

Summary
Statistical analyses of genome sequence-derived protein sequence data can identify amino acid residues that interact between proteins or between domains of a protein. These statistical methods are based on evolution-directed amino acid variation responding to structural and functional constraints in proteins. The identified residues form a basis for determining structure and folding of proteins as well as inferring mechanisms of protein function. When applied to two-component systems, several research groups have shown they can be used to identify the amino acid interactions between response regulators and histidine kinases and the specificity therein. Recently, statistical studies between the HisKA and HATPase-ATP binding domains of histidine kinases identified amino acid interactions for both the inactive and active catalytic states of such kinases. The identified interactions generated a model structure for the domain conformation of the active state. This conformation requires an unwinding of a portion of the C-terminal helix of the HisKA domain that destroys the inactive state residue contacts and suggests how signal-binding determines the equilibrium between the inactive and active states of histidine kinases. The rapidly accumulating protein sequence databases from genome, metagenome and microbiome studies are an important resource for functional and structural understanding of proteins and protein complexes in microbes.
Keywords: histidine kinase, two-component system, signal transduction, co-evolution, domain-domain interaction
Introduction
Two-component signal transduction mechanisms are woven in the fabric of cellular regulation in bacteria, plants and lower eukaryotic forms of life (Wuichet et al., 2010). These innate systems play a wide variety of roles from sensing the environment to controlling the cell cycle, cellular behavior and development. Countless publications have been written on the wide variety of phenotypes and genes controlled by these systems, firmly establishing their cellular roles (Bourret & Silversmith, 2010). Despite the copious information enumerating the physiological outputs from signal transduction, only a handful of the input signals, controlling the many thousands of two-component systems identified in bacteria, are known. Moreover, the biochemical mechanisms, through which these signals control enzymatic functions and regulate output of the system, are not fully understood as well.
The fundamental units of a two-component signal transduction system are the sensor histidine kinase and the response regulator to which it is mated. The sensor histidine kinases, in general, consist of several domains including a “sensor” domain(s) and a catalytic portion characterized by two structurally conserved domains, the HisKA (aka DHp) four-helix bundle dimerization domain and the ATP-binding HATPase (aka CA) domain (Fig. 1). There are innumerable variations on the structures of signal binding sensor domains reflecting the multiplicity of kinds and types of signals recognized by histidine kinases (Szurmant et al., 2007). For the purposes of this treatise, sensor domains will be viewed solely as undefined signal binding entities that induce conformational changes in the catalytic domains. The major questions addressed in here are how the two domains, comprising the catalytic portions of histidine kinases, rearrange into the active and inactive states in response to signals and how does the binding of signals induce this conformational switch.
Figure 1. The two-component system signal transduction cycle.
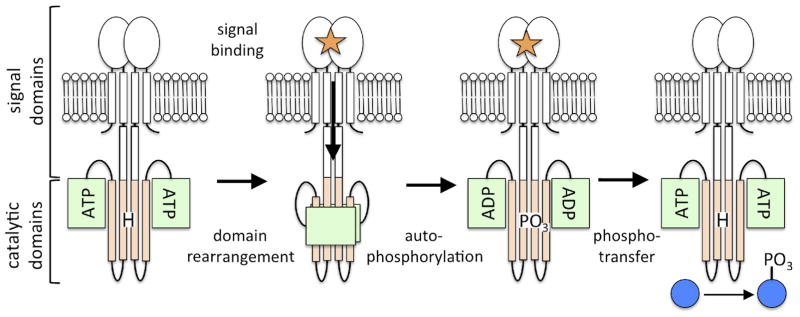
The sensor histidine kinase is typically membrane embedded and features variable numbers and types of signal domains (white) at its N-terminus. The catalytic domains at the C-terminus include the four-helix bundle HisKA domain (tan), featuring the histidine site of phosphorylation (H) and the ATP-binding HATPase domain (green). In the presence of an activation signal (orange star) a conformational change is transduced to the catalytic domains initiating domain rearrangement and subsequent autophosphorylation of the histidine residue. The phosphoryl group is transferred from the kinase to the response regulator protein (blue), which generates the appropriate response to the signal, typically by acting as a transcription factor.
A major advance in understanding biochemical mechanisms in signal transduction has come from the application of new statistical methods for analyses of protein sequences, accumulated from genome sequencing, to identify amino acid residue positions responsible for interaction between signaling proteins (White et al., 2007, Burger & van Nimwegen, 2008, Skerker et al., 2008, Weigt et al., 2009). These approaches build on earlier ones employed on a variety of individual proteins to infer folding and function (Suel et al., 2003, Lockless & Ranganathan, 1999, Altschuh et al., 1987, Atchley et al., 2000, Göbel et al., 1994). Employing these methods to determine how protein domains interact and transfer information in the sensor histidine kinases of two-component signal transduction has revealed a model of the structure of the active domain configuration and has revealed how signals switch the kinase between the active and inactive conformations (Dago et al., 2012). The evolutionary basis underlying how statistical methods reveal this information and its interpretation is described here.
Evolutionary variation identifies interacting amino acid residues
Statistical analyses of protein sequence alignments can be employed to distinguish pairs of amino acid residues that interact within proteins or between proteins from those that don’t. Since microbes have existed for several billion years, all the amino acid positions of a given protein will have been tested with all of the 20 amino acids along a functional trajectory. As a result for a 100 amino acid protein a subset of the possible 20100 sequences have been probed. Deleterious changes in residue positions of the protein will have been eliminated if these changes affected the function of the protein and the competitive advantage of an organism in its milieu. By the present day, the testing of all amino acids in every position of a protein by evolution results in each position being comprised of amino acids that are consistent with the protein’s structure and function or, at least, not deleterious to both.
Statistical programs exploit the evolution driven amino acid variation at each position of a protein by comparing it to variation at another position, to infer function. If amino acids at two different positions in a protein do not interact, they will commonly evolve independently from one another and a random assortment of amino acids will be found when comparing the two positions. There are of course exceptions, for instance residues that line a ligand binding site or an active site might have to co-evolve despite not being in direct contact (McLaughlin et al., 2012). Conversely, two amino acids that interact in a protein because of structure or function considerations will have some pairs of amino acids at the two positions that are incompatible with interaction. This is a consequence of the need for contact compatibility between the two amino acids in charge, size and hydrophobicity. Incompatible pairs will be eliminated by evolution resulting in a deviation from randomness when comparing the variation at the two positions. Thus the concept here is covariant pairing, not unlike A:T and G:C pairing in nucleic acids, but the pairing code is much more ambiguous. What the mathematics does is identify two positions subject to pairing and evolving together. The earlier local covariance statistical approaches (Lockless & Ranganathan, 1999, Altschuh et al., 1987, Göbel et al., 1994, Szurmant et al., 2008, White et al., 2007) may be used to directly identify such positions but these methods are complicated by spurious indirect correlations (Szurmant & Weigt, 2012, Morcos et al., 2011, Weigt et al., 2009). Such problems have been eliminated by use of direct coupling analysis, DCA, which amends local covariance with global statistical inference methods (Lunt et al., 2010, Weigt et al., 2009).
In practice, the amino acid sequences of proteins of identical structure and function, identified in genome sequencing data, are used to build alignments of several hundred examples of the same protein from different, evolved bacterial species. These alignments may be probed for amino acid interactions within the proteins defined by the alignment, or two different protein alignments may be probed for amino acid interactions between proteins. When proteins are compared against all of the others in the alignment, a position by position comparison reveals the variation at, for example, position X compared to position Y or at any of the other positions. An informative example of a protein compared to itself may be found in the supplement in (Weigt et al., 2009). In this study of response regulators using DCA, the top 60 scores were residue pairs that make contact within the fold of the protein and those residues that interact when response regulators form a dimer, the structure of which is well characterized (Gao & Stock, 2009, Toro-Roman et al., 2005). This study revealed that DCA statistical methodology may be used to define and describe the folding of a protein. This has now been demonstrated in various recent studies applying DCA or closely related algorithms to protein folding and de novo structure prediction (Sułkowska et al., 2012, Nugent & Jones, 2012, Hopf et al., 2012, Marks et al., 2011).
DCA (Procaccini et al., 2011, Lunt et al., 2010, Weigt et al., 2009) and other related covariance analysis programs (White et al., 2007, Burger & van Nimwegen, 2008, Skerker et al., 2008) have been employed to analyze protein alignments and extract the interaction residues between the histidine kinase HisKA and response regulator domains, first identified in the co-crystal structure of Spo0B-Spo0F (Zapf et al., 2000) and, more recently, in HK853-RR468 (Casino et al., 2009). Some extensive site-directed mutagenesis experiments to rewire the partner preference between sensor kinase and response regulator RR proofed consistent with the computational efforts (Skerker et al., 2008). More recently co-variance analysis combined with mutagenesis experiments identified the residues that mediate histidine kinase homo-dimerization specificity (Ashenberg et al., 2011). These studies in their entirety confirmed that the covariance-based methods were capable of identifying amino acids responsible for interaction and specificity between two individual proteins with high fidelity. However the question arose how well this method would work to reveal the interaction residues between two protein domains tethered together by a linker such as is found in multi-domain histidine kinases.
Application of DCA to domain interactions in histidine kinases
A major unanswered question in histidine kinase biochemical mechanisms is how the binding of signals to the sensor domain influences the spatial relationships of the catalytic domains to control autophosphorylation (Stewart, 2010). The two catalytic domains in question are the HisKA and HATPase domains connected by a flexible linker. Crystallographic experiments revealed the spatial relationships of these two domains in the inactive state of HK853 (Marina et al., 2005). In the inactive state, the HATPase domain is not in the correct position to present ATP to the histidine residue on the HisKA domain. The phosphorylation active state of histidine kinases with ATP, bound by the HATPase domain, positioned to react with the conserved histidine of the HisKA domain, has not been captured in a crystal; perhaps because of instability in transient active structures. DCA analyses designed to reveal this latter structure were undertaken based on two premises; (A) the structure of the active phosphorylation complex is likely to be highly ordered and conserved among histidine kinases given the requirement to bring together two domains, a histidine residue and ATP into juxtaposition, and (B) the active structure will require considerable domain rearrangement from the inactive state, suggesting a comparison of the spatial arrangement of the two states will indicate how signals control the switch between the two structures.
The results of DCA studies on >13,000 histidine kinase catalytic domains identified five high ranking inter-domain contacts between the HisKA and HATPase domains (Dago et al., 2012). Two of these contacts were found in the crystal structures of the inactive state of these two domains (Marina et al., 2005). This suggests that the observed spatial relationship of these domains in the inactive state crystal should be considered as a physiologically relevant structure representative for most HisKA-type histidine kinases and not a potential artifact of crystallization. The other three inter-domain contacts were not consistent with this inactive state structure and were presumed to be contacts made in the active configuration of these domains. The problem now arose whether this supposition was true and how to prove or disprove it.
Two independent methods were applied to generate an active configuration and test its validity. A constrained docking approach (Schug et al., 2010, Schug et al., 2009) was used to obtain a structural model consistent with the three inter-domain contacts and the ATP-His distance, which was further refined by unrestrained molecular dynamics simulations. This putative active structure of the two domains was tested in mutagenesis experiments that placed non-compatible amino acid pairings at the contact positions under the premise they should prevent the interaction and destroy autophosphorylation. This expectation was realized using the Bacillus subtilis KinA histidine kinase; non-compatible amino acid pairs inactivated the kinase in vitro and in vivo. Moreover, an inactive hybrid kinase consisting of KinA HisKA domain with the closely related KinD HATPase domain was restored to activity by replacing potential interacting residues with compatible pairs. This combination of diverse methods generated a physically and biologically tested structure for the active state of histidine kinases (Dago et al., 2012).
The two state on/off model of histidine kinase catalytic domains
It is clear, as predicted, that considerable rearrangement of the two catalytic domains occurs when shifting between the inactive and active states in histidine kinases. In both states the HisKA and HATPase domains are sequestered in their relative spatial configurations by state-specific residue contacts between the domains (Fig. 2). The equilibrium position of the two domains is specific for each kinase and dependent on the relative strength of the state-specific residue contacts in the absence of signal. Signal ligands have to exert their effects by modifying the equilibrium between the two states.
Figure 2. The two state on/off model of histidine kinase catalytic domains.

The histidine kinase transitions between an inactive (left) and an active (right) confirmation. The inactive conformation is stabilized by a set of co-evolving contact residue pairs (blue bars) between the HisKA (tan) and the HATPase (green) domains that were identified by the DCA analysis by Dago et al., 2012. We speculate that the presence of an activation signal (orange star) destabilizes the inactive state contacts by partially unwinding the C-terminal end of the HisKA helix (center), thus allowing a second set of co-evolving residue pairs (red dots) to drive the formation of the active state. An experimental structure of the inactive kinase state (Marina et al., 2005) and a structural model of the active (Dago et al., 2012) kinase state are displayed and the location of the two sets of coevolving residue pairs are highlighted in both structures.
The question of how the kinase domains shift from one state to another upon binding signal is implied by comparison of the structures of the two states. The computational active state model is characterized by a bend or partial unwinding of the C-terminal helix of the HisKA domain that would destroy the inactive state-specific residue contacts between the two domains. We speculate that signal binding to sensor domains is responsible for this unwinding that mitigates the inactive-state contacts, thereby forcing the two domains into the active configuration. The very high relative concentration of the two domains as a consequence of being tethered together suggests there is little likelihood of a stable unbound state. Since signaling domains of all types are contiguous with the N-terminal helix of the HisKA domain, we propose that signals, upon binding, must induce a torque or otherwise strain the internal interactions between the two helices of the HisKA domain. In signal-activated kinases this results in the partial unwinding of the C-terminal helix that weakens the inactive state-specific contacts between the two catalytic domains. How kinases accomplish this signal-induced shift may differ from one to the other, but the proposed feature common to all is the change in the inactive state-specific contacts upon signal binding.
The active state model for sensor kinases described by (Dago et al., 2012) utilized the sequences of HisKA-type sensor kinase. About 80% of all sensor kinases are described by the HisKA Pfam sequence model. Minor classes of HisKA-like domains are identified by HisKA_2, HisKA_3 His_kinase and HWE_HK Pfam sequence models. Recent structural characterizations of DesK as a representative for HisKA_3-type kinases suggests that the structure of the autophosphorylation complex differs from that of the HisKA-type kinases (Trajtenberg et al., 2010, Albanesi et al., 2009). Here, the ATP-binding domain appears to be rotated by roughly 180° when compared to the HK853 active state model, consistent with docking and mutagenesis studies. It can thus be speculated that the differences in sequence that distinguish the various Pfam classes of histidine kinases are reflective of modifications in the structures of the autophosphorylation and or phosphotransfer complexes. Nonetheless, the two state on/off model appears to be conserved for all sensor kinases.
Setting the default mode of the histidine kinase
This two state model for the spatial arrangements of the HisKA and HATPase domains of histidine kinases explains one of their innate properties. Histidine kinases exist as either signal-activated or signal-inhibited as the “default” or non-signal mode (e.g., (Wei et al., 2012). The signal-activated mode as described above envisions signals weakening the inactive state specific contacts but signal binding could have the opposite effect, as well, and strengthen the inactive state specific contacts in signal-inhibited kinases. Thus the default mode of a kinase is determined by how the signal sensing domains affect the equilibrium between the spatial relationships of the two catalytic domains in the absence of signal.
Perspectives
Application of statistical methods to histidine kinase based signal transduction has shown that the interaction residues between two proteins, between two domains of a single protein, the folding points and dimerization sites within a single domain are amenable to discovery. These statistical methods may be applied to any protein or pair of proteins for which evolution has resulted in sufficient variation. One application of particular interest would be identifying cellular multi-protein complexes in silico. Biological interaction studies, i.e. two-hybrid analyses, identify protein complexes but such methods are unable to reveal structure. This contrasts to DCA methods that can precisely predict structure of interacting proteins in addition to simply identifying protein pairing.
Acknowledgments
The authors were supported by grant GM19416 from the National Institute of General Medical Sciences, National Institutes of Health.
References
- Albanesi D, Martin M, Trajtenberg F, Mansilla MC, Haouz A, Alzari PM, de Mendoza D, Buschiazzo A. Structural plasticity and catalysis regulation of a thermosensor histidine kinase. Proc Natl Acad Sci U S A. 2009;106:16185–16190. doi: 10.1073/pnas.0906699106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuh D, Lesk AM, Bloomer AC, Klug A. Correlation of coordinated amino acid substitutions with function in viruses related to tobacco mosaic virus. J Mol Biol. 1987;193:693–707. doi: 10.1016/0022-2836(87)90352-4. [DOI] [PubMed] [Google Scholar]
- Ashenberg O, Rozen-Gagnon K, Laub MT, Keating AE. Determinants of homodimerization specificity in histidine kinases. J Mol Biol. 2011;413:222–235. doi: 10.1016/j.jmb.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atchley WR, Wollenberg KR, Fitch WM, Terhalle W, Dress AW. Correlations among amino acid sites in bHLH protein domains: an information theoretic analysis. Mol Biol Evol. 2000;17:164–178. doi: 10.1093/oxfordjournals.molbev.a026229. [DOI] [PubMed] [Google Scholar]
- Bourret RB, Silversmith RE. Two-component signal transduction. Curr Opin Microbiol. 2010;13:113–115. doi: 10.1016/j.mib.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger L, van Nimwegen E. Accurate prediction of protein-protein interactions from sequence alignments using a Bayesian method. Mol Syst Biol. 2008;4:165. doi: 10.1038/msb4100203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casino P, Rubio V, Marina A. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell. 2009;139:325–336. doi: 10.1016/j.cell.2009.08.032. [DOI] [PubMed] [Google Scholar]
- Dago AE, Schug A, Procaccini A, Hoch JA, Weigt M, Szurmant H. Structural basis of histidine kinase autophosphorylation deduced by integrating genomics, molecular dynamics, and mutagenesis. Proc Natl Acad Sci USA. 2012;109:E1733–1742. doi: 10.1073/pnas.1201301109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Stock AM. Biological insights from structures of two-component proteins. Annu Rev Microbiol. 2009;63:133–154. doi: 10.1146/annurev.micro.091208.073214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göbel U, Sander C, Schneider R, Valencia A. Correlated mutations and residue contacts in proteins. Proteins. 1994;18:309–317. doi: 10.1002/prot.340180402. [DOI] [PubMed] [Google Scholar]
- Hopf TA, Colwell LJ, Sheridan R, Rost B, Sander C, Marks DS. Three-Dimensional Structures of Membrane Proteins from Genomic Sequencing. Cell. 2012;149:1607–1621. doi: 10.1016/j.cell.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- Lunt B, Szurmant H, Procaccini A, Hoch JA, Hwa T, Weigt M. Inference of direct residue contacts in two-component signaling. Methods Enzymol. 2010;471:17–41. doi: 10.1016/S0076-6879(10)71002-8. [DOI] [PubMed] [Google Scholar]
- Marina A, Waldburger CD, Hendrickson WA. Structure of the entire cytoplasmic portion of a sensor histidine-kinase protein. Embo J. 2005;24:4247–4259. doi: 10.1038/sj.emboj.7600886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks DS, Colwell LJ, Sheridan R, Hopf TA, Pagnani A, Zecchina R, Sander C. Protein 3D structure computed from evolutionary sequence variation. PLoS One. 2011;6:e28766. doi: 10.1371/journal.pone.0028766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin RN, Jr, Poelwijk FJ, Raman A, Gosal WS, Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012;491:138–142. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, Zecchina R, Onuchic JN, Hwa T, Weigt M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci U S A. 2011;108:E1293–1301. doi: 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent T, Jones DT. Accurate de novo structure prediction of large transmembrane protein domains using fragment-assembly and correlated mutation analysis. Proc Natl Acad Sci U S A. 2012;109:E1540–1457. doi: 10.1073/pnas.1120036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procaccini A, Lunt B, Szurmant H, Hwa T, Weigt M. Dissecting the specificity of protein-protein interaction in bacterial two-component signaling: orphans and crosstalks. PLoS One. 2011;6:e19729. doi: 10.1371/journal.pone.0019729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug A, Weigt M, Hoch JA, Onuchic JN, Hwa T, Szurmant H. Computational modeling of phosphotransfer complexes in two-component signaling. Methods Enzymol. 2010;471:43–58. doi: 10.1016/S0076-6879(10)71003-X. [DOI] [PubMed] [Google Scholar]
- Schug A, Weigt M, Onuchic JN, Hwa T, Szurmant H. High resolution protein complexes from integrating genomic information with molecular simulation. Proc Natl Acad Sci U S A. 2009;106:22124–22129. doi: 10.1073/pnas.0912100106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerker JM, Perchuk BS, Siryaporn A, Lubin EA, Ashenberg O, Goulian M, Laub MT. Rewiring the specificity of two-component signal transduction systems. Cell. 2008;133:1043–1054. doi: 10.1016/j.cell.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RC. Protein histidine kinases: assembly of active sites and their regulation in signaling pathways. Curr Opin Microbiol. 2010;13:133–141. doi: 10.1016/j.mib.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- Sułkowska JI, Morcos F, Weigt M, Hwa T, Onuchic JN. Genomics-Aided Structure Prediction. Proc Natl Acad Sci U S A. 2012;109:10340–10345. doi: 10.1073/pnas.1207864109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szurmant H, Bobay BG, White RA, Sullivan DM, Thompson RJ, Hwa T, Hoch JA, Cavanagh J. Co-evolving motions at protein-protein interfaces of two-component signaling systems identified by covariance analysis. Biochemistry. 2008;47:7782–7784. doi: 10.1021/bi8009604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szurmant H, Weigt M. Genetic covariance. In: Maloy S, Hughes K, editors. Brenner’s Encyclopedia of Genetics. Waltham: Academic Press; 2012. in press. [Google Scholar]
- Szurmant H, White RA, Hoch JA. Sensor complexes regulating two-component signal transduction. Curr Opin Struct Biol. 2007;17:706–715. doi: 10.1016/j.sbi.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro-Roman A, Wu T, Stock AM. A common dimerization interface in bacterial response regulators KdpE and TorR. Protein Sci. 2005;14:3077–3088. doi: 10.1110/ps.051722805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajtenberg F, Grana M, Ruetalo N, Botti H, Buschiazzo A. Structural and enzymatic insights into the ATP binding and autophosphorylation mechanism of a sensor histidine kinase. J Biol Chem. 2010;285:24892–24903. doi: 10.1074/jbc.M110.147843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Ng WL, Cong J, Bassler BL. Ligand and antagonist driven regulation of the Vibrio cholerae quorum-sensing receptor CqsS. Mol Microbiol. 2012;83:1095–1108. doi: 10.1111/j.1365-2958.2012.07992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigt M, White RA, Szurmant H, Hoch JA, Hwa T. Identification of direct residue contacts in protein-protein interaction by message passing. Proc Natl Acad Sci U S A. 2009;106:67–72. doi: 10.1073/pnas.0805923106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RA, Szurmant H, Hoch JA, Hwa T. Features of protein-protein interactions in two-component signaling deduced from genomic libraries. Methods Enzymol. 2007;422:75–101. doi: 10.1016/S0076-6879(06)22004-4. [DOI] [PubMed] [Google Scholar]
- Wuichet K, Cantwell BJ, Zhulin IB. Evolution and phyletic distribution of two-component signal transduction systems. Curr Opin Microbiol. 2010;13:219–225. doi: 10.1016/j.mib.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapf J, Sen U, Madhusudan, Hoch JA, Varughese KI. A transient interaction between two phosphorelay proteins trapped in a crystal lattice reveals the mechanism of molecular recognition and phosphotransfer in signal transduction. Structure. 2000;8:851–862. doi: 10.1016/s0969-2126(00)00174-x. [DOI] [PubMed] [Google Scholar]