

Abstract
A single Mendelian trait has been mapped to the human Y chromosome: Y-linked hearing impairment. The molecular basis of this disorder is unknown. Here, we report the detailed characterization of the DFNY1 Y chromosome and its comparison with a closely related Y chromosome from an unaffected branch of the family. The DFNY1 chromosome carries a complex rearrangement, including duplication of several noncontiguous segments of the Y chromosome and insertion of ∼160 kb of DNA from chromosome 1, in the pericentric region of Yp. This segment of chromosome 1 is derived entirely from within a known hearing impairment locus, DFNA49. We suggest that a third copy of one or more genes from the shared segment of chromosome 1 might be responsible for the hearing-loss phenotype.
Main Text
The human Y chromosome is transmitted directly from father to son for most of its length, generating a male-specific pattern of inheritance that is distinct from the rest of the genome. Y-chromosomal phenotypic traits should thus be readily recognizable from their male-line inheritance, and early studies claimed to identify several such traits, including notable examples such as "hairy ears." However, as long ago as 1957, critical examination of these claims failed to find support for any of the 17 traits under consideration,1 a finding reinforced by subsequent molecular-genetic analysis of "hairy ears" itself.2 This perhaps surprising lack of Y-chromosomal traits can be understood at least in part as the consequence of two factors. First, when a reference sequence for the male-specific region of the human Y chromosome was generated in 2003,3 it revealed that the chromosome carries few male-specific genes and codes for only 23 distinct proteins. Subsequent work has found that three of these are absent from about 2% of South Asian men, whose Y chromosomes thus code for just 20 male-specific proteins.4 Second, the Y chromosome has specialized functions in male sex determination and fertility, in which Mendelian variation would be unlikely to be heritable. Thus, in early 2004 it was possible to write “pedigree analysis has yet to reveal a single Y-linked gene.”5 Later that year, however, Y-linked hearing impairment (DFNY1, MIM 400043) was reported in a Chinese family6 and remains the sole documented Mendelian disorder showing Y-linkage in humans. Its basis thus seems likely to be unusual and is of considerable interest. Here, we investigate this basis by examining the sequence of the DFNY1 Y chromosome and comparing it with the Y chromosome of a closely related but unaffected branch of the family.
In the seven-generation DFNY1 pedigree reported in 2004, all adult males were affected.6 Subsequently, the pedigree was traced back two more generations, and additional male-line descendants of an earlier ancestor were identified.7 Strikingly, their hearing was unaffected (Figure 1). We had previously demonstrated the identity of the Y chromosomes from the two branches of the family at 67 Y-STR loci, and we therefore reasoned that the phenotypic difference between the branches must be associated with a genetic variant carried specifically by the Y chromosome of the affected branch. We sorted a representative Y chromosome from each branch by flow cytometry and then sequenced it. Only four base-substitutional differences were identified between the chromosomes.8 Three of these had arisen on the unaffected branch. The fourth had arisen on the affected branch and segregated with the phenotype, but it provided a poor candidate for the causal mutation because it lay outside any annotated gene. Although this analysis could not evaluate the repeated regions of the chromosome, the known repeated genes are involved mainly in spermatogenesis,3,9 so in the current study we have explored the possibility that the causal mutation might not be a point mutation. This study was approved by the sample donors, who provided written informed consent, and by the Committee of Medical Ethics of the Chinese PLA General Hospital, Beijing, China.
Figure 1.
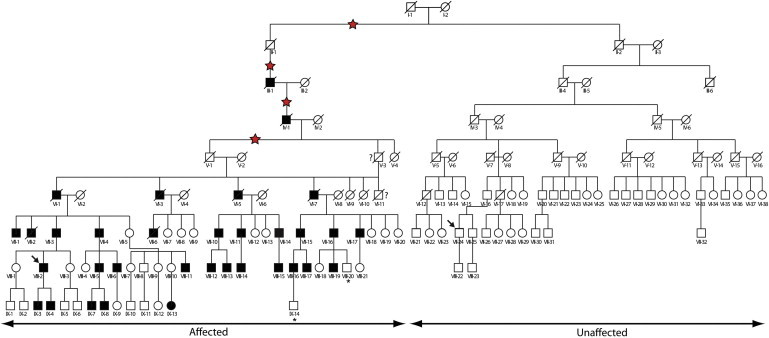
The DFNY1 Pedigree
Males, squares; females, circles; diagonal line, deceased. Filled symbols indicate hearing impairment (including from family records); question marks indicate two individuals whose hearing phenotype is unknown; and asterisks indicate individuals who are on the affected branch but who were below the age of onset of symptoms at the time of examination. Arrows indicate the two individuals whose Y chromosomes were sequenced. The structural rearrangement occurred during one of the four meioses marked by red stars. Spouses are omitted in generations VII-IX and in generation VI on the unaffected branch.
We examined the relative read depth along the chromosome by aligning high-quality duplicate-filtered reads to the chrY reference sequence by using Sequence Search and Alignment by Hashing Algorithm 2 (SSAHA2)10 and comparing the numbers of reads per 10 kb bin between Y chromosomes from the affected individual VIII-2 (Figure 1) and the unaffected individual VII-24 (Figure 1). This revealed three discontinuous segments duplicated in the affected chromosome, and all were derived from the limited region between the TSPY1 (MIM 480100) gene cluster ending at ∼9.3 Mb and the centromeric gap at ∼10.1 Mb (Figure 2A). These duplications were confirmed by conventional oligonucleotide array comparative genomic hybridization (CGH) with an Agilent 1 million array covering the whole genome and a custom NimbleGen 385K array spanning positions chrY: 2,000,000–10,715,000 (1,990,000–10,105,000 hg19) at high (∼20 bp) resolution (Figure 2B). Because they included part of the TSPY1 gene cluster, we verified an increase in TSPY1 gene number by qPCR (Table S1 in the Supplemental Data available online) and pulsed-field gel electrophoresis (PFGE; Figure S1). These analyses showed that the duplication was shared by all affected family members tested and was absent from all unaffected members and also that the duplication lay in a separate restriction fragment from the original TSPY1 cluster and was thus noncontiguous. Although the combined evidence confirmed a complex duplication of the 9.3–10.1 Mb region, it provided no information about where the duplicated sequences were inserted. Metaphase fluorescence in situ hybridization (FISH) with a TSPY1 probe showed a single signal (not shown), so we used fiber-FISH as described previously11 for higher resolution. Y-chromosomal probes were BAC clones spanning most of ∼9.3–10.1 Mb of the reference sequence: RP11-334D2 (including TSPY1), RP11-182H20, RP11-155J5, RP11-160K17, and RP11-108I14 (including centromeric sequences). The results (Figure 2C) showed that the unaffected chromosome was organized in the same way as the reference sequence, which can be summarized as TSPY1-182H20-centromere. In contrast, the affected chromosome (Figure 2D) was interrupted within the 182H20 sequence to produce the organization TSPY1-partial 182H20-gap-centromeric duplication-TSPY1 duplication-182H20-centromere. The fiber-FISH results thus demonstrated that the duplicated Y sequences were reinserted locally in a rearranged form, but they also revealed the presence of a segment that did not hybridize to any probe from the TSPY1-centromere region.
Figure 2.
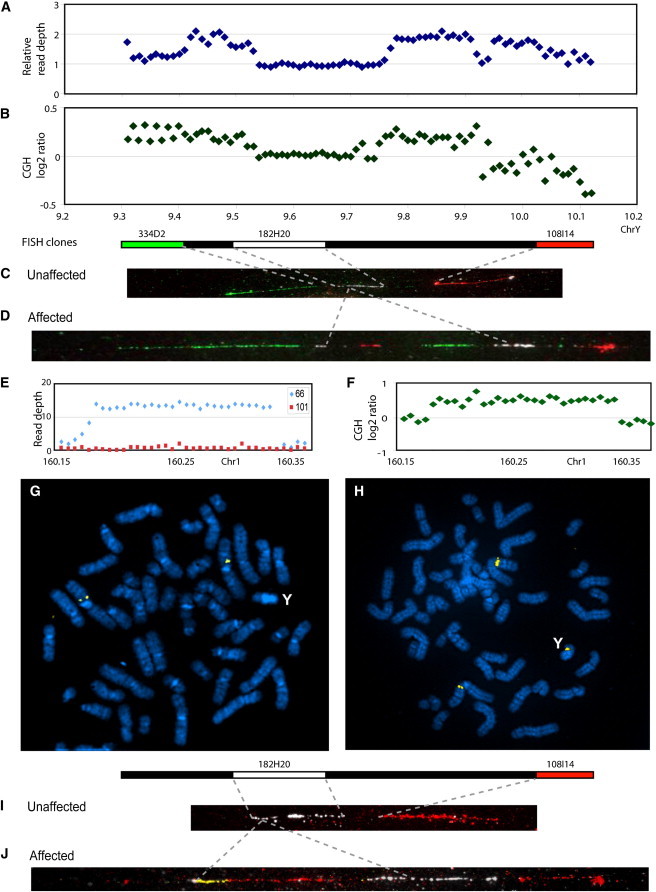
Characterization of the Structural Rearrangement Carried by the DFNY1 Y Chromosome
(A) Relative read depth (affected/unaffected) in 10 kb bins mapped to the Y chromosome reference sequence between 9.3 and 10.1 Mb. Note the assembly gaps at each end of this region.
(B) CGH log2 ratio (affected/unaffected) for the same region.
(C) Fiber-FISH of the three BAC clones indicated to the unaffected Y chromosome, showing that it carries the reference structure in this region. Both 334D2 and 108I14 detect tandem arrays larger than the size of the BAC clone.
(D) Fiber-FISH of the same BAC clones to the affected Y chromosome. This chromosome carries an insertion that interrupts the 182H20-hybridizing region and contains partial duplications of both 334D2- and 108I14-hybridizing sequences.
(E) Absolute read depth of unaffected and affected Y chromosome reads to the 160.15–160.35 Mb region of the chromosome 1 reference sequence.
(F) CGH log2 ratio (affected/unaffected) for the same chromosome 1 region.
(G) Metaphase FISH of the chromosome 1 BAC clone 179G5 on the unaffected Y chromosome (yellow). Hybridization is seen only at the reference location on chromosome 1.
(H) Metaphase FISH of the same chromosome 1 probe on the affected Y chromosome (yellow). Hybridization is seen at an additional locus on the Y chromosome.
(I) Fiber-FISH of the two Y BAC clones indicated plus the chromosome 1 clones 574F21, 179G5 and 1365F20 to the unaffected Y chromosome. No chromosome 1 signal is detected on the Y.
(J) Fiber-FISH of the same clones to the unaffected Y chromosome. The chromosome 1 clones detect a signal on the Y between the partial 182H20 signal and the 108I14 signal. Genome coordinates refer to GRCh37/hg19.
In order to identify these unknown sequences, we performed thermal asymmetric interlaced PCR (TAIL-PCR)12 on the segment extending from the 182H20 breakpoint into the gap by using the primers shown in Table S2. In consecutive amplifications, this procedure pairs nested primers specific to the known sequence with degenerate primers expected to prime in the unknown flanking sequence. Candidate junction products are recognized because their size differences in consecutive reactions reflect the known primer positions. The adjacent sequences, confirmed with breakpoint-specific primers (Table S3 and Figures S2 and S3), were derived from chromosome 1 (160.16 Mb), and examination of the read depth, CGH profiles (Figures 2E and 2F), and sequence suggested the involvement of a contiguous region of ∼160 kb; such involvement was supported by the identification of a second more complex chromosome 1-Y junction at 160.32 Mb. The translocation of chromosome 1 sequences to the affected Y was confirmed by metaphase FISH (Figures 2G and 2H), and their location within the gap in the Y duplication was confirmed by fiber-FISH (Figures 2I and 2J) with BACs RP11-574F21, RP11-179G5, and W12-1365F20 from chromosome 1 as probes. The combined data thus revealed a complex duplication structure consisting of both a chromosome 1 fragment and multiple segments of Y DNA (Table S4 and Figures S2 and S3). None of the sequenced breakpoints exhibited extensive lengths of homology, but all revealed microhomology, a pattern indicative of FoSTeS (fork stalling and template switching), which is a mechanism that combines disparate genomic fragments during replication.13
The duplicated structure observed here has not been reported elsewhere and must have arisen during one of just four meioses, which encompass the meiosis in which the DFNY1 mutation itself occurred (Figure 1). It is therefore likely to be causal. The duplicated Y-chromosomal sequences code for only one known protein, TSPY1, from a tandem array of ∼10 TSPY1 genes, whereas the chromosome 1 sequences carry five RefSeq genes (CASQ1 [MIM 114250], PEA15 [MIM 603434], DCAF8 [MIM NA], PEX19 [MIM 600279], and COPA [MIM 601924]) and the 5′ end of another gene, NCSTN (MIM 605254) (exons 1–3; Figure 3). Mechanisms by which the duplication might lead to the deafness phenotype include an increase in gene copy number, inappropriate expression resulting from novel flanking DNA, or formation of an altered product through truncation, fusion, or point mutation. No expression data are available from the DFNY1 family, and no nonsynonymous mutations were found in the chromosome 1 RefSeq genes (Table S5). TSPY1 copy number is polymorphic within the population,14 and larger numbers of TSPY1 copies have been found without reported deafness, including in 47,XYY individuals; one study associated high TSPY1 copy number with a different phenotype, infertility.15 Increased TSPY1 copy number thus provides a poor candidate for deafness. In contrast, the chromosome 1 region lies entirely within an approximately 900 kb interval, DFNA49 (MIM %608372), previously associated with hearing loss; the causal mutation in this interval remains unknown.16 We therefore propose that the same gene or genes might underlie both the DFNA49 and DFNY1 phenotypes. The most parsimonious mechanism would be dosage-sensitivity of one or more of these genes, such that increased expression resulting from three copies leads to hearing loss, although other mechanisms are not excluded.
Figure 3.
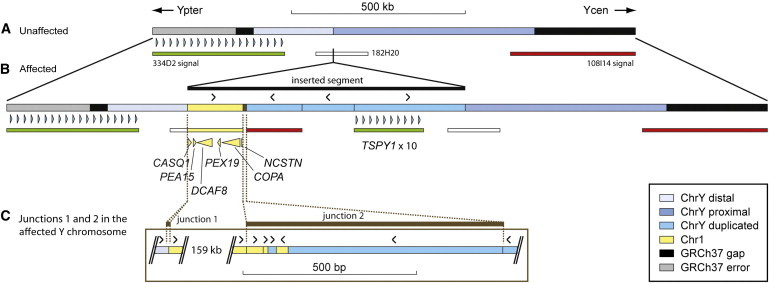
Structure and Gene Content of the Unaffected and Affected Y Chromosomes
(A) The structure of the unaffected (VII-24 in Figure 1) Y chromosome. Grey and black: errors and gaps, respectively, in the GRCH37 assembly. Blue: region matching the reference sequence at the level of resolution used. Shown below are part of the TSPY1 array (blue arrowheads) and the location of BAC clone signals identified in Figure 2.
(B) Structure of the affected (VIII-2 in Figure 1) Y chromosome. This chromosome contains a segment that is not in the unaffected chromosome; this segment is derived from duplications of sequences from both chromosome 1 (yellow) and the Y chromosome (blue). Again, the gene content and BAC signals are shown below. The arrowheads above indicate the orientation of the duplicated segments relative to the reference sequence.
(C) Detailed view of the two chromosome 1-Y junctions studied at the sequence level (Figures S2 and S3), showing the difference between the simple structure of junction 1 and the complex structure of junction 2.
We found the cause of the human Y-linked Mendelian disorder was associated with insertion of chromosome 1 sequences rather than mutation of a Y-chromosomal gene. This is consistent with the observations that neither duplication (47,XYY karyotype) nor deletion (45,X karyotype) of the entire Y chromosome and thus of all its genes is associated with hearing impairment, implying that loss of function or duplication of a Y-chromosomal gene is unlikely to underlie the DFNY1 phenotype. The complexity of the rearrangement is also consistent with its rarity: Although male-line deafness should be an easily recognizable phenotype, to our knowledge only a single additional family with a similar phenotype has been reported.17 The relationship between the two families is unknown; the second family is also Chinese, but from a different ethnic group (Tujia instead of Han). However, the audiological characteristics are similar, and on the basis of current knowledge it is impossible to exclude a common origin. Despite the rarity of this specific rearrangement, we nevertheless view the acquisition of autosomal sequences by the Y chromosome as a standard part of Y-chromosomal evolution, usually neutral and occasionally reaching fixation, although in the DFNY1 case it is disadvantageous. Indeed, it is noteworthy that the Y chromosome carries a fixed insertion of ∼100 kb from chromosomal region 1q43 in proximal Yq,18 close to the DFNY1 insertion. This observation raises the question of whether proximity in the nucleus might favor DNA transfer between these two chromosomes.19 The proposed DFNY1 mutational mechanism, FoSTeS, has previously been associated only with intrachromosomal rearrangements,13 so the current study expands its influence to include interchromosomal rearrangements; it also highlights the need for a better understanding of the neglected relationship between copy-number variation and hearing impairment.20
Acknowledgments
We thank the DFNY1 family for participating in this project, Rebecca Curley for early CGH experiments, Oliver Dovey for help with Southern hybridization, and the Sanger Faculty Small Sequencing Projects Group for capillary sequencing. This work was supported by a Joint Project from The Royal Society and the Natural Science Foundation of China key Project (No. 30830104), by a Beijing Nature Science Technology Major Project (grant No. 070002), and by The Wellcome Trust (098051).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
NCBI RefSeqGene, http://www.ncbi.nlm.nih.gov/refseq/rsg/
Accession Numbers
The GenBank accession numbers for the two junction sequences reported in this paper are KC489797–KC489798.
References
- 1.Stern C. The problem of complete Y-linkage in man. Am. J. Hum. Genet. 1957;9:147–166. [PMC free article] [PubMed] [Google Scholar]
- 2.Lee A.C., Kamalam A., Adams S.M., Jobling M.A. Molecular evidence for absence of Y-linkage of the hairy ears trait. Eur. J. Hum. Genet. 2004;12:1077–1079. doi: 10.1038/sj.ejhg.5201271. [DOI] [PubMed] [Google Scholar]
- 3.Skaletsky H., Kuroda-Kawaguchi T., Minx P.J., Cordum H.S., Hillier L., Brown L.G., Repping S., Pyntikova T., Ali J., Bieri T. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–837. doi: 10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- 4.Jobling M.A., Lo I.C., Turner D.J., Bowden G.R., Lee A.C., Xue Y., Carvalho-Silva D., Hurles M.E., Adams S.M., Chang Y.M. Structural variation on the short arm of the human Y chromosome: Recurrent multigene deletions encompassing Amelogenin Y. Hum. Mol. Genet. 2007;16:307–316. doi: 10.1093/hmg/ddl465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Page D.C. 2003 Curt Stern Award address. On low expectation exceeded: Or, the genomic salvation of the Y chromosome. Am. J. Hum. Genet. 2004;74:399–402. doi: 10.1086/382659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q.J., Lu C.Y., Li N., Rao S.Q., Shi Y.B., Han D.Y., Li X., Cao J.Y., Yu L.M., Li Q.Z. Y-linked inheritance of non-syndromic hearing impairment in a large Chinese family. J. Med. Genet. 2004;41:e80. doi: 10.1136/jmg.2003.012799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Q.J., Rao S.Q., Zhao Y.L., Liu Q.J., Zong L., Han M.K., Han D.Y., Yang W.Y. The large Chinese family with Y-linked hearing loss revisited: Clinical investigation. Acta Otolaryngol. 2009;129:638–643. doi: 10.1080/00016480802216347. [DOI] [PubMed] [Google Scholar]
- 8.Xue Y., Wang Q., Long Q., Ng B.L., Swerdlow H., Burton J., Skuce C., Taylor R., Abdellah Z., Zhao Y., Asan Human Y chromosome base-substitution mutation rate measured by direct sequencing in a deep-rooting pedigree. Curr. Biol. 2009;19:1453–1457. doi: 10.1016/j.cub.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jobling M.A., Tyler-Smith C. The human Y chromosome: An evolutionary marker comes of age. Nat. Rev. Genet. 2003;4:598–612. doi: 10.1038/nrg1124. [DOI] [PubMed] [Google Scholar]
- 10.Ning Z., Cox A.J., Mullikin J.C. SSAHA: A fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korbel J.O., Urban A.E., Affourtit J.P., Godwin B., Grubert F., Simons J.F., Kim P.M., Palejev D., Carriero N.J., Du L. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y.-G., Chen Y. High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. Biotechniques. 2007;43:649–650. doi: 10.2144/000112601. 652, 654 passim. [DOI] [PubMed] [Google Scholar]
- 13.Hastings P.J., Ira G., Lupski J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathias N., Bayés M., Tyler-Smith C. Highly informative compound haplotypes for the human Y chromosome. Hum. Mol. Genet. 1994;3:115–123. doi: 10.1093/hmg/3.1.115. [DOI] [PubMed] [Google Scholar]
- 15.Vodicka R., Vrtel R., Dusek L., Singh A.R., Krizova K., Svacinova V., Horinova V., Dostal J., Oborna I., Brezinova J. TSPY gene copy number as a potential new risk factor for male infertility. Reprod. Biomed. Online. 2007;14:579–587. doi: 10.1016/s1472-6483(10)61049-8. [DOI] [PubMed] [Google Scholar]
- 16.Moreno-Pelayo M.A., Modamio-Høybjør S., Mencía A., del Castillo I., Chardenoux S., Fernández-Burriel M., Lathrop M., Petit C., Moreno F. DFNA49, a novel locus for autosomal dominant non-syndromic hearing loss, maps proximal to DFNA7/DFNM1 region on chromosome 1q21-q23. J. Med. Genet. 2003;40:832–836. doi: 10.1136/jmg.40.11.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu S., Yan J., Wang X., Dong J., Chen P., Wang C., Chen G. The audiological characteristics of a hereditary Y-linked hearing loss in a Chinese ethnic Tujia pedigree. Int. J. Pediatr. Otorhinolaryngol. 2011;75:202–206. doi: 10.1016/j.ijporl.2010.10.035. [DOI] [PubMed] [Google Scholar]
- 18.Wimmer R., Kirsch S., Rappold G.A., Schempp W. Direct evidence for the Homo-Pan clade. Chromosome Res. 2002;10:55–61. doi: 10.1023/a:1014222311431. [DOI] [PubMed] [Google Scholar]
- 19.Bolzer A., Kreth G., Solovei I., Koehler D., Saracoglu K., Fauth C., Müller S., Eils R., Cremer C., Speicher M.R., Cremer T. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 2005;3:e157. doi: 10.1371/journal.pbio.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman T.B., Griffith A.J. Human nonsyndromic sensorineural deafness. Annu. Rev. Genomics Hum. Genet. 2003;4:341–402. doi: 10.1146/annurev.genom.4.070802.110347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.