

Abstract
Anophthalmia and microphthalmia (A/M) are early-eye-development anomalies resulting in absent or small ocular globes, respectively. A/M anomalies occur in syndromic or nonsyndromic forms. They are genetically heterogeneous, some mutations in some genes being responsible for both anophthalmia and microphthalmia. Using a combination of homozygosity mapping, exome sequencing, and Sanger sequencing, we identified homozygosity for one splice-site and two missense mutations in the gene encoding the A3 isoform of the aldehyde dehydrogenase 1 (ALDH1A3) in three consanguineous families segregating A/M with occasional orbital cystic, neurological, and cardiac anomalies. ALDH1A3 is a key enzyme in the formation of a retinoic acid gradient along the dorso-ventral axis during early eye development. Transitory expression of mutant ALDH1A3 open reading frames showed that both missense mutations reduce the accumulation of the enzyme, potentially leading to altered retinoic acid synthesis. Although the role of retinoic acid signaling in eye development is well established, our findings provide genetic evidence of a direct link between retinoic-acid-synthesis dysfunction and early-eye-development anomalies in humans.
Main Text
Anophthalmia and microphthalmia (A/M) are rare errors of eye development (with a combined prevalence of 3:100,000 to 30:100,000).1 Whereas true anophthalmia refers to the histological absence of ocular tissue in the orbit, microphthalmia ranges from extreme cases with only tiny or no visible remnants of globe in the orbit (also called “clinical anophthalmia”) to simple microphthalmia with structurally normal small eyes (axial length at least 2 SD below the mean for an individual of that age).1 A/M may affect one or both eyes. Both errors are associated with systemic anomalies in at least 50% of individuals,2 a large proportion of such individuals presenting with defined syndromes.1,3–5 Learning difficulties are described in one-fifth of individuals with A/M and/or coloboma.6 This frequency is probably higher in individuals with A/M when isolated colobomia cases are excluded. Both genetic and environmental factors contribute to A/M. Monogenic A/M is inherited as autosomal-dominant (AD), autosomal-recessive (AR), or X-linked traits. Both AD and AR forms are characterized by clinical and genetic heterogeneity.3 To date, mutations in genes critical for normal eye development have been identified in 20%–40% of A/M cases, some genes being involved in simple, complex, and syndromic A/M.7–10 With the exception of SOX2 mutations, which underlie 10%–15% of severe A/M cases, mutations in other genes are, individually, uncommon causes of the disease.3,10–12
Using a combination of the Affymetrix GeneChip Human Mapping 10K 2.0 Array and microsatellite markers, we performed homozygosity mapping in a multiplex inbred Pakistani pedigree with multiple loops of consanguinity (Figure 1) in whom Sanger sequencing failed to detect mutations in GDF6 (MIM 601147), FOXE3 (MIM 601094), OTX2 (IM 600037), PAX6 (MIM 607108), RAX (MIM 601881), SOX2 (MIM 184429), and VSX2 (CHX10, [MIM 142993]). Informed consent was obtained from each individual participating in this study, which was approved by Le Comité de Protection des Personnes Ile de France II or by the Cambridgeshire 1 Multicenter Research Ethics Committee (04/Q0104/129). Considering that A/M-causing mutations are rare, we assumed that affected individuals of the two available nuclear families (IV1, IV4, and IV5; Figure 1) were most likely homozygous for the same disease-causing mutation and surrounding SNP markers. This strategy defined three regions with LOD scores ≥ 3 (2p24.2–p24.1, 5.5Mb; 8q24, 0.7Mb; and 15q26.3, 3.8Mb) and one region with a LOD score close to 3 (4p14–p11, 10.1Mb) (Figure 1A). Analysis of highly informative microsatellite markers in each candidate region allowed us to show that an apparent linkage on chromosomes 2p24, 8q24, and 4p14–p11 resulted from uninformative SNP markers (data not shown). Conversely, homozygosity for informative markers of the 15q26.3 locus was confirmed (Figure 1B).
Figure 1.
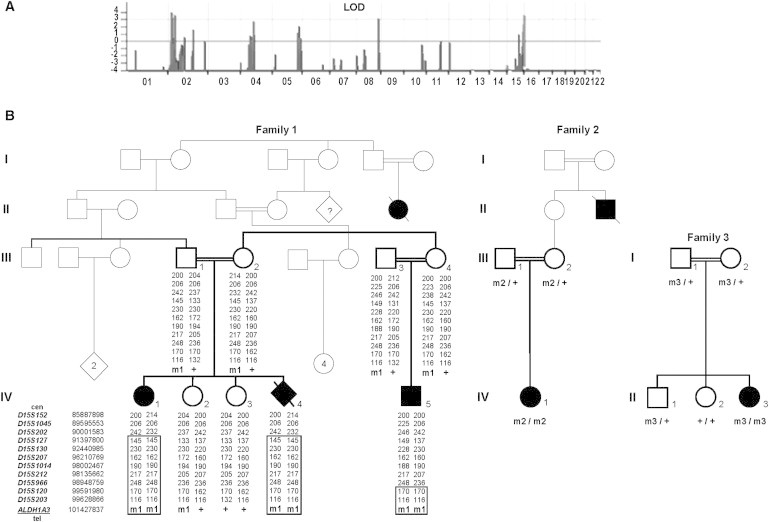
Linkage Analysis, Pedigree, and Segregation Analysis in A/M Families with Homozygous ALDH1A3 Mutations
(A) Full parametric linkage analysis of family 1 using a combination of Affymetrix GeneChip Human Mapping 10K 2.0 Arrays. Parametric LOD scores were calculated with the MERLIN software program.
(B) Pedigree, haplotype, and/or segregation analyses of ALDH1A3 mutations in the three A/M families. The positions of microsatellite markers of the 15q26.3 region based on hg19 assembly of the human genome were obtained from the: Human (Homo sapiens) Genome Browser Gateway at UCSC. ml, c.265C>T (p.Arg89Cys); m2, c.1477G>C (p.Ala493Pro); m3, c.475+1G>T; +, wild-type allele.
The critical interval on 15q26.3 spanned 3.8 Mb and contained 31 genes. To identify the disease-causing mutation, we subjected the DNA of the index case, IV1, to whole-exome sequencing by using the SureSelectXT Human All Exon V3 50 Mb target-enrichment kit (Agilent Technologies, Massy, France) in accordance with the manufacturer’s recommendations. Each genomic DNA fragment was sequenced with the use of the paired-end strategy and an average read length of 75 bases (Illumina HISEQ, Illumina, San Diego, CA, USA). Image analysis and base calling were performed with the Illumina Sequence Control Software (SCS) with Real Time Analysis (RTA) version 1.9 and default parameters were used. Sequences were aligned to the human genome reference sequence (hg19 assembly), and SNPs were called on the basis of allele calls and read depth with the use of the CASAVA pipeline (Consensus Assessment of Sequence and Variation 1.8, Illumina). Genetic variation annotation was performed with an in-house pipeline (Plateforme Bioinformatique Paris Descartes, Paris, France).
Considering that A/M-causing mutations are uncommon, we searched for homozygous variants absent in the dbSNP132, Exome Variant Server, 1000 Genomes, and in-house databases or with allelic frequencies <0.01. We found no homozygosity for consensus splice-site changes, nonsense mutations, or insertions or deletions in coding regions upon whole-genome analysis. We subsequently selected nonsynonymous changes predicted to be “damaging” or “possibly damaging” using the Polyphen and SIFT programs available through our in-house analysis pipeline. This led to the selection of 28 variants in 27 genes. The only gene mapping in the 3.8 Mb interval on chromosome 15q26.3, ALDH1A3 (NM_000693.2 [MIM600463]), harbored a homozygote missense mutation, c.265C>T (p.Arg89Cys) (Table S1, available online). The mutation was confirmed by Sanger sequencing, and familial analysis confirmed the biparental transmission of the mutation and segregation with the disease (Figure 1).
ALDH1A3 encodes a retinaldehyde dehydrogenase (ALDH1A3; also referred to as RALDH1A3, RALDH3, or ADLH6) involved in retinoic acid synthesis through the oxidation of retinaldehyde. It plays a pivotal role in retinoic acid signaling in eye development.13–17 Thus, ALDH1A3 was regarded a strong candidate gene by virtue of both its localization and its function.
We performed Sanger sequencing of all 13 exons and the intron-exon boundaries of ALDH1A3 (Table S2) in a series of 28 additional A/M index individuals born to consanguineous parents with no mutation in GDF6, FOXE3, OTX2, PAX6, RAX, SOX2, or VSX2. We identified homozygous ALDH1A3 mutations in 2 of 28 simplex individuals (Figure 1). The first simplex individual, a girl born to Turkish parents, harbored a homozygous missense mutation, c.1477G>C (p.Ala493Pro) (IV1, family 2). The second individual, a girl born to Moroccan parents, harbored a homozygous splice-site mutation, c.475+1G>T (II3, family 3). Biparental transmission was confirmed in the two families.
The three mutations were absent in the SNP databases, in the Exome Variant Server, and in 200 control chromosomes. The mutations were analyzed with Alamut Mutation Interpretation software, a decision-support system for mutation interpretation based on Align DGVD, Polyphen-2, SIFT, SpliceSiteFinder-like, MaxEntScan, NNSPLICE, and Human Splicing Finder. The c.475+1G>T mutation is predicted to abolish the splice-donor site of intron 4. This is expected to result in an in-frame skipping of exon 5, which contributes to the nicotinamide adenine dinucleotide (NAD) binding pocket (exon 5 codes for residues 159–179; NAD binding pocket: residues 8–135 and 159–170; Protein Data Bank [PDB] 1BXS). The c.265C>T (p.Arg89Cys) mutation affects an amino acid conserved from human to worm and in the ALDH1A1 and ALDH1A2 paralogs. The c.1477G>C (p.Ala493Pro) mutation changes a residue conserved from human to frog. In fish, fly, and C. elegans, the p.Ala493 residue is replaced by a leucine or a glycine residue. The replacement of the positively charged p.Arg89 by a cysteine residue and the replacement of p.Ala495 by a proline residue were both predicted to be deleterious by the Alamut Interpretation software.
The three-dimensional structure of the tetrameric human ALDH1A3 (residues 20–511; Figure S1A) was modeled by comparative protein modeling methods and energy minimization with the use of the Swiss-Model program in the automated mode.18–20 The 2.35 Å coordinate set for the tetrameric sheep liver class 1 aldehyde dehydrogenase with bound NAD (PDB code 1BXS) was used as a template for modeling the human ALDH1A3 protein (71.3% sequence identity). Swiss-PdbViewer 3.7 was used for structural insight into ALDH1A3 substitutions. The p.Arg89 residue is located between two helixes in a conserved loop of the NAD binding domain. Molecular modeling of the wild-type (WT) protein suggests that the p.Arg89 amino acid is involved in the stabilization of the tetramer through its interaction with p.Asn511 residue at a distance of 5 Å in another subunit (Figure S1B). This latter residue is located in a loop connecting the two β sheets of the monomeric oligomerization domain. Thus, the p.Arg89Cys substitution most likely affects the stability of the tetramer. The p.Ala493 residue is located in a small helix between two β sheets involved in the oligomerization domain of the enzyme (Figure S1C). The introduction of a proline at position 493 in the helix is expected to introduce an elbow, leading to an incorrect position of the two β sheets relative to each other and hampering the tetramerization of the enzyme.
Using site-directed mutagenesis (QuikChange II Site-Directed Mutagenesis Kit, Agilent), we introduced the c.265C>T (p.Arg89Cys) and c.1477G>C (p.Ala493Pro) A/M mutations into a pCMV6-entry eukaryote expression vector that encodes the full ALDH1A3 open reading frame and is fused to c-Myc (Origen, Rockville, MD, USA). We also generated a construct harboring a mutation that affected the p.Trp180 (c.538T>G; p.Trp180Gly) residue. This residue corresponds to the p.Trp168 amino acid, which is directly involved in the fixation of NAD in the sheep ALDH1 and does not interfere with the oligomerization of the protein. Primers used for mutagenesis are shown in Table S3. We assessed the effect of the ALDH1A3 substitutions on mRNA and protein levels in human embryonic kidney cells (HEK293 cells) transiently cotransfected with WT or mutant ALDH1A3 constructs, and pCGA-GFP (Addgene, Cambridge, MA, USA) to normalize the data. RT-qPCR using primers specific to the c-Myc tag and the GFP normalizer (Table S4) showed no significant difference in expression between WT and mutant ALDH1A3 mRNA (Figure S2). Immunoblot analysis revealed that the c-Myc-tagged ALDH1A3 p.Arg89Cys and p.Ala493Pro mutant protein levels were strongly reduced compared to the WT and p.Trp180Gly mutant proteins, suggesting that the p.Arg89Cys and p.Ala493Pro mutant proteins might be unstable and subject to proteasomal degradation, leading to an absence or low level of high-molecular-weight complexes (Figure 2).
Figure 2.
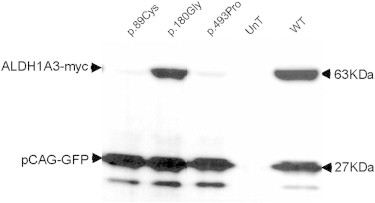
Immunoblot Analysis of Mutant and Wild-Type ALDH1A3 Proteins
HEK293 cells were transfected with pCMV6-Entry-ALDH1A3-WT-cMyc, pCMV6-Entry-ALDH1A3-Arg89CYS-aMyc, pCMV6-Entry-ALDH1A3-Trp180Gly-cMyc, and pCMV6-Entry-ALDH1A3-ALA493PRO-cMyc plasmids, respectively. The pCAG-GFP plasmid was systematically cotransfected with the pCMV6-Entry-ALDH1A3 constructs. Untransfected (UnT) cells served as controls. Total proteins were extracted and run (50 μg) on a NuPAGE 4%–12% Bis-TrisGel (Life Technologies, Cergy Pontoise, France). c-Myc-tagged proteins and GFP were detected with the use of mouse anti-cMyc (1:1,000, Santa Cruz, San Diego, CA, USA) and mouse anti-GFP (1:1,000, Roche, Meylan, France) primary antibodies, respectively, and with rabbit anti-mouse IgG-HRP (2 mg/ml, 1:5,000; Abcam, Paris, France) as a secondary antibody. Immunoblots were revealed with the use of SuperSignalWest Dura Extended Duration Substrate (Thermo Scientific, Courtaboeuf, France) and the Chemidoc XRS+ Imaging System (Bio-Rad, Marnes-la-Coquette, France). Immunoblot images were acquired and analyzed with the Image Lab software 3.0.1 build 18 (Bio-Rad). Transfections and immunoblots were performed in triplicate. The figure shows the result of one experiment. A drastic reduction in the amount of the p.Arg89Cys (p.89Cys) and p.Ala493Pro (p.493Pro) mutant proteins is shown as compared to the wild-type (WT) and p.Trp180Gly (p.180Gly) mutant proteins.
Together, our findings suggest that the synthesis of retinoic acid by ALDH1A3 is impaired in the affected individuals (n = 5) of the three families who consistently presented severe bilateral clinical anophthalmia (extreme microphthalmia with no visible ocular structure). In family 1, the A/M index individual (IV1) was born after full-term normal delivery and was in the 2nd centile for weight (2.59 kg), 3rd centile for length (46.5 cm), and 10th centile for head circumference (33.5 cm). She exhibited small optic nerves and a small optic chiasm upon a cerebral MRI at 1 week of age. Autism was diagnosed at the age of 3 years. She has two healthy sisters. Her mother’s last pregnancy before the study was terminated at 23 weeks of gestation after ultrasonography detection of apparent bilateral anophthalmia with normal brain structures (IV4) detected on ultrasonography. Her maternal cousin (IV5) was born at 34 weeks of gestation by Caesarian section and had a birth weight of 2 kg. He had severe bilateral microphthalmia with cysts (a rudimentary globe on the left and a grossly abnormal globe associated with a cyst on the right), moderate pulmonary and supravalvar pulmonary stenosis, and a moderately sized atrial septal defect. His growth progressed along the 2nd–9th centile for weight and length and 0.4th centile for head circumference. At the age of 4 years, he has a possible diagnosis of autism. The proband in family 2 (IV1) has left and extreme right microphthalmia. She had no other health problems and displays normal intelligence. A review of the family’s medical history revealed that a maternal uncle died at the age of 1 month with bilateral clinical anophthalmia, but we were unable to obtain additional clinical details. Finally, the proband in family 3, who had a homozygous c.475+1G>T mutation was born after a full term and normal delivery and had a birth weight of 3.5 kg (II3; family 3). She presented with severe right microphthalmia and severe left microphthalmia with cyst. An MRI showed dysplastic globes, a hypoplastic chiasm and optic nerves, and a normal remainder of the brain. She had no other health problems and displays normal intelligence.
In the mouse, Aldh1a3, along with Aldh1a1 and Aldh1a2, contributes to the synthesis of retinoic acid, which functions as ligand for nuclear receptors that directly regulate gene expression crucial for embryonic eye development.16,17 Aldh1a1, Aldh1a2, and Aldh1a3 are expressed with unique, nonoverlapping, spatiotemporal patterns in embryonic eyes, leading to tissue- and time-restricted retinoic acid synthesis during development.16,17 ALDH1A3 is a key enzyme in the formation of a retinoic acid gradient along the dorso-ventral axis during early eye development. It is required for the complete invagination of the ventral optic cup and closure of the choroid fissure.15–17,21,22 It also contributes to correct axonal projections of retinal cells into the brain.21,22 Interestingly, the phenotype of A/M individuals with ALDH1A3 mutations is consistent with abnormal closures of the choroid fissure and abnormal optic nerve development. Indeed, evidence of cysts in at least two individuals suggests that globe induction—and probably invagination—is present, whereas closure of the choroid fissure may be lacking. In addition, available MRIs showed hypoplastic optic nerves (n = 2).
In addition to being a key player in the developing sensory neuroepithelia of the eye, in the mouse, Aldh1a3 plays a role in the development of the nose and ear and in discrete sites within the CNS.16,17 However, knockout of Aldh1a3 causes malformations restricted to ocular and nasal regions.21,22 Aldh1a3−/− mutant embryos begin the process of optic cup formation, but they exhibit ventral retina shortening associated with lens rotation and persistence of primary vitreous.16,21 In addition, the knockout causes choanal atresia, which is responsible for respiratory distress and the death of Aldh1a3−/− mutants at birth.21 Individuals homozygous for ALDH1A3 mutations had ocular malformations but no nasal defects. Unlike the Aldh1a3 knockout mouse, the affected individuals of the three families harbor mutations expected to result in the production of mutant proteins. We cannot exclude the possibility that the complete loss of function of both ALDH1A3 alleles is lethal in humans.
Two out of the four living affected individuals had autism (IV1 and IV5; family 1). The available cerebral MRI of individual IV1 revealed normal cerebellar and cerebral features. Autism and intellectual disability are not infrequent features in individuals affected with A/M.6 However, given that two A/M individuals had normal intelligence, autism may be unrelated to altered ALDH1A3 function in the CNS. Similarly, it is difficult to decide whether the cardiac anomalies noted in an individual (IV5, family 1) are caused by ALDH1A3 mutations. Nevertheless, it is worth remembering that extraocular anomalies are not uncommon in A/M and that environmental or epigenetic factors have been proposed to explain that some genes are involved in variable phenotypes.12 Additional studies will hopefully allow a more accurate clinical definition of A/M caused by ALDH1A3 mutations.
In summary, we report that mutations in ALDH1A3 cause bilateral severe microphthalmia, possibly in association with heart anomalies and autism. Interestingly, the role of retinoic acid synthesis from vitamin A in eye development is well established. Yet, the deciphering of the genetic causes underlying A/M in humans has resulted in the identification of a number of master control genes for the growth and development of eyes, downstream of retinoic acid signaling. The previous example of a direct link between vitamin A deficiency and severe hereditary developmental eye defect in humans is STRA6 [MIM 610745], the gene that encodes the membrane receptor for retinol-binding protein, which mediates cellular uptake of vitamin A,23 and mutations in this gene can cause nonsyndromic and syndromic A/M.8,9 Our data provide evidence of a direct link between retinoic acid synthesis dysfunction and eye anomalies in humans. Finally, considering that eye defects in mouse aldh1a mutants can be rescued by maternal dietary retinoic acid supplementation,22 this raises the possibility of using this strategy to prevent A/M in conceptus that harbor ALDH1A3 mutations.
Acknowledgments
We are grateful to the families for their participation in the study. We thank A. Martin and L. Harrison for research co-ordination, A. Salt and R. Collin (Moorfields Eye Hospital, London) for clinical support, and D. Robinson (Wessex Regional Genetics Laboratory, Salisbury), N. Graham (Southampton University Hospitals Trust, Southampton), E. Leclerc and B. Leloire (CRB-CHU Biobanque de Picardie, Amiens), and Soraya Sin (Laboratoire d’Oncogénétique, Hôpital René Huguenin-Institut Curie, St Cloud) for technical assistance. This work was supported by grants from Retina France, the Clinical Research Hospital Program of the French Ministry of Health (PHRC 09 109 01), the Academy of Medical Sciences/Health Foundation (NR), VICTA (Visually Impaired Children Taking Action), MACS (Microphthalmia and Anophthalmia Children’s Society), and the Hampshire and Isle of Wight Comprehensive Local Research Network.
Contributor Information
Josseline Kaplan, Email: josseline.kaplan@inserm.fr.
Jean-Michel Rozet, Email: jean-michel.rozet@inserm.fr.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Plateforme Bioinformatique Paris Descartes (BIPD), http://mendel.necker.fr/polyweb/index.html
Alamut Interpretation Software 2.0, http://alamut.interactive-biosoftware.com
Exome Variant Server, http://evs.gs.washington.edu/EVS
Genome Browser, http://genome.ucsc.edu
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Swiss-PdbViewer 3.7, http://www.expasy.org/spdbv
1000 Genomes, http://www.1000genomes.org
References
- 1.Verma A.S., Fitzpatrick D.R. Anophthalmia and microphthalmia. Orphanet J. Rare Dis. 2007;2:47. doi: 10.1186/1750-1172-2-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tucker S., Jones B., Collin R. Systemic anomalies in 77 patients with congenital anophthalmos or microphthalmos. Eye (Lond.) 1996;10:310–314. doi: 10.1038/eye.1996.65. [DOI] [PubMed] [Google Scholar]
- 3.Bardakjian T.M., Schneider A. The genetics of anophthalmia and microphthalmia. Curr. Opin. Ophthalmol. 2011;22:309–313. doi: 10.1097/ICU.0b013e328349b004. [DOI] [PubMed] [Google Scholar]
- 4.Slavotinek A.M. Eye development genes and known syndromes. Mol. Genet. Metab. 2011;104:448–456. doi: 10.1016/j.ymgme.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaham O., Menuchin Y., Farhy C., Ashery-Padan R. Pax6: a multi-level regulator of ocular development. Prog. Retin. Eye Res. 2012;31:351–376. doi: 10.1016/j.preteyeres.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Morrison D., FitzPatrick D., Hanson I., Williamson K., van Heyningen V., Fleck B., Jones I., Chalmers J., Campbell H. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J. Med. Genet. 2002;39:16–22. doi: 10.1136/jmg.39.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williamson K.A., Hever A.M., Rainger J., Rogers R.C., Magee A., Fiedler Z., Keng W.T., Sharkey F.H., McGill N., Hill C.J. Mutations in SOX2 cause anophthalmia-esophageal-genital (AEG) syndrome. Hum. Mol. Genet. 2006;15:1413–1422. doi: 10.1093/hmg/ddl064. [DOI] [PubMed] [Google Scholar]
- 8.Chassaing N., Golzio C., Odent S., Lequeux L., Vigouroux A., Martinovic-Bouriel J., Tiziano F.D., Masini L., Piro F., Maragliano G. Phenotypic spectrum of STRA6 mutations: from Matthew-Wood syndrome to non-lethal anophthalmia. Hum. Mutat. 2009;30:E673–E681. doi: 10.1002/humu.21023. [DOI] [PubMed] [Google Scholar]
- 9.Chassaing N., Ragge N., Kariminejad A., Buffet A., Ghaderi-Sohi S., Martinovic J., Calvas P. Mutation analysis of the STRA6 gene in isolated and non-isolated anophthalmia/microphthalmia. Clin. Genet. 2012;9999 doi: 10.1111/j.1399-0004.2012.01904.x. [DOI] [PubMed] [Google Scholar]
- 10.Ragge N.K., Brown A.G., Poloschek C.M., Lorenz B., Henderson R.A., Clarke M.P., Russell-Eggitt I., Fielder A., Gerrelli D., Martinez-Barbera J.P. Heterozygous mutations of OTX2 cause severe ocular malformations. Am. J. Hum. Genet. 2005;76:1008–1022. doi: 10.1086/430721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ragge N.K., Lorenz B., Schneider A., Bushby K., de Sanctis L., de Sanctis U., Salt A., Collin J.R., Vivian A.J., Free S.L. SOX2 anophthalmia syndrome. Am. J. Med. Genet. A. 2005;135:1–7. doi: 10.1002/ajmg.a.30642. discussion 8. [DOI] [PubMed] [Google Scholar]
- 12.Bakrania P., Robinson D.O., Bunyan D.J., Salt A., Martin A., Crolla J.A., Wyatt A., Fielder A., Ainsworth J., Moore A. SOX2 anophthalmia syndrome: 12 new cases demonstrating broader phenotype and high frequency of large gene deletions. Br. J. Ophthalmol. 2007;91:1471–1476. doi: 10.1136/bjo.2007.117929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grün F., Hirose Y., Kawauchi S., Ogura T., Umesono K. Aldehyde dehydrogenase 6, a cytosolic retinaldehyde dehydrogenase prominently expressed in sensory neuroepithelia during development. J. Biol. Chem. 2000;275:41210–41218. doi: 10.1074/jbc.M007376200. [DOI] [PubMed] [Google Scholar]
- 14.Graham C.E., Brocklehurst K., Pickersgill R.W., Warren M.J. Characterization of retinaldehyde dehydrogenase 3. Biochem. J. 2006;394:67–75. doi: 10.1042/BJ20050918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cvekl A., Wang W.L. Retinoic acid signaling in mammalian eye development. Exp. Eye Res. 2009;89:280–291. doi: 10.1016/j.exer.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duester G. Keeping an eye on retinoic acid signaling during eye development. Chem. Biol. Interact. 2009;178:178–181. doi: 10.1016/j.cbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuhrmann S. Eye morphogenesis and patterning of the optic vesicle. Curr. Top. Dev. Biol. 2010;93:61–84. doi: 10.1016/B978-0-12-385044-7.00003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnold K., Bordoli L., Kopp J., Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 19.Kiefer F., Arnold K., Künzli M., Bordoli L., Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37(Database issue):D387–D392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peitsch M.C. Protein modeling by E-mail. Nature Biotechnology. 1995;13:658–660. [Google Scholar]
- 21.Dupé V., Matt N., Garnier J.M., Chambon P., Mark M., Ghyselinck N.B. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc. Natl. Acad. Sci. USA. 2003;100:14036–14041. doi: 10.1073/pnas.2336223100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molotkov A., Molotkova N., Duester G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development. 2006;133:1901–1910. doi: 10.1242/dev.02328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawaguchi R., Yu J., Honda J., Hu J., Whitelegge J., Ping P., Wiita P., Bok D., Sun H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. 2007;315:820–825. doi: 10.1126/science.1136244. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.