIHU-06 (to the Brain and Spine Institute, Paris).
Contributor Information
Alexis Brice, Email: alexis.brice@upmc.fr.
Giovanni Stevanin, Email: giovanni.stevanin@upmc.fr.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org/index.html
Ensembl Genome Browser, http://www.ensembl.org/index.html
Genomes Management Application (GEM.app) database, https://genomics.med.miami.edu/gem-app/
Mutalyzer, https://mutalyzer.nl/index
Mutation Taster, http://www.mutationtaster.org/
NHLBI Exome Variant Server Exome Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
References
- 1.Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. doi: 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- 2.Schüle R., Schöls L. Genetics of hereditary spastic paraplegias. Semin. Neurol. 2011;31:484–493. doi: 10.1055/s-0031-1299787. [DOI] [PubMed] [Google Scholar]
- 3.Finsterer J., Löscher W., Quasthoff S., Wanschitz J., Auer-Grumbach M., Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 2012;318:1–18. doi: 10.1016/j.jns.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 4.Boukhris A., Feki I., Elleuch N., Miladi M.I., Boland-Augé A., Truchetto J., Mundwiller E., Jezequel N., Zelenika D., Mhiri C. A new locus (SPG46) maps to 9p21.2-q21.12 in a Tunisian family with a complicated autosomal recessive hereditary spastic paraplegia with mental impairment and thin corpus callosum. Neurogenetics. 2010;11:441–448. doi: 10.1007/s10048-010-0249-2. [DOI] [PubMed] [Google Scholar]
- 5.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mencarelli C., Martinez-Martinez P. Ceramide function in the brain: when a slight tilt is enough. Cell. Mol. Life Sci. 2012 doi: 10.1007/s00018-012-1038-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matern H., Gartzen R., Matern S. Beta-glucosidase activity towards a bile acid glucoside in human liver. FEBS Lett. 1992;314:183–186. doi: 10.1016/0014-5793(92)80970-r. [DOI] [PubMed] [Google Scholar]
- 10.Matern H., Heinemann H., Legler G., Matern S. Purification and characterization of a microsomal bile acid beta-glucosidase from human liver. J. Biol. Chem. 1997;272:11261–11267. doi: 10.1074/jbc.272.17.11261. [DOI] [PubMed] [Google Scholar]
- 11.Boot R.G., Verhoek M., Donker-Koopman W., Strijland A., van Marle J., Overkleeft H.S., Wennekes T., Aerts J.M.F.G. Identification of the non-lysosomal glucosylceramidase as beta-glucosidase 2. J. Biol. Chem. 2007;282:1305–1312. doi: 10.1074/jbc.M610544200. [DOI] [PubMed] [Google Scholar]
- 12.Yildiz Y., Matern H., Thompson B., Allegood J.C., Warren R.L., Ramirez D.M.O., Hammer R.E., Hamra F.K., Matern S., Russell D.W. Mutation of beta-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J. Clin. Invest. 2006;116:2985–2994. doi: 10.1172/JCI29224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsuji S., Choudary P.V., Martin B.M., Stubblefield B.K., Mayor J.A., Barranger J.A., Ginns E.I. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher’s disease. N. Engl. J. Med. 1987;316:570–575. doi: 10.1056/NEJM198703053161002. [DOI] [PubMed] [Google Scholar]
- 14.Kolter T., Winau F., Schaible U.E., Leippe M., Sandhoff K. Lipid-binding proteins in membrane digestion, antigen presentation, and antimicrobial defense. J. Biol. Chem. 2005;280:41125–41128. doi: 10.1074/jbc.R500015200. [DOI] [PubMed] [Google Scholar]
- 15.van Weely S., Brandsma M., Strijland A., Tager J.M., Aerts J.M. Demonstration of the existence of a second, non-lysosomal glucocerebrosidase that is not deficient in Gaucher disease. Biochim. Biophys. Acta. 1993;1181:55–62. doi: 10.1016/0925-4439(93)90090-n. [DOI] [PubMed] [Google Scholar]
- 16.Walden C.M., Sandhoff R., Chuang C.-C., Yildiz Y., Butters T.D., Dwek R.A., Platt F.M., van der Spoel A.C. Accumulation of glucosylceramide in murine testis, caused by inhibition of beta-glucosidase 2: implications for spermatogenesis. J. Biol. Chem. 2007;282:32655–32664. doi: 10.1074/jbc.M702387200. [DOI] [PubMed] [Google Scholar]
- 17.Aureli M., Bassi R., Loberto N., Regis S., Prinetti A., Chigorno V., Aerts J.M., Boot R.G., Filocamo M., Sonnino S. Cell surface associated glycohydrolases in normal and Gaucher disease fibroblasts. J. Inherit. Metab. Dis. 2012;35:1081–1091. doi: 10.1007/s10545-012-9478-x. [DOI] [PubMed] [Google Scholar]
- 18.Dekker N., Voorn-Brouwer T., Verhoek M., Wennekes T., Narayan R.S., Speijer D., Hollak C.E.M., Overkleeft H.S., Boot R.G., Aerts J.M.F.G. The cytosolic β-glucosidase GBA3 does not influence type 1 Gaucher disease manifestation. Blood Cells Mol. Dis. 2011;46:19–26. doi: 10.1016/j.bcmd.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 19.van der Spoel A.C., Jeyakumar M., Butters T.D., Charlton H.M., Moore H.D., Dwek R.A., Platt F.M. Reversible infertility in male mice after oral administration of alkylated imino sugars: a nonhormonal approach to male contraception. Proc. Natl. Acad. Sci. USA. 2002;99:17173–17178. doi: 10.1073/pnas.262586099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bone W., Walden C.M., Fritsch M., Voigtmann U., Leifke E., Gottwald U., Boomkamp S., Platt F.M., van der Spoel A.C. The sensitivity of murine spermiogenesis to miglustat is a quantitative trait: a pharmacogenetic study. Reprod. Biol. Endocrinol. 2007;5:1. doi: 10.1186/1477-7827-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferreirinha F., Quattrini A., Pirozzi M., Valsecchi V., Dina G., Broccoli V., Auricchio A., Piemonte F., Tozzi G., Gaeta L. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J. Clin. Invest. 2004;113:231–242. doi: 10.1172/JCI20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soderblom C., Stadler J., Jupille H., Blackstone C., Shupliakov O., Hanna M.C. Targeted disruption of the Mast syndrome gene SPG21 in mice impairs hind limb function and alters axon branching in cultured cortical neurons. Neurogenetics. 2010;11:369–378. doi: 10.1007/s10048-010-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin E., Yanicostas C., Rastetter A., Naini S.M.A., Maouedj A., Kabashi E., Rivaud-Péchoux S., Brice A., Stevanin G., Soussi-Yanicostas N. Spatacsin and spastizin act in the same pathway required for proper spinal motor neuron axon outgrowth in zebrafish. Neurobiol. Dis. 2012;48:299–308. doi: 10.1016/j.nbd.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Fox M.A., Sanes J.R. Synaptotagmin I and II are present in distinct subsets of central synapses. J. Comp. Neurol. 2007;503:280–296. doi: 10.1002/cne.21381. [DOI] [PubMed] [Google Scholar]
- 25.Wood J.D., Landers J.A., Bingley M., McDermott C.J., Thomas-McArthur V., Gleadall L.J., Shaw P.J., Cunliffe V.T. The microtubule-severing protein Spastin is essential for axon outgrowth in the zebrafish embryo. Hum. Mol. Genet. 2006;15:2763–2771. doi: 10.1093/hmg/ddl212. [DOI] [PubMed] [Google Scholar]
- 26.Lin P., Li J., Liu Q., Mao F., Li J., Qiu R., Hu H., Song Y., Yang Y., Gao G. A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42) Am. J. Hum. Genet. 2008;83:752–759. doi: 10.1016/j.ajhg.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fassier C., Hutt J.A., Scholpp S., Lumsden A., Giros B., Nothias F., Schneider-Maunoury S., Houart C., Hazan J. Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat. Neurosci. 2010;13:1380–1387. doi: 10.1038/nn.2662. [DOI] [PubMed] [Google Scholar]
- 28.Southgate L., Dafou D., Hoyle J., Li N., Kinning E., Critchley P., Németh A.H., Talbot K., Bindu P.S., Sinha S. Novel SPG11 mutations in Asian kindreds and disruption of spatacsin function in the zebrafish. Neurogenetics. 2010;11:379–389. doi: 10.1007/s10048-010-0243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song Y., Wang M., Mao F., Shao M., Zhao B., Song Z., Shao C., Gong Y. Knockdown of pnpla6 protein results in motor neuron defects in zebrafish. Dis. Model Mech. 2012 doi: 10.1242/dmm.009688. Published online November 16, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Overkleeft H.S., Renkema G.H., Neele J., Vianello P., Hung I.O., Strijland A., van der Burg A.M., Koomen G.J., Pandit U.K., Aerts J.M. Generation of specific deoxynojirimycin-type inhibitors of the non-lysosomal glucosylceramidase. J. Biol. Chem. 1998;273:26522–26527. doi: 10.1074/jbc.273.41.26522. [DOI] [PubMed] [Google Scholar]
- 31.Cox T.M. Gaucher disease: understanding the molecular pathogenesis of sphingolipidoses. J. Inherit. Metab. Dis. 2001;24(Suppl 2):106–121. doi: 10.1023/a:1012496514170. discussion 87–88. [DOI] [PubMed] [Google Scholar]
- 32.Nietupski J.B., Pacheco J.J., Chuang W.-L., Maratea K., Li L., Foley J., Ashe K.M., Cooper C.G.F., Aerts J.M.F.G., Copeland D.P. Iminosugar-based inhibitors of glucosylceramide synthase prolong survival but paradoxically increase brain glucosylceramide levels in Niemann-Pick C mice. Mol. Genet. Metab. 2012;105:621–628. doi: 10.1016/j.ymgme.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 33.Sun Y., Ran H., Liou B., Quinn B., Zamzow M., Zhang W., Bielawski J., Kitatani K., Setchell K.D.R., Hannun Y.A., Grabowski G.A. Isofagomine in vivo effects in a neuronopathic Gaucher disease mouse. PLoS ONE. 2011;6:e19037. doi: 10.1371/journal.pone.0019037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun Y., Liou B., Xu Y.-H., Quinn B., Zhang W., Hamler R., Setchell K.D.R., Grabowski G.A. Ex vivo and in vivo effects of isofagomine on acid β-glucosidase variants and substrate levels in Gaucher disease. J. Biol. Chem. 2012;287:4275–4287. doi: 10.1074/jbc.M111.280016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stevanin G., Ruberg M., Brice A. Recent advances in the genetics of spastic paraplegias. Curr. Neurol. Neurosci. Rep. 2008;8:198–210. doi: 10.1007/s11910-008-0032-z. [DOI] [PubMed] [Google Scholar]
- 36.Edvardson S., Hama H., Shaag A., Gomori J.M., Berger I., Soffer D., Korman S.H., Taustein I., Saada A., Elpeleg O. Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am. J. Hum. Genet. 2008;83:643–648. doi: 10.1016/j.ajhg.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tesson C., Nawara M., Salih M.A., Rossignol R., Zaki M.S., Al Balwi M., Schüle R., Mignot C., Obre E., Bouhouche A. Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am. J. Hum. Genet. 2012;91:1051–1064. doi: 10.1016/j.ajhg.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schüle R., Siddique T., Deng H.-X., Yang Y., Donkervoort S., Hansson M., Madrid R.E., Siddique N., Schöls L., Björkhem I. Marked accumulation of 27-hydroxycholesterol in SPG5 patients with hereditary spastic paresis. J. Lipid Res. 2010;51:819–823. doi: 10.1194/jlr.M002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.