

Abstract
Syndromes associated with multiple mtDNA deletions are due to different molecular defects that can result in a wide spectrum of predominantly adult-onset clinical presentations, ranging from progressive external ophthalmoplegia (PEO) to multisystemic disorders of variable severity. The autosomal-dominant form of PEO is genetically heterogeneous. Recently, causative mutations have been reported in several nuclear genes that encode proteins of the mtDNA replisome machinery (POLG, POLG2, and C10orf2) or that are involved in pathways for the synthesis of deoxyribonuclotides (ANT1 and RRM2B). Despite these findings, putative mutations remain unknown in half of the subjects with PEO. We report the identification, by exome sequencing, of mutations in DNA2 in adult-onset individuals with a form of mitochondrial myopathy featuring instability of muscle mtDNA. DNA2 encodes a helicase/nuclease family member that is most likely involved in mtDNA replication, as well as in the long-patch base-excision repair (LP-BER) pathway. In vitro biochemical analysis of purified mutant proteins revealed a severe impairment of nuclease, helicase, and ATPase activities. These results implicate human DNA2 and the LP-BER pathway in the pathogenesis of adult-onset disorders of mtDNA maintenance.
Main Text
mtDNA is replicated and maintained in multiple copies in each cell by a specific nuclear-gene-encoded set of proteins.1 Although some members of the replication machinery, such as the mtDNA polymerase POLG2 and the hexameric ring-shaped helicase C10orf2 (also known as Twinkle),3 have been identified, the roles of other proteins thought to be relevant for mtDNA synthesis and repair are still unclear. Inherited disorders of mtDNA maintenance are associated with most deleterious mitochondrial diseases, which affect people of all ages. In adults, defects in proteins involved in mtDNA replication, deoxyribonucleotide metabolism, and mitochondrial fusion can lead to the accumulation of multiple mtDNA deletions in postmitotic tissues, such as skeletal muscle. Most affected individuals develop progressive external ophthalmoplegia (PEO [MIM 157640, 609283, 609286, and 610131]), which is often accompanied by ptosis and mitochondrial myopathy; multisystemic involvement is not uncommon.4
Despite the discovery of causative mutations in several genes, a firm diagnosis can be achieved, at best, in half of patients.5 To identify genetic defects in individuals who showed signs of mtDNA instability, we performed whole-exome next-generation sequencing6 in a family in which two siblings (probands 1 [P1] and 2 [P2]) developed progressive myopathy with evidence of muscle mitochondrial dysfunction, as disclosed by histochemical analysis of skeletal muscle biopsy (Figure S1, available online).
P1 is a 62-year-old male who first came to our attention when he was 49 years old, when we diagnosed a preexisting suspected myopathy. He had always been thin with a harmonically decreased muscle bulk and reported that his muscle strength had declined progressively over the last 20 years. A neurological examination revealed an alert person with normal cognitive function, decreased facial expressions, moderate diffuse muscle atrophy, relatively mild weakness with a predominant lower-limb-girdle distribution, an anserine gait with dorsolumbar hyperlordosis, and a positive Gowers’ sign. At the age of 54 years, he developed exertional dyspnea with obstructive sleep apnea and a patent foramen ovale with minimal shunt. Serum creatine-phosphokinase (CPK) levels were moderately increased (245 IU/l; normal values are 21–195 IU/l). Electromyography (EMG) showed neurogenic signs in the lower-limb muscles and myogenic signs in the proximal upper-limb muscles.
P2 is the 45-year-old sister of P1. During childhood, she was easily fatigued and fell down frequently. She had always been thin and slender and experienced cramps in her calf muscles. She had a major depressive episode at the age of 22 years during a postpartum period. The depression remained a primary clinical problem over the following years, and she was treated with bupropion, bromazepam, and valproic acid. At the age of 32 years, she presented with hyperthyroidism, which was successfully treated with tapazole. Four years later, she had episodic obstructive dyspnea and bronchial asthma, which were treated with steroids. At the age of 40 years, she manifested upper- and lower-limb-girdle muscle weakness with difficulty climbing stairs and standing from sitting or recumbent positions. She also complained of myalgia and muscle cramps. Neurological examination showed mild eyelid ptosis and ophthalmoparesis, limb-girdle muscle weakness, a positive Gowers’ sign, and difficulty walking on the tiptoes.
Written informed consent was obtained from the probands, and investigations were carried out according to the guidelines of the Ethical Committee of Istituto Di Ricovero e Cura a Carattere Scientifico Foundation Ca’ Granda Ospedale Maggiore Policlinico in Milan and in agreement with Italian and European Union laws. Standard Sanger sequencing had previously ruled out the involvement of POLG, SLC25A4 (MIM 103220), C10orf2 (also known as PEO1), POLG2 (MIM 604983), OPA1 (MIM 605290), RRM2B (MIM 604712), TK2 (MIM 188250), MFN2 (MIM 608507), and DGUOK (MIM 601465). Total DNA was extracted from muscle by means of standard procedures. The accumulation of multiple mtDNA deletions was confirmed by Southern-blot (Figure 1A) and long-PCR (Figure 1B) techniques, as previously described.7 Muscle mtDNA content in our probands did not differ from that of age-matched control individuals or from that of groups of individuals with molecularly confirmed forms of autosomal-dominant or -recessive PEO (Figure 1C).
Figure 1.
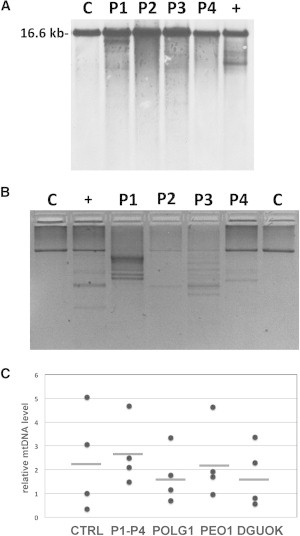
Molecular Findings in Muscle Tissue of Probands with DNA2 Mutations
(A) Southern-blot analysis of muscle mtDNA derived from probands 1–4 revealed the presence of multiple bands. “C” indicates control samples that display only a single band corresponding to WT mtDNA, and “+” indicates a positive control (POLG-mutated subject).
(B) Long PCR. This panel shows an agarose-electrophoresis separation of the WT mtDNA long-PCR amplified fragment (approximately 8,520 bp). Variably abundant extrabands due to mtDNA-deleted molecules from muscle DNA are observed in all the probands, but not in age-matched controls (“C”).
(C) Quantitative PCR analysis of mtDNA content in patients’ muscle. Real-time quantitative PCR analysis was performed with primers and probes for human MTCYB (MIM 516020, mtDNA) and APP (MIM 104760, nDNA). Primer sequences and PCR conditions are available upon request. Results were analyzed with a Student’s t test. The mtDNA copy number was determined by quantitative real-time PCR in skeletal-muscle samples of our probands (P1–P4, n = 4), age-matched control individuals (n = 4), and three groups of individuals with autosomal-dominant or -recessive PEO and mutations in POLG (n = 4), PEO1 (n = 4), or DGUOK (n = 4); no significant difference was observed. All determinations were performed in triplicate. Values are normalized to a control sample. Horizontal bars indicate mean values.
The genomic DNA of P1, P2, and an elder healthy brother was collected, and the exome targets were captured with the NimbleGen Sequence Capture 2.1M Human Exome v.1.0 Array (Roche NimbleGen) and sequenced on the GAIIx platform (Illumina). The two 82 bp paired-end sequence reads were first trimmed of any possible B-quality blocks from the end of the reads by removal of any pair that had a read smaller than 36 bp. The quality scores were then converted to the Sanger phred score with Emboss (version 6.3.1) and aligned to the human reference sequence GRCh37/hg19 (24 chromosomes; RefSeq accession numbers NC_000001 to NC_000024). The reference alignment analysis was performed with CLC Genomics Workbench v.4.7.1. The mapped reads were used for the analysis of SNPs and deletion-insertion polymorphisms (DIPs). The list of SNPs and DIPs was filtered with the Phred quality scores of the position and surrounding bases.
As a result of variant calling, 14,634 SNPs were identified. These were then filtered for variants that were not listed in dbSNP or the 1000 Genomes Project and that were within gene regions. Of the resulting variants, 100 were shared by P1 and P2, but not by their elder healthy brother, and 75 were nonsynonymous changes. All variants were subjected to a scan of known genes involved in mitochondrial disorders. Seventeen nucleotide substitutions either were not confirmed by Sanger sequencing or were found in control subjects. Finally, in the DNA2 (MIM 601810) coding sequence (RefSeq NM_001080449.1, chromosomal region 10q21.3), we identified the heterozygous c.851G>A mutation (exon 5), which is predicted to result in the amino acid change p.Arg284His. Sanger sequencing confirmed the finding in both probands (Figure 2A), whereas their healthy 64-year-old brother did not carry this variant.
Figure 2.
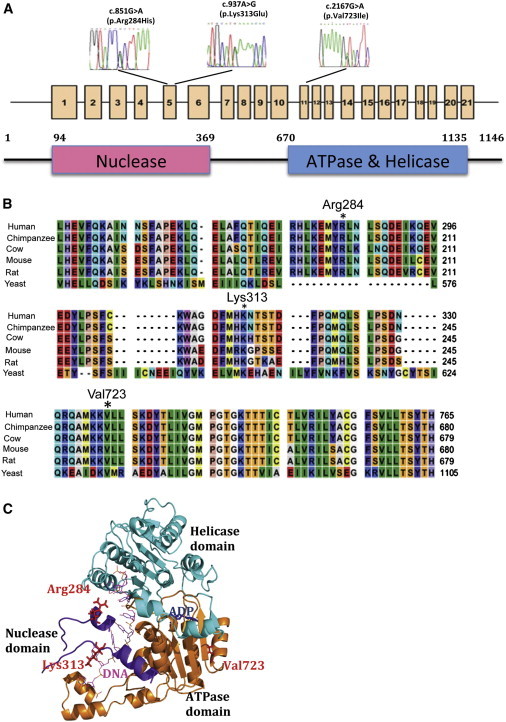
DNA2 Variants Occur at Conserved Residues Located in Important Structural Elements of Human DNA2
(A) A scheme of DNA2 includes the location of the identified mutations in the coding sequence. Exons are numbered. A Diagram representing human DNA2 shows the functional domains conserved in this enzyme.
(B) Multiple-sequence alignment of nuclease and ATPase domains for the DNA2 mammalian and yeast orthologous protein sequences with the use of CLC Bio Main Workbench v.5.1. The tabular format of six orthologs of DNA2 sequences is color coded to display the conservation at each position in the alignment.
(C) Homology model of DNA2 with ADP (blue) binding in the catalytic center of the ATPase domain (brown) interacts with ssDNA (pink). The positions of three residues, Arg284, Lys313, and Val723, are illustrated as red sticks. The nuclease and helicase domains are displayed in purple and cyan, respectively.
We next performed a mutational screening of DNA2 in a cohort of undiagnosed adult subjects (n = 44) with muscle mtDNA deletions. In two additional individuals, probands 3 (P3) and 4 (P4), we identified missense variants c.937A>G (exon 5) and c.2167G>A (exon 12), which lead to protein changes p.Lys313Glu and p.Val723Ile, respectively. Both subjects presented with adult-onset mitochondrial disease that consisted of ptosis and progressive myopathy. P3 also developed a mild weakness of the upper- and lower-limb-girdle muscles with occasional exertional dyspnea. EMG and Desmedt tests were normal, as were the levels of acetylcholine-receptor antibodies, CPK, aldolase, and thyroid hormones. The clinical, histological, and molecular features of the affected subjects with the mutations are summarized in Table 1 and Figure S1. None of the identified mutations were found in 460 Italian or 180 European control chromosomes or by analysis of data from >10,000 chromosomes included in the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server.
Table 1.
Clinical, Histochemical, and Molecular Data of Affected Subjects
| Subject | Sex | Family History | Age at Exam (Years) | Age at Onset (Years) | Primary Neurological Symptoms | Additional Clinical Symptoms | EMG | Muscle Histology and Histochemistrya | mtDNA Deletions (Southern Blot) | mtDNA Deletions (Long PCR) | DNA2 Analysis |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | male | yes | 49 | childhood | limb-girdle weakness | hypotonia, hypostenia, exertional dyspnea | M/N | SDH+, COX– (5%) | ++ | ++ | c.851G>A (p.Arg284His) |
| 2 | female | yes | 45 | childhood | limb-girdle weakness | depression, hyperthyroidism, episodic dyspnea, mild ptosis, ophthalmoplegia | M/N | COX− (4%), type II fiber hypotrophy | ++ | + | c.851G>A (p.Arg284His) |
| 3 | female | no | 57 | 20 | slight limb-girdle weakness, ptosis | myalgia, dysphoria, exertional dyspnea | normal | COX− (5%), RRF | + | ++ | c.937A>G (p.Lys313Glu) |
| 4 | female | no | 55 | 35 | lower-limb weakness | diplopia, myalgia, mild ophthalmoplegia | normal | SDH+, COX− (2%) | + | + | c.2167G>A (p.Val723Ile) |
The following abbreviations are used: EMG, electromyography; M, myogenic pattern; N, neurogenic pattern; SDH, succinate dehydrogenase; COX, cytochrome c oxidase; +, small amount; ++, moderate amount; and RRF, ragged red fiber.
The percentages in parentheses indicate the proportion of COX− fibers.
Muscle from P1 and unaffected controls was homogenized, and total RNA was extracted in accordance with the Eurozol protocol (Euroclone). First-strand cDNA was synthesized from total RNA with random hexamer primers with the First-Strand cDNA Synthesis kit (GE Healthcare). The resulting RT-PCR products were analyzed by electrophoresis and sequenced, but there was no evidence of distinct DNA2 splicing variants.
The expression of DNA2 was quantified in eight control human tissue samples contained in the Human Total Master Panel II (Clontech Laboratories) by quantitative real-time PCR analysis by means of the ΔΔCt method on a 7500 System (Software 2.01, Applied Biosystems). DNA2 mRNA was evaluated with the Taqman gene-expression assay (Hs01055246_m1), and GAPDH (MIM 138400; Hs99999905_m1) and RNA18S (Hs99999901_s1) were used as control housekeeping genes. Real-time PCR analysis showed relatively high DNA2 mRNA levels in brain, cerebellum, kidney, and heart tissue, but, perhaps surprisingly, a lower level of expression in skeletal muscle (Figure S2). The vulnerability of skeletal muscle to DNA2 mutations might be due to low expression levels, which most likely reflects a low basal-muscle activity combined with a high demand for mitochondrial-gene-encoded respiratory-chain proteins and mtDNA content. The balance between DNA2 activity and demand for its product in muscle is probably finely tuned and near threshold levels.
Human DNA2 is a member of the nuclease/helicase family.8 It was recently found in mammalian mitochondria, where it participates in the removal of RNA primers during mtDNA replication;9 however, whether it is exclusively localized to the mitochondria is debated.10 Human DNA2 interacts with POLG and stimulates its catalytic activity and most likely serves as an alternative to the Twinkle helicase.9 As a member of the nuclease/helicase family, human DNA2 contains conserved nuclease, ATPase, and helicase domains.8,11 To understand the pathologic role of these human DNA2 variants, we first verified whether they are conserved in DNA2 from different species. We found that the protein sequence alignments revealed that the Arg284 residue is conserved in mammalian DNA2, whereas the Lys313 and Val723 residues are conserved in both mammalian and yeast DNA2 (Figure 2B). We then employed the Schrödinger Suite 2012 Prime homology-modeling tool and three-dimensional X-ray crystallography structures of the yeast Upf1-RNA complex (Protein Data Bank [PDB] 2XZL) and human Upf1 helicase-ADP complex (PDB 2GK6) as templates to create a homolog model for the DNA2-ssDNA-ADP complex (Figure 2C). This model revealed that all three domains of human DNA2 interact with the DNA molecule (Figure 2C). More importantly, the Arg284 and Lys313 residues are located in the nuclease domain, and the side chain of these two residues is in direct contact with the ssDNA molecule (Figure 2C), suggesting that p.Arg284His and p.Lys313Glu might affect the DNA2 substrate binding and impair its nuclease and helicase activities. On the other hand, the Val723 residue is located in the interface between the ATPase domain and the helicase domain (Figure 2B), implicating that the p.Val723Ile alteration might affect the interaction between these two domains and the ATP-dependent helicase activity of DNA2.
To further assess the impact of the variants identified in our probands, we transiently expressed wild-type (WT) and recombinant forms of human DNA2 (Figure S3) as previously described.12 Nuclease activity, which is important for efficient processing of the RNA primer intermediates that occur in the strand-coupled replication mechanism of mtDNA, was assayed according to a previously described protocol.8 In brief, WT and altered forms of human DNA2 were incubated with 0.25 pmol DNA substrate in reaction buffer that contained 50 mM HEPES-KOH (pH 7.5), 4 mM MgCl2, 2 mM DTT, and 0.25 mg/ml BSA. The reactions were stopped with 2× stop buffer (95% deionized formamide, 10 mM EDTA, 0.1% bromophenol blue, and 0.1% xylene cyanol). The nuclease products were separated on a 15% denatured urea gel. As expected, p.Arg284His and p.Lys313Glu, located in the nuclease domain, affected nuclease activity; there was a complete loss of nuclease activity for p.Arg284His and a significant reduction for the p.Lys313Glu variant. The p.Val723Ile alteration, which affects the helicase domain, was also associated with defective nuclease activity (30% compared to the WT protein) (Figure 3A).
Figure 3.
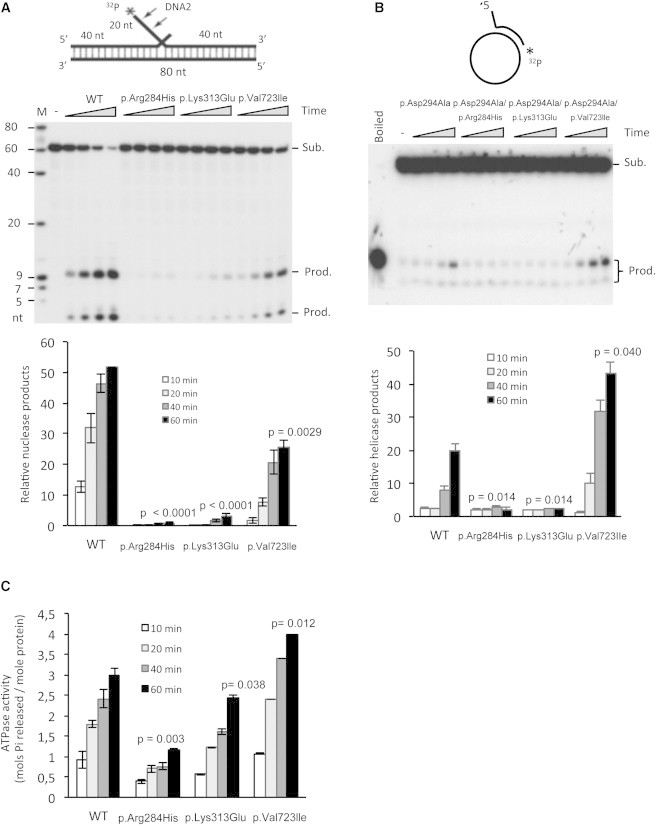
Changes in Enzymatic Activities of Altered Forms of DNA2
(A) Endonuclease activities of WT and altered forms of DNA2. In the top panel is the DNA double-flap substrate. WT, p.Arg284His, p.Lys313Glu, or p.Val723Ile human DNA2 (0.8 nM) was incubated with 0.25 pmol 5′ end 32P-labeled flap DNA substrate. Reactions were carried out at 37°C for 10, 20, 40, and 60 min. Arrows indicate the cleavage sites. In the bottom panel is the quantification of DNA2 endonuclease activity. Error bars indicate SEM.
(B) Helicase activities of WT and altered forms of DNA2. In the top panel is the DNA helicase substrate. DNA2 was first altered for the elimination of nuclease activity (p.Asp294Ala) so that helicase activity could be assayed. A helicase mixture containing 1 fmol 3′-end-labeled duplex substrate and 100 nM purified recombinant p.Asp294Ala, p.Asp294Ala/p.Arg284His, p.Asp294Ala/p.Lys313Glu, or p.Asp294Ala/p.Val723Ile DNA2 was incubated at 37°C for 10, 20, 40, and 60 min. In the bottom panel is the quantification of DNA2 helicase activity. Error bars indicate SEM.
(C) ATPase activity of WT and altered forms of DNA2. ATPase activity of WT, p.Arg284His, p.Lys313Glu, or p.Val723Ile forms of human DNA2 was measured with an ATPase assay kit. A reaction mixture containing 30 nM recombinant protein and 1 μg ssDNA was incubated at 37°C for 10, 20, 40, and 60 min. In each panel, the p value was calculated by a Student’s t test. Error bars indicate SEM.
We next evaluated helicase activity, which was determined after inactivation of the nuclease domain by site-directed mutagenesis, as described previously.12 In brief, the reaction mixtures contained 50 mM Tris-HCl (pH 7.5), 25 mM NaCl, 2 mM DTT, 0.25 mg/ml BSA, 4 mM MgCl2, 4 mM ATP, and 32P-labeled helicase substrate. The substrate was prepared as described previously.13 The 3′ ends of the substrate were labeled with terminal transferase (Roche). The reaction was terminated with 5× stop solution (60 mM EDTA, 40% sucrose, 0.6% SDS, 0.25% bromophenol blue, and 0.25% xylene cyanole FF) at the indicated time points. The reaction was loaded on an 8% native polyacrylamide gel.
The p.Arg284His and p.Lys313Glu altered forms of DNA2 displayed a severe impairment in helicase activity; DNA2 with the p.Val723Ile variant displayed enhanced helicase activity (Figure 3B). This anomalous unbalance in helicase activity might be detrimental to affected tissues, as previously demonstrated in mice in which the Twinkle protein was overexpressed.14
These findings suggest that the activity of the nuclease and helicase domains is likely to be coupled. Genetic experiments performed previously on the yeast ortholog yDna2 revealed that a mutant with helicase activity, but not nuclease activity, was more sensitive to double-strand breaks than a helicase- and nuclease-deficient double mutant.15 Because both nuclease and helicase activities rely upon an ATP-dependent mechanism, we used the Innova Biosciences ATPase assay kit to assess the function of ATPase in WT and altered recombinant forms of human DNA2. The reaction mixture (20 μl) contained 30 nM purified DNA2, 40 mM Tris-HCl, 5 mM MgCl2, 25 mM NaCl, 2.5 mM DTT, 0.1 mg/ml BSA, 0.5 mM ATP, and 1 μg oligonucleotide (ssDNA, 40 bases). The reaction was collected at the indicated time points and stopped by the addition of 5 μl PiColorLock Gold mix for 5 min; the mixture was further incubated for 30 min at room temperature after the addition of 2 μl stabilizer. The absorbance was measured with a NanoVue Plus spectrophotometer at 650 nm.
Biochemical analysis revealed a significant reduction of ATPase activity in the p.Arg284His and p.Lys313Glu altered proteins, which was expected because of the overlap of the nuclease and ATPase domains. No difference was observed between the WT protein and the p.Val723Ile altered protein (Figure 3C).
Because the human DNA2 variants were heterozygous, we sought to determine whether they had heterozygous effects or dominant-negative effects on its function. To this purpose, we mixed the WT DNA2 with equal amounts of WT, p.Arg284His, p.Lys313Glu, or p.Val723Ile forms of the protein in order to model the situation in normal and different mutant cells. The product of the reaction with WT/p.Arg284His, WT/p.Lys313Glu, or WT/p.Val723Ile proteins was approximately half of that of the reaction with WT/WT proteins (Figures 4A and 4B), thus indicating that in this in vitro condition, WT DNA2 could not compensate for the detrimental effect on the nuclease activity of p.Arg284His, p.Lys313Glu, or p.Val723Ile altered proteins.
Figure 4.
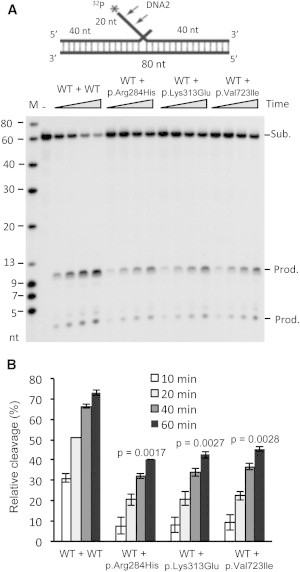
Heterogeneity Effects of the Nuclease Activity of Altered Forms of DNA2
(A) 0.2 nM WT DNA2 was mixed with 0.2 nM WT or p.Arg284His, p.Lys313Glu, or p.Val723Ile forms of DNA2. The DNA2 mixture was incubated with 0.25 pmol 5′ end 32P-labeled flap DNA substrates. Reactions were carried out at 37°C for 10, 20, 40, and 60 min.
(B) Quantification of DNA2 cleavage products. Values are means ± SEM of three assays. In each panel, the p value was calculated by a Student’s t test.
DNA2 is critical for long-patch base-excision repair (LP-BER), the predominant pathway for repairing small DNA lesions induced by oxidation, alkylation, and spontaneous hydrolysis.16 Evidence that LP-BER acts on mtDNA is suggested by the mitochondrial localization of several factors, such as flap endonuclease 1 (FEN1),17 that facilitate this process. Together with FEN1, DNA2 is thought to be involved in the repair of oxidative lesions in DNA molecules.18 This role seems especially important for mitochondria, in which there is a significant threat of oxidative DNA damage.
Heterozygous mutations associated with autosomal-dominant or sporadic PEO with multiple mtDNA deletions have been described in several genes.4 In our subject cohort, mutations in POLG remained the major cause of multiple-mtDNA-deletion disorders; POLG mutations accounted for approximately 19% of all individuals presenting with mitochondrial disease and were followed by those in PEO1 (18% of our PEO and multiple-mtDNA-deletion cohort), SLC25A4 (6.7%), and OPA1 (2.2%). We did not detect any mutations in RRM2B, although it appears to be relevant in other populations.19 Mutations in DNA2 occurred in three independent probands in our cohort. These accounted for 6.8% of genetically undiagnosed subjects (n = 44) carrying multiple mtDNA deletions and 3.3% of all probands (n = 90) carrying multiple mtDNA deletions as confirmed by Southern-blot and long-PCR analyses. These results suggest that DNA2 is an appropriate candidate for molecular screening in subjects with sporadic or familial PEO with ascertained instability of muscle mtDNA. Further studies of DNA2 and other proteins involved in mtDNA-repair pathways will improve our understanding of mtDNA replication and the clinical consequences of its impairment.
Acknowledgments
We wish to thank the patients and their families for their support and collaboration. This work was supported by the following grants: Telethon project GTB07001ER, Eurobiobank project QLTR-2001-02769, Telethon grant GUP09004 to G.P.C., and National Institutes of Health grant R01CA085344 to B.S. The financial support of Associazione Amici del Centro Dino Ferrari, University of Milan is gratefully acknowledged.
Contributor Information
Binghui Shen, Email: bshen@coh.org.
Giacomo Pietro Comi, Email: giacomo.comi@unimi.it.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
NHLBI Exome Sequencing Project Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Protein Data Bank, http://www.rcsb.org
References
- 1.Wanrooij S., Falkenberg M. The human mitochondrial replication fork in health and disease. Biochim. Biophys. Acta. 2010;1797:1378–1388. doi: 10.1016/j.bbabio.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 2.Stumpf J.D., Copeland W.C. Mitochondrial DNA replication and disease: Insights from DNA polymerase γ mutations. Cell. Mol. Life Sci. 2011;68:219–233. doi: 10.1007/s00018-010-0530-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sen D., Nandakumar D., Tang G.Q., Patel S.S. Human mitochondrial DNA helicase TWINKLE is both an unwinding and annealing helicase. J. Biol. Chem. 2012;287:14545–14556. doi: 10.1074/jbc.M111.309468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Copeland W.C. Defects in mitochondrial DNA replication and human disease. Crit. Rev. Biochem. Mol. Biol. 2012;47:64–74. doi: 10.3109/10409238.2011.632763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Virgilio R., Ronchi D., Hadjigeorgiou G.M., Bordoni A., Saladino F., Moggio M., Adobbati L., Kafetsouli D., Tsironi E., Previtali S. Novel Twinkle (PEO1) gene mutations in mendelian progressive external ophthalmoplegia. J. Neurol. 2008;255:1384–1391. doi: 10.1007/s00415-008-0926-3. [DOI] [PubMed] [Google Scholar]
- 6.Gilissen C., Hoischen A., Brunner H.G., Veltman J.A. Unlocking Mendelian disease using exome sequencing. Genome Biol. 2011;12:228. doi: 10.1186/gb-2011-12-9-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeviani M., Gellera C., Pannacci M., Uziel G., Prelle A., Servidei S., DiDonato S. Tissue distribution and transmission of mitochondrial DNA deletions in mitochondrial myopathies. Ann. Neurol. 1990;28:94–97. doi: 10.1002/ana.410280118. [DOI] [PubMed] [Google Scholar]
- 8.Budd M.E., Campbell J.L. A yeast gene required for DNA replication encodes a protein with homology to DNA helicases. Proc. Natl. Acad. Sci. USA. 1995;92:7642–7646. doi: 10.1073/pnas.92.17.7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng L., Zhou M., Guo Z., Lu H., Qian L., Dai H., Qiu J., Yakubovskaya E., Bogenhagen D.F., Demple B., Shen B. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol. Cell. 2008;32:325–336. doi: 10.1016/j.molcel.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duxin J.P., Dao B., Martinsson P., Rajala N., Guittat L., Campbell J.L., Spelbrink J.N., Stewart S.A. Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol. Cell. Biol. 2009;29:4274–4282. doi: 10.1128/MCB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bae S.H., Seo Y.S. Characterization of the enzymatic properties of the yeast dna2 Helicase/endonuclease suggests a new model for Okazaki fragment processing. J. Biol. Chem. 2000;275:38022–38031. doi: 10.1074/jbc.M006513200. [DOI] [PubMed] [Google Scholar]
- 12.Kim J.H., Kim H.D., Ryu G.H., Kim D.H., Hurwitz J., Seo Y.S. Isolation of human Dna2 endonuclease and characterization of its enzymatic properties. Nucleic Acids Res. 2006;34:1854–1864. doi: 10.1093/nar/gkl102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masuda-Sasa T., Imamura O., Campbell J.L. Biochemical analysis of human Dna2. Nucleic Acids Res. 2006;34:1865–1875. doi: 10.1093/nar/gkl070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ylikallio E., Tyynismaa H., Tsutsui H., Ide T., Suomalainen A. High mitochondrial DNA copy number has detrimental effects in mice. Hum. Mol. Genet. 2010;19:2695–2705. doi: 10.1093/hmg/ddq163. [DOI] [PubMed] [Google Scholar]
- 15.Budd M.E., Campbell J.L. Interplay of Mre11 nuclease with Dna2 plus Sgs1 in Rad51-dependent recombinational repair. PLoS ONE. 2009;4:e4267. doi: 10.1371/journal.pone.0004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robertson A.B., Klungland A., Rognes T., Leiros I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell. Mol. Life Sci. 2009;66:981–993. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y., Zhang H., Veeraraghavan J., Bambara R.A., Freudenreich C.H. Saccharomyces cerevisiae flap endonuclease 1 uses flap equilibration to maintain triplet repeat stability. Mol. Cell. Biol. 2004;24:4049–4064. doi: 10.1128/MCB.24.9.4049-4064.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Copeland W.C., Longley M.J. DNA2 resolves expanding flap in mitochondrial base excision repair. Mol. Cell. 2008;32:457–458. doi: 10.1016/j.molcel.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fratter C., Raman P., Alston C.L., Blakely E.L., Craig K., Smith C., Evans J., Seller A., Czermin B., Hanna M.G. RRM2B mutations are frequent in familial PEO with multiple mtDNA deletions. Neurology. 2011;76:2032–2034. doi: 10.1212/WNL.0b013e31821e558b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.