

Abstract
Autosomal-recessive cerebellar ataxia (ARCA) comprises a large and heterogeneous group of neurodegenerative disorders with more than 20 different forms currently recognized, many of which are also associated with increased tone and some of which have limb spasticity. Gaucher disease is a lysosomal storage disease resulting from a defect in the enzyme acid β-glucosidase 1. β-glucosidase 2 is an enzyme with similar glucosylceramidase activity but to date has not been associated with a monogenic disorder. We studied four unrelated consanguineous families of Tunisian decent diagnosed with cerebellar ataxia of unknown origin. We performed homozygosity mapping and whole-exome sequencing in an attempt to identify the genetic origin of their disorder. We were able to identify mutations responsible for autosomal-recessive ataxia in these families within the gene encoding β-glucosidase 2, GBA2. Two nonsense mutations (c.363C>A [p.Tyr121∗] and c.1018C>T [p.Arg340∗]) and a substitution (c.2618G>A [p.Arg873His]) were identified, probably resulting in nonfunctional enzyme. This study suggests GBA2 mutations are a cause of recessive spastic ataxia and responsible for a form of glucosylceramide storage disease in humans.
Main Text
Autosomal-recessive cerebellar ataxias (ARCA) belong to the large group of disorders known as the inherited ataxias.1 More than 20 different forms of ARCA are currently recognized.2 Symptoms start in childhood and include balance abnormalities, incoordination, and dysarthria. Cerebellum and/or spinocerebellar tracts are involved.3,4 Many of these individuals with recessive ataxia can additionally present with increased tone in the limbs and some, often later in the disease, develop pronounced limb spasticity with clinical signs such as the Hoffman sign and have extensor planters. These individuals and families are often called spastic ataxias.5 Five main pathogenic mechanisms may be distinguished: defective DNA repair, abnormal protein folding and degradation, channelopathies, and mitochondrial and metabolic defects.6 Metabolic ataxias include lipid metabolism, peroxysomal, and storage diseases.6 Although mutations in a number of genes have been identified as causes of ARCA, there are still many families and individuals remaining with an unknown etiology.
In this study, we performed homozygosity mapping and whole-exome sequencing in an attempt to identify the genetic origin of autosomal-recessive ataxia in three Tunisian families. This work was approved by the local ethics committee and by the Office of Human Subjects Research at the National Institutes of Health and all affected individuals gave informed consent. We initially studied three unrelated consanguineous families (C, E, and I) of Tunisian decent in which seven individuals were previously diagnosed with autosomal-recessive cerebellar ataxia. All affected individuals presented with childhood- or juvenile-onset cerebellar ataxia and brisk tendon reflexes as discussed below. Blood was collected from the affected individuals and six unaffected members of families C, E, and I. The pedigrees of these families are shown in Figure 1 and the clinical features are summarized in Table 1. Genomic DNA was extracted from peripheral blood lymphocytes by standard protocols. Linkage to 16 known loci for cerebellar ataxia has been previously excluded in these families and the Friedreich ataxia expansion was not detected.7
Figure 1.
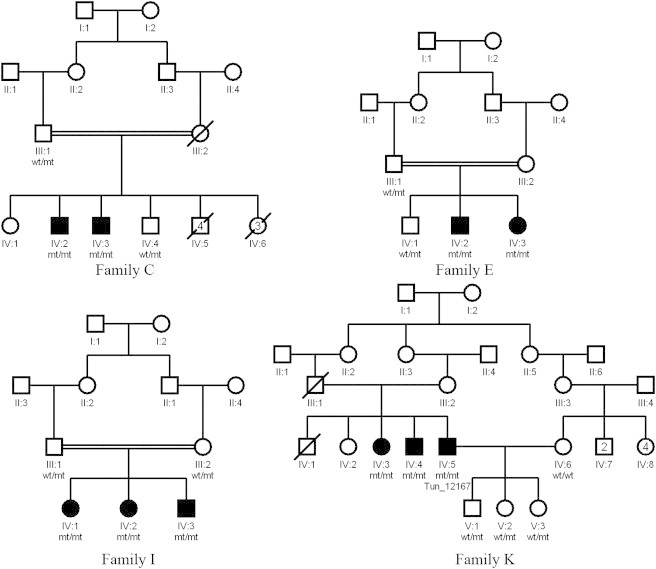
Pedigrees of Four Families with ARCA
Affected family members are shaded. Males are represented by squares, females are represented by circles. Mutation status is denoted as follows: WT, wild-type; mt, mutant.
Table 1.
Clinical Data Summary
| Family | C | E | I | K | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ID | C.IV:2 | C.IV:3 | E.IV:2 | E.IV:3 | I.IV.1 | I.IV.2 | I.IV:3 | K.IV.3 | K.IV.4 | K.IV:5 |
| Gender | M | M | M | F | F | F | M | F | M | M |
| Initial cerebellar signs | GA | GA,D,Ny | GA,D,Ny | GA | GA,D,Ny | GA | GA | GA | GA | GA |
| Limb ataxia | P | P | P | P | P | P | P | N/A | P | P |
| Age at onset (years) | 4 | 4 | 10 | 8 | 5 | 3 | 6 | 14 | 15 | 15 |
| Age at last exam (years) | 35 | 33 | 41 | 37 | 25 | 25 | 12 | 32 | 34 | 40 |
| DD (years) | 31 | 29 | 31 | 29 | 20 | 22 | 6 | 18 | 19 | 25 |
| ICARS score/100 | 36 | 32 | 53 | 28 | 21 | 10 | 6 | N/A | 33 | 29 |
| ICARS subsection scoresa | 12, 22, 2, 0 | 6, 14, 2, 0 | 21, 22, 4, 6 | 6, 22, 0, 0 | 9, 10, 1, 1 | 19, 11, 4, 0 | 2, 4, 0, 0 | N/A | 12, 17, 4, 0 | 12, 15, 2, 0 |
| ICARS score/DD | 1.16 | 1.1 | 1.7 | 0.97 | 1.05 | 1.43 | 1 | N/A | 1.74 | 1.16 |
| TR knee | brisk | brisk | brisk | brisk | brisk | brisk | brisk | brisk | brisk | brisk |
| Ankle | brisk | brisk | brisk | brisk | brisk | brisk | brisk | brisk | brisk | brisk |
| Pyr S Babinski | P | P | P | P | P | P | P | P | P | P |
| Spasticity | P | P | P | P | P | P | A | P | P | P |
| DSD position error | A | P | P | P | P | P | P | P | P | P |
| Vibration sense | P | A | P | A | N/A | N/A | N/A | N/A | N/A | N/A |
| Distal limb amyotrophy | A | A | A | A | A | A | P | N/A | A | A |
| Skel. def. PC | A | A | PC | PC | A | A | A | A | A | A |
| Scoliosis | P | A | P | A | A | A | A | A | A | A |
| Sphincter disturbance | A | A | P | A | A | A | A | N/A | A | P |
| EMG at last exam | neurogenic plot | neurogenic plot | neurogenic plot | neurogenic plot | N/A | N/A | severe axonal sensory motor neuropathy | axonal sensory neuropathy | severe axonal sensory motor neuropathy | severe sensory neuropathy |
| ENG | central vestibular syndrome | central vestibular syndrome | central vestibular syndrome | central vestibular syndrome | N/A | N/A | N/A | N/A | N/A | N/A |
| EP SEP | N/A | N/A | N/A | N/A | Abn | Abn | N/A | N/A | N/A | Abn |
| VEP | N/A | N/A | N/A | N/A | N | N | N/A | N | N/A | Abn |
| AEP | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N | N/A | N |
| Cholesterol | N | N | N | N | N/A | N/A | N/A | N | N/A | N |
| Vit. E (N: 7–15 mg/L) | 6.48 | 6.07 | N/A | N/A | 9 | 6.91 | 5.82 | N/A | N/A | N/A |
| Vit. A (N: 0.3–0.7 mg/L) | 0.41 | 0.42 | N/A | N/A | 0.27 | 0.23 | 0.45 | N/A | N/A | N/A |
| Other clinical signs | none | none | mild intellectual disability, bilateral Hoffman sign | none | none | head tremor | Hoffman sign, hepato-megaly | polycystic kidney | intermittent head tremor | intermittent trunk ataxia and head tremor, bilateral Hoffman sign, right visual loss |
Abbreviations are as follows: A, absent; Abn, abnormal; AEP, auditory evoked potentials; D, dysarthria; DD, disease duration; DSD, deep sensory disturbances; EMG, electromyography; ENG, electronystagmogram; EP, evoked potentials; F, female; GA, gait ataxia; ICARS, International Cooperative Ataxia Rating Scale; M, male; N, normal; N/A, not assessed; Ny, nystagmus; P, present; PC, pes cavus; Pyr S, pyramidal sign; Skel. def., skeletal deformities; TR, tendon reflexes; SEP, somatosensory evoked potentials; VEP, visual evoked potentials; Vit, vitamin.
Separate ICARS scores are as follows: (postural and gait disturbance)/34, (limb ataxia)/52, (dysarthria)/8, (oculomotor disorders)/6.
High-density SNP genotyping is a rapid and effective method for mapping autozygous regions of the genome,8 so we undertook genome-wide SNP genotyping in all available affected and unaffected individuals belonging to families C, E, and I. Genotyping was performed with the OmniExp-12,v1.0 DNA Analysis BeadChip (Illumina Inc., San Diego, CA) according to the manufacturer’s instructions. Because the families studied were highly consanguineous, SNP array data was subjected to homozygosity mapping with the Homozygosity Mapper software by using only homozygous stretches of 15 alleles or longer.9 For each family, we searched for shared homozygous regions present among the affected individuals and absent within the unaffected family members. This revealed one large region of homozygosity on chromosome 9 (Figure S1 available online), spanning from rs3936927 (chr9: 31,066,983 bp) to rs4646770 (chr9: 38,383,171 bp); notably, although disease in each of the families segregated with this region, the genotypes observed between families E and I were identical, suggesting a shared common founder. APTX (MIM 606350), a gene mutated in ataxia with oculomotor apraxia type 1 (AOA1 [MIM 208920]), was present in this candidate region, but Sanger sequencing of APTX in the index individuals did not reveal mutations.
In an attempt to rapidly identify the underlying genetic mutation, we performed whole-exome sequencing on the DNA of each of the seven affected individuals belonging to families C, I, and E according to the Nimblegen protocol (Nimblegen v2.0, Roche Nimblegen, Indianapolis, IN). Prior to sequencing, DNA templates were bridge amplified to form clonal clusters inside a flowcell via the cBot cluster generation process. The flowcells were then loaded into the next-generation sequencer Illumina HiSeq 2000. Paired end sequence reads were aligned with BWA against the reference human genome (UCSC hg18).10 Duplicate read removal, format conversion, and indexing were performed with Picard. The Genome Analysis Toolkit was used to recalibrate base quality scores, perform local realignments around possible indels, and to call and filter the variants.11,12 Overall, more than 200 million sequencing reads were produced for each sample, covering more than 12 billion bases. Approximately 98% of these were aligned to the human reference genome (hg18). On average, 92% of exome capture baits had at least 10× depth and 87% at least 30× depth. Variants previously identified in the 1000 Genomes project or within neurological normal control exome sequence within the same laboratory were excluded. VCF tools were used to annotate gene information for the remaining novel variants.11 Single-nucleotide variants that are nonsynonymous, stop loss or gain, or within essential splice sites and indels that were nonreference homozygous and shared among affected family members were prioritized as potential candidates for causal variants. These prioritized, potentially novel shared protein coding variants were then visually inspected with the Integrative Genomics Viewer (IGV).12 After filtering to identify potentially novel variants, less than 15 homozygous coding variants and less than 25 homozygous indels were left per family.
In both families E and I, the same homozygous single-nucleotide change (c.1018C>T) was identified in exon 5 of GBA2, leading to a stop codon formation (p.Arg340∗) with a predicted premature protein termination. In family C, another homozygous variant (c.2618G>A) was found in exon 17 of GBA2 resulting in the substitution of arginine for histidine at residue 873 (p.Arg873His). This variant is predicted to alter protein structure with Polyphen2 software analysis and affect a conserved residue (Figure 2). Nucleotide and protein positions of identified variants in GBA2 are based on RefSeq accession numbers NM_020944.2 and NP_065995.1 from the National Center for Biotechnology Information. Variant positions within the cDNA are numbered with the A of the translation initiation codon as position 1. Sanger sequencing of these exons was performed with ABI BigDye Terminator Cycle Sequencing Kit on an ABI 3730 sequencer. This verified that these variants segregated with disease within the families and further showed that none of these variants were present in 50 Tunisian neurologically normal controls or in 330 African, European, and Asian controls from the Human Gene Diversity Panel (HGDP). We interpreted these results to strongly suggest GBA2 mutations as the cause of ataxia in these families.
Figure 2.
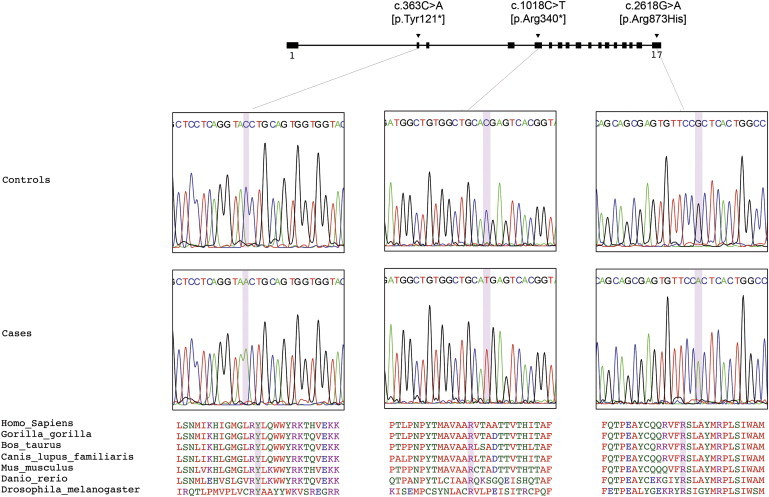
GBA2 Mutations Detected
Illustration of GBA2, which comprises 17 exons. The three mutations identified are located in exons 2, 5, and 17. In the middle portion of the figure, sequence chromatograms are shown for each of the different mutations identified. Amino acid alignment via ClustalW is shown in the lower portion of the figure and illustrates the sequence conservation across species.
Next, we screened 21 Tunisian individuals diagnosed with cerebellar ataxia of unknown origin. These samples were exome sequencing with the Truseq exome enrichment protocol according to the manufacturer’s instructions (Illumina, San Diego, CA). Alignment and variant calling was performed as described above. A third homozygous variant, c.363C>A, was identified in exon 2 of GBA2 in one of the samples (sample ID# Tun_12167) leading to a stop codon formation (p.Tyr121∗) predicted to cause a very early truncation of the protein. This variant was absent in 50 Tunisian neurologically normal controls and in 330 controls from HGDP. It affects an amino acid highly conserved across species (Figure 2). Further investigation revealed two additional affected family members and this variant demonstrated segregation with disease (family K) (Figure 1).
The initial clinical presentation was progressive cerebellar ataxia in all affected individuals (six males and four females). Onset was during childhood or early adulthood with kinetic and static cerebellar ataxia as the presenting feature associated with cerebellar dysarthria. Three of the individuals exhibited head tremor (K.IV:5, K.IV.4, I.IV:3). All individuals developed a pyramidal syndrome that consisted of spasticity in the lower limbs, brisk reflexes, extensor plantars, and Hoffman sign. In one individual, there was also external ophthalmoparesis with a jerky pursuit and slow saccades. Pes cavus and scoliosis were present in three of the individuals. One affected individual also exhibited mild intellectual disability (E.IV:2). The course of the disease was slowly progressive with an international cooperative ataxia rating scale (ICARS) score/disease duration nearing 1 to 1.5.
With disease progression, affected individuals developed an axonal sensory or sensory-motor neuropathy confirmed by nerve conduction studies. The neuropathy was variable in type and in severity. Cerebral and spinal MRI were normal in most of the individuals. However, in individual K.IV:3, the MRI showed global cerebral, cerebellar, corpus callosum, and spinal cord atrophy. No abnormalities were noted in blood sugar, vitamin A, cholesterol, triglycerides, CK, AFP levels, lipid or protein electrophoresis, or electrocardiogram in all screened individuals. We noted normal or subnormal vitamin E levels in the individuals. Individual I.IV:3 presented hepatomegaly, a frequently observed clinical feature in type 1 Gaucher disease,13 but other Gaucher features were absent in this and the other affected individuals. The overall phenotype of these Tunisian individuals is typical of the known recessive ataxias, with the addition of pathological spasticity and an axonal neuropathy.
Gaucher disease (MIM 606463) is the most common lysosomal storage disease. It results from a defect in the acid β-glucosidase 1 leading to the accumulation of glycosylceramide in lysosomes of macrophages in different organs, typically involving spleen, kidney, lungs, brain, and bone marrow;14 neuronopathic forms of Gaucher disease include progressive neurodegenerative disease. β-glucosidase 2, which harbors similar glucosylceramidase activity to β-glucosidase 1, is encoded by GBA2 (MIM 609471) and is not deficient in individuals with Gaucher disease.15 β-glucosidase 2 cleaves the sphingolipid glucosylceramide into glucose and ceramide.16 Degradation of glucosylceramide by β-glucosidase 2 leads to further metabolism of the generated ceramide to sphingomyelin.15 β-glucosidase 2 is a ubiquitous enzyme detected in all tissues and expressed maximally in brain, heart, skeletal muscle, kidney, and placenta.17 It is present in the endoplasmic reticulum (ER)16 and/or in the plasma membrane.15,18 Nonfunctional copies of GBA2 would lead to the accumulation of glycolipids and an ER storage disease.16
The tissue distribution of GBA2 mRNA is different between human and mice. The mouse Gba2 mRNA is abundant in the testis but not the human mRNA.16 Gba2 knockout mice display misshapen spermatozoa leading to an impaired fertility.16 However, one of our individuals (Tun_12167) has three children, which confirms that the glycosylceramide metabolism is less important in spermatogenesis in humans15 and suggests that β-glucosidase 2 may play a different role in humans. Homozygous Gba2 knockout mice showed no obvious neurological signs,16 although it is worth noting that the Gba2 knockout mice lacked only exons 5 to 10 and retained 50% of normal glucosidase activity. We hypothesize that the accumulation of glucosylceramides in the ER and/or plasma membrane did not reach a critical threshold necessary to drive neurological symptoms. It has also been shown that the Gba2 activity increases more than 3-fold during the neuronal differentiation19 and suggested that the accumulation of glucosylceramides in the ER of neuronal cells would change calcium homeostasis and lead to neurological symptoms.20
In summary, we describe families where ataxia was the presenting feature, but later, spasticity became very pronounced involving initially the lower limbs and later also the upper limbs. All affected individuals were homozygous for three different mutations in GBA2. Two of these (p.Arg340∗ and p.Tyr121∗) are nonsense mutations leading to a protein truncated by 67% and 86%, respectively. The resulting enzyme is very likely to be nonfunctional. The third mutation is a substitution (p.Arg873His) occurring within the COOH-terminal (residues 500–886), a region apparently very conserved across different species implying its importance in the function of the protein.17 Thus, we have shown that mutations in GBA2 are a likely cause of this disorder. The mutation mechanism is likely to be loss of function which, based on mouse work, will lead to an accumulation of glucosylceramide and subsequently the clinical phenotype.
Acknowledgments
The authors thank J. Hammer for his contribution in the correction of the manuscript and his help with the figures. We also thank the affected individuals and their families for taking part in this work. This work was supported in part by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, part of the Department of Health and Human Services (project number ZIA AG000958-09) and the National Ataxia Foundation. We are also grateful to the Medical Research Council (MRC), the Brain Research Trust, and NORD for funding to H.H., L.V.S., and A.S. and also the MRC/Wellcome Trust Parkinson’s disease consortium grant and the UCLH/UCL Department of Health’s NIHR Biomedical Research Centres funding scheme.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ClustalW2 - Multiple Sequence Alignment, http://www.ebi.ac.uk/Tools/msa/clustalw2/
HomozygosityMapper software, http://www.homozygositymapper.org/
National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net/
References
- 1.Fogel B.L., Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol. 2007;6:245–257. doi: 10.1016/S1474-4422(07)70054-6. [DOI] [PubMed] [Google Scholar]
- 2.Embiruçu E.K., Martyn M.L., Schlesinger D., Kok F. Autosomal recessive ataxias: 20 types, and counting. Arq. Neuropsiquiatr. 2009;67:1143–1156. doi: 10.1590/s0004-282x2009000600036. [DOI] [PubMed] [Google Scholar]
- 3.Palau F., Espinós C. Autosomal recessive cerebellar ataxias. Orphanet J. Rare Dis. 2006;1:47. doi: 10.1186/1750-1172-1-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sailer A., Houlden H. Recent advances in the genetics of cerebellar ataxias. Curr. Neurol. Neurosci. Rep. 2012;12:227–236. doi: 10.1007/s11910-012-0267-6. [DOI] [PubMed] [Google Scholar]
- 5.de Bot S.T., Willemsen M.A.A.P., Vermeer S., Kremer H.P.H., van de Warrenburg B.P.C. Reviewing the genetic causes of spastic-ataxias. Neurology. 2012;79:1507–1514. doi: 10.1212/WNL.0b013e31826d5fb0. [DOI] [PubMed] [Google Scholar]
- 6.De Michele G., Coppola G., Cocozza S., Filla A. A pathogenetic classification of hereditary ataxias: is the time ripe? J. Neurol. 2004;251:913–922. doi: 10.1007/s00415-004-0484-2. [DOI] [PubMed] [Google Scholar]
- 7.Bouhlal Y., El-Euch-Fayeche G., Amouri R., Hentati F. Distinct phenotypes within autosomal recessive ataxias not linked to already known loci. Acta Myol. 2005;24:155–161. [PubMed] [Google Scholar]
- 8.Gibbs J.R., Singleton A. Application of genome-wide single nucleotide polymorphism typing: simple association and beyond. PLoS Genet. 2006;2 doi: 10.1371/journal.pgen.0020150. e150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seelow D., Schuelke M., Hildebrandt F., Nürnberg P. HomozygosityMapper–an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37 doi: 10.1093/nar/gkp369. W593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., 1000 Genomes Project Analysis Group The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lachmann R.H., Wight D.G., Lomas D.J., Fisher N.C., Schofield J.P., Elias E., Cox T.M. Massive hepatic fibrosis in Gaucher’s disease: clinico-pathological and radiological features. QJM. 2000;93:237–244. doi: 10.1093/qjmed/93.4.237. [DOI] [PubMed] [Google Scholar]
- 14.Grabowski G.A., Gatt S., Horowitz M. Acid beta-glucosidase: enzymology and molecular biology of Gaucher disease. Crit. Rev. Biochem. Mol. Biol. 1990;25:385–414. doi: 10.3109/10409239009090616. [DOI] [PubMed] [Google Scholar]
- 15.Boot R.G., Verhoek M., Donker-Koopman W., Strijland A., van Marle J., Overkleeft H.S., Wennekes T., Aerts J.M.F.G. Identification of the non-lysosomal glucosylceramidase as beta-glucosidase 2. J. Biol. Chem. 2007;282:1305–1312. doi: 10.1074/jbc.M610544200. [DOI] [PubMed] [Google Scholar]
- 16.Yildiz Y., Matern H., Thompson B., Allegood J.C., Warren R.L., Ramirez D.M.O., Hammer R.E., Hamra F.K., Matern S., Russell D.W. Mutation of beta-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J. Clin. Invest. 2006;116:2985–2994. doi: 10.1172/JCI29224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matern H., Boermans H., Lottspeich F., Matern S. Molecular cloning and expression of human bile acid beta-glucosidase. J. Biol. Chem. 2001;276:37929–37933. doi: 10.1074/jbc.M104290200. [DOI] [PubMed] [Google Scholar]
- 18.Overkleeft H.S., Renkema G.H., Neele J., Vianello P., Hung I.O., Strijland A., van der Burg A.M., Koomen G.J., Pandit U.K., Aerts J.M. Generation of specific deoxynojirimycin-type inhibitors of the non-lysosomal glucosylceramidase. J. Biol. Chem. 1998;273:26522–26527. doi: 10.1074/jbc.273.41.26522. [DOI] [PubMed] [Google Scholar]
- 19.Aureli M., Gritti A., Bassi R., Loberto N., Ricca A., Chigorno V., Prinetti A., Sonnino S. Plasma membrane-associated glycohydrolases along differentiation of murine neural stem cells. Neurochem. Res. 2012;37:1344–1354. doi: 10.1007/s11064-012-0719-z. [DOI] [PubMed] [Google Scholar]
- 20.Walden C.M., Sandhoff R., Chuang C.-C., Yildiz Y., Butters T.D., Dwek R.A., Platt F.M., van der Spoel A.C. Accumulation of glucosylceramide in murine testis, caused by inhibition of beta-glucosidase 2: implications for spermatogenesis. J. Biol. Chem. 2007;282:32655–32664. doi: 10.1074/jbc.M702387200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.