

Abstract
Multiple systems and regulatory strategies interact to control cardiac homeostasis. In fact, regulated systems, feedback controls, and redundant control mechanisms dominate in whole animals. Accordingly, molecular and cellular tools and techniques must be utilized in complex models with multiple systems and regulatory strategies to fully appreciate the physiological context. Currently, these techniques are mainly performed under conditions remote from the normal in vivo condition; thus, the extrapolation of molecular changes to the in vivo situation and the facilitation of translational aspect of the findings are limited. A major obstacle has been the reliance on preparations that do not mimic the clinical or physiological situation. This is particularly true regarding measurements of cardiac function in mice. To address these concerns, we used a permanently implanted Doppler ultrasonic flow probe on the ascending aorta and coronary artery occluder for repeated measurements of ascending aortic blood flow (cardiac output) in conscious mice, at rest and during exercise, before and during coronary artery occlusion/reperfusion and infarction. The conscious mouse model permits detailed monitoring of within-animal changes in cardiac function during myocardial ischemia, reperfusion, and infarction in an intact, complex model free of the confounding influences of anesthetics, surgical trauma, and restraint stress. Results from this study suggest that previous protocols may have overestimated resting baseline values and underestimated cardiac output reserve. Using these procedures in currently available spontaneous or engineered mouse mutants has the potential to be of major importance for advancing the concepts and methods that drive cardiovascular research.
Keywords: photomicrographs, histological sections, electrocardiogram, myocardial ischemia
numerous molecular and cellular tools and techniques are currently used to advance our understanding of cardiac physiology and pathophysiology. These molecular tools and interventions are mainly performed in reduced preparations and, thus, are void of regulated systems, feedback controls, and redundant control mechanisms operating in whole animals. Accordingly, the physiological context of the reductionist approach can be lost because the system is far more complex than the reductionist preparations allow (26). Specifically, multiple systems and regulatory strategies interact to regulate cardiac homeostasis.
Our challenge, therefore, is to integrate the molecular intervention with the assessment of change in physiological function in an intact system (4, 22). Specifically, molecular and cellular tools and techniques must be linked to physiological function in complex models with multiple systems and regulatory strategies to fully appreciate the physiological context. Currently, these techniques are mainly performed under conditions remote from the normal in vivo condition, where the extrapolation of molecular changes to the in vivo situation and the facilitation of the translational aspect of the findings are limited (22).
A major obstacle has been the reliance on preparations that do not mimic the clinical or physiological situation. This is particularly true regarding measurements of cardiac function in mice. To overcome this obstacle, we recently described, for the first time, the use of an intact, conscious, and unrestrained mouse model of myocardial ischemia/reperfusion and infarction (33). The conscious mouse model permitted occlusion and reperfusion of the left anterior descending coronary artery in an intact, complex model, free of the confounding influences of anesthetics and surgical trauma. One major limitation of that model was the failure to measure cardiac function during myocardial ischemia, reperfusion, and infarction. Measurements of cardiac function during myocardial ischemia and reperfusion and infarction have not been conducted in conscious mice and would be important for advancing the concepts and ideas that drive cardiovascular research.
A second limitation of the earlier model (33) was an inability to assess cardiac function during exercise before and after the myocardial ischemia, reperfusion, and infarction protocol. An exercise protocol should be used whenever possible in the study of cardiovascular function, since exertion may reveal cardiovascular phenotypes that are not apparent under resting conditions (12). That is, more can be learned about how a system operates when it is forced to perform than when it is at rest (40). Therefore, to extend the earlier work (33), we used a permanently implanted Doppler ultrasonic flow probe on the ascending aorta and coronary artery occluder for repeated measurements of ascending aortic blood flow (cardiac output) in conscious mice, at rest, and during exercise, before and during coronary artery occlusion/reperfusion and infarction. Specifically, we describe, for the first time, repeated measurement of cardiac output at rest and during exercise in an intact, conscious, unrestrained mouse model of myocardial ischemia/reperfusion and infarction. The conscious mouse model permits continuous and repeated measurements of cardiac output during occlusion and reperfusion of the left anterior descending coronary artery (LAD) in an intact, complex model free of the confounding influences of anesthetics, surgical trauma, and restraint stress (41). Furthermore, the myocardial ischemia and reperfusion protocol can be initiated after the resolution of the inflammation that occurs during the initial surgical preparation.
MATERIALS AND METHODS
Animals
Experimental procedures and protocols were reviewed and approved by the Animal Care and Use Committee of Wayne State University and complied with The American Physiological Society's Guiding Principles in the Care and Use of Animals. The procedures were initiated in 16 male C57BL/6 mice (3 to 4 mo of age). Four mice died during the initial protocol, and the hearts from these animals were harvested and used for histological assessment. One mouse had instrumentation failure (occluder failed), and this animal served as an 8-wk time control. In the remaining 11 intact, conscious, and unrestrained mice, resting cardiac output, the ECG, and heart rate were recorded before, during, and each week for 8 wk after the myocardial ischemia, reperfusion, and infarction protocol. In addition, cardiac output, the ECG, and heart rate were recorded during a graded exercise test before the myocardial ischemia, reperfusion, and infarction protocol and after 8 wk of reperfusion in 6 of the same 11 intact, conscious, and unrestrained mice.
All surgical procedures were performed using aseptic surgical techniques. Mice were anesthetized with pentobarbital sodium (50 mg/kg ip), atropinized (0.05 mg/kg ip), intubated, and prepared for aseptic surgery. Supplemental doses of pentobarbital sodium (10–20 mg/kg ip) were administered if the mice regained the blink reflex or responded during the surgical procedures.
Thoracotomy Procedures
The hearts were approached via a left thoracotomy through the second intercostal space, as recently described (33). Subsequently, the sleeve of the pericardium that extends onto the ascending aorta was dissected free of surrounding tissue and a 1.6-mm Silastic-type Doppler ultrasonic flow probe (Iowa Doppler Products, Iowa City, IA) was positioned around the ascending aorta. The flow probe wires were tunneled subcutaneously and exteriorized at the back of the neck. Subsequently, a coronary artery occluder was passed around the proximal segment of the LAD, as recently described (33). The two ends of the occluder were passed through guide tubing fashioned from a “modified” mouse thoracic carotid artery catheter (Braintree Scientific) and the guide tubing with the two ends of the occluder were exteriorized at the back of the neck, filled with a sterile petrolatum ophthalmic ointment (Puralube), and sealed with a stainless-steel pin to prevent a pneumothorax (33). The local anesthetic, bupivicaine, was injected subcutaneously at the incision site. Subsequently, ECG electrodes (DataSciences International, standard lead coupler kit: 276–0031-001) were sutured subcutaneously in a modified lead II configuration (33). The ECG wires were tunneled subcutaneously and exteriorized at the back of the neck.
All animals remained on the feedback-based temperature control system and ventilator until fully recovered from the anesthesia. Once the animals regained consciousness, they were placed in a “rodent recovery cage” (Thermocare Intensive Care Unit, Braintree Scientific, Braintree, MA). Animals were returned to the housing room when they had fully recovered from the anesthesia and gained the ability to maintain body temperature. The nonsteroidal anti-inflammatory agent ketoprofen (5 mg/kg sc) and the antibiotic cefazolin (10 mg/kg sc) were continued for 2 days. At least 10 days were allowed for recovery. During the recovery period, the mice were handled, weighed, and acclimatized to the laboratory and investigators.
Experimental Procedures
Heart rate, ECG, and cardiac output at rest and during exercise.
A minimum of 10 days was allowed for recovery. Following recovery, the mice (n = 6) were brought to the laboratory and allowed to adapt to the environment for ∼2 h to ensure stable hemodynamic conditions. After the stabilization period, beat-by-beat, steady-state hemodynamic variables were recorded over 10–15 s. Subsequently, each mouse ran continuously, without aversive stimuli on a motorized treadmill at 5, 10, 15, 20, and 25 m/min on a 10% grade for ∼3 min at each workload (34). The steady-state cardiac output and heart rate responses were recorded during the 3rd min at workloads 5–25 m/min. Although no aversive stimuli were used, all mice required gentle coaxing, (i.e., tapping on the hindquarters) during the highest workload (34).
Induction of myocardial infarction.
A minimum of 1 wk was allowed between the graded exercise test and the induction of myocardial ischemia, reperfusion, and infarction. On the day of the protocol, conscious, unrestrained mice (n = 11) were studied in their home cages (33). The temperature within the cage was monitored and maintained near the thermoneutral zone for mice (46) by use of a circulating water pad under the cage and a Presto HeatDish Plus Parabolic Heater. Cardiac output and the ECG were recorded by taping the leads to single-stranded stainless-steel wires from a miniature fluid and electric swivel (Alice King Chatham Medical Arts, Hawthorne, CA). Mice were allowed to adapt to the laboratory environment for ∼2 h to ensure stable hemodynamic conditions. After the stabilization period, beat by beat, steady-state cardiac output, heart rate, and ECG parameters were recorded over 10–15 s. Subsequently, the left anterior descending coronary artery was temporarily occluded by use of the occluder, as recently described (33). Specifically, acute coronary artery occlusion was performed by pulling up on the occluder that was around the LAD and securing it to the modified catheter with a vascular clip (Schwartz straight smooth clip). The coronary artery occlusion was maintained for 45 min, followed by 2 h and 8 wk of reperfusion. Following the 2 h of reperfusion, the mice were returned to the housing room. The mice were returned to the laboratory each week for the following 8 wk to determine their resting values for cardiac output and heart rate. At week 8, the graded exercise protocol was repeated.
Subsequently, the mice were deeply anesthetized with pentobarbital sodium (90 mg/kg ip), and the hearts were harvested, sectioned, and processed with Masson Trichrome Stain kit (Sigma: HT15) to visualize the infarct scar. Using this staining protocol (33), we calculated the infarct size scores using the length measurement approaches for infarct size, and left ventricular chamber size and wall thickness were calculated.
Preparation of heart sections.
The hearts were embedded in optimal cutting temperature compound and sliced transversely from the apex to the base at 10-μm thickness with the use of a cryostat. An interval of 300 μm was maintained between each section. All sections were thaw mounted on Superfrost Plus slides (33).
Measurements.
All histological sections were examined with an Olympus BH-2 microscope using a 1× objective. Our primary end points were infarct size, chamber size, and left ventricular wall thickness. To achieve these endpoints, every section from apex to base (300 μm apart) totaling 14–15 sections per heart was quantified from the digital photomicrographs using image analysis software (Image J 1.45s) (47).
Data analysis.
All physiological recordings were sampled at 4 kHz, and the data were expressed as means ± SE. Separate one-way ANOVAs with repeated measures were used to determine differences in body weight (Fig. 1), cardiac output, stroke volume, and heart rate before occlusion of the LAD, at 20 and 45 min of occlusion and following 2 h and 8 wk of reperfusion (Fig. 5), as well as cardiac output, stroke volume, and heart rate at rest before occlusion of the LAD, and each week for 8 wk of reperfusion (Fig. 6). Dunnett's post hoc multiple-comparison procedure was used to compare each of the time points with the single control value.
Fig. 1.

Body weights before the myocardial ischemia, reperfusion and infarction protocol (Control) and each week for 8 wk after the protocol in 11 intact, conscious, and unrestrained C57BL/6 mice (black bars), as well as one fully instrumented, sham-occluded mouse (time control; gray bars) are shown. *P ≤ 0.05, vs. control.
Fig. 5.

Cardiac output (A), stroke volume (B), and heart rate (C) before occlusion of the left anterior descending coronary artery (control), at 20 and 45 min of occlusion and following 2 h and 8 wk of reperfusion in 11 intact, conscious, and unrestrained mice are presented. Cardiac output and stroke volume were significantly reduced after the ischemia, reperfusion, and the infarction protocol at all the points. Heart rate was elevated only during 20 and 45 min of occlusion. *P ≤ 0.05, vs. control.
Fig. 6.

Ascending aortic blood flow (cardiac output; A), stroke volume (B), and heart rate (C) at rest before occlusion of the left anterior descending coronary artery (control) and each week for 8 wk of reperfusion (black bars) in 11 intact, conscious, and unrestrained mice, as well as one fully instrumented, sham-occluded (time control; gray bars) mouse are shown. Cardiac output and stroke volume were reduced at week 1 of reperfusion and remained reduced for 8 wk. Heart rate was not significantly different during the 8-wk reperfusion period. *P ≤ 0.05, vs. control.
A two-way ANOVA with repeated measures was used to determine changes in cardiac output, stroke volume, and heart rate at rest and during the graded exercise test in the same 6 mice before occlusion of the LAD and after 8 wk of reperfusion (Fig. 7). The Holm-Sidak post hoc procedure was used for post hoc pairwise comparisons.
Fig. 7.

Ascending aortic blood flow (cardiac output; A), stroke volume (B), and heart rate (C) at rest and during the graded exercise test in 6 of the 11 mice, before occlusion of the left anterior descending coronary artery (●) and in the same 6 mice after 8 wk of reperfusion (○) are presented. Cardiac output was reduced at each workload following 8 wk of reperfusion. Heart rate was not significantly different during the graded exercise test after 8 wk of reperfusion. *P ≤ 0.05, control vs. 8 wk of reperfusion.
Unpaired t-tests were used to determine differences in left ventricular chamber size and left ventricular wall thickness in noninfarcted hearts and infarcted hearts following 8 wk of reperfusion (Fig. 9).
Fig. 9.

A: average infarct size (●) and individual infarct sizes (horizontal lines) following 8 wk of reperfusion in 11 mice are presented. B: the average (○) and individual (horizontal lines) left ventricular chamber sizes from the five noninfarcted hearts, as well as the 11 hearts following 8 wk of reperfusion (● and horizontal lines). Left ventricular chamber size was significantly larger after 8 wk of reperfusion. C: average (○) and individual (horizontal lines) left ventricular wall thickness from the 5 noninfarcted hearts as well as 11 hearts following 8 wk of reperfusion (● and horizontal lines). Left ventricular wall thickness was significantly smaller after 8 wk of reperfusion. *P ≤ 0.05; noninfarcted vs. 8 wk of reperfusion.
Calculations.
Cardiac output was evaluated using an ultrasonic range-gated pulsed Doppler flow meter (17). Blood flow (Q̇), in milliliters per minute, was calculated by Q̇ = kHz of Doppler frequency shift × K, where K = 1.24 × d2, [d is the cuff diameter of the flow probe, (19)]. When this system is used, the Doppler shift frequency (kHz) is directly proportional to blood flow. Stroke volume (SV), in microliters per beat, was calculated by SV (μl/beat) = cardiac output (μl/min) / heart rate (beats/min).
RESULTS
The myocardial ischemia, reperfusion, and infarction protocol was initiated in 16 male intact, conscious, and unrestrained mice. Four mice died following instrumentation or during the initial protocol, and the hearts from these animals were used for histological assessment. One mouse had instrumentation failure (occluder failed), and this animal served as an 8-wk time control.
Figure 1 shows body weight before the myocardial ischemia, reperfusion, and infarction protocol and each week for 8 wk after the protocol in 11 intact, conscious, and unrestrained C57BL/6 mice, as well as one fully instrumented, sham-occluded (time control) mouse. Body weights were significantly higher between week 3 and 8 compared with the control body weight.
Original recordings of ascending aortic blood flow (cardiac output) and the electrocardiogram (ECG) in intact, conscious, and unrestrained mice are shown in Figs. 2, 3, and 4. Figure 2 shows recordings at rest and during a graded exercise test before occlusion of the LAD and after 8 wk of reperfusion in the same mouse. Although cardiac output increased during exercise both before and after 8 wk of reperfusion, the increase was significantly reduced after 8 wk of reperfusion. Note the loss of R wave amplitude following 8 wk of reperfusion. Figure 3 shows recordings of cardiac output and the ECG at the onset of occlusion of the LAD. The onset of occlusion (indicated by the dotted line) resulted in rapid reductions in cardiac output and changes in the ECG (peaked T wave and S-T segment elevation) within seconds of pulling on the suture, documenting coronary artery occlusion. Figure 4 shows recordings of cardiac output and the ECG before occlusion of the LAD (control), at 45 min of occlusion, and following 2 h and 8 wk of reperfusion. Coronary artery occlusion reduced cardiac output at all time points. Note the ST-segment elevation at 45 min of occlusion and the pronounced Q-wave and loss of R-wave amplitude following 2 h and after 8 wk of reperfusion, respectively.
Fig. 2.

Original recordings of ascending aortic blood flow (cardiac output) and the ECG at rest and during a graded exercise test before occlusion of the left anterior descending coronary artery (Control) (A) and after 8 wk of reperfusion (B) in the same mouse are shown in Fig. 2. Although cardiac output increased during exercise both before and after 8 wk of reperfusion, the increase was significantly reduced after 8 wk of reperfusion. Note the loss of R wave amplitude following 8 wk of reperfusion.
Fig. 3.

Original recordings of ascending aortic blood flow (cardiac output) (top) and the ECG (bottom) at the onset of occlusion of the left anterior descending coronary artery are shown. The onset of occlusion (indicated by the dashed line) resulted in rapid reductions in cardiac output and changes in the ECG (peaked T wave and S-T segment elevation) within seconds of pulling on the suture, documenting coronary artery occlusion.
Fig. 4.

Original recordings of ascending aortic blood flow (cardiac output) (top) and the ECG (bottom) before occlusion of the left anterior descending coronary artery (control), at 45 min of occlusion and following 2 h and 8 wk of reperfusion are shown. Coronary artery occlusion reduced cardiac output at all time points. Note the ST-segment elevation at 45 min of occlusion and the pronounced Q-wave and loss of R wave amplitude following 2 h and 8 wk of reperfusion, respectively.
In Fig. 5, cardiac output (A), stroke volume (B), and heart rate (C) before occlusion of the LAD (control), at 20 and 45 min of occlusion and following 2 h and 8 wk of reperfusion in 11 intact, conscious, and unrestrained mice are presented. Resting cardiac output averaged 9.5 ± 0.6 ml/min before occlusion of the LAD and significantly decreased to 5.6 ± 0.5 and 5.6 ± 0.7 ml/min at 20 and 45 min of occlusion, respectively. Subsequently, cardiac output increased to 6.8 ± 0.6 and 5.7 ± 0.7 ml/min in the same mice after 2 h and 8 wk of reperfusion, respectively (40% decrease in cardiac output in the same mice after 8 wk of reperfusion). The cardiac output values at 2 h and 8 wk of reperfusion were significantly lower from the cardiac output values recorded before occlusion of the LAD.
Changes in stroke volume were similar to changes in cardiac output. Specifically, resting stroke volume averaged 20 ± 1.3 μl/beat before occlusion of the LAD and significantly decreased to 9.3 ± 0.8 and 9.2 ± 1.0 μl/beat at 20 and 45 min of occlusion, respectively. Subsequently, stroke volume increased to 13.1 ± 1.0 and 11.6 ± 1.4 μl/beat in the same mice after 2 h and 8 wk of reperfusion, respectively (42% decrease in stroke volume in the same mice after 8 wk of reperfusion). The stroke volume values at 2 h and 8 wk of reperfusion were significantly lower from the stroke volume values recorded before occlusion of the LAD.
Heart rate averaged 463 ± 7 bpm before occlusion of the LAD and increased significantly to 608 ± 14 and 605 ± 12 bpm at 20 and 45 min of occlusion, respectively. Subsequently, heart rate decreased to 510 ± 22 bpm at 2 h of reperfusion in the same mice. Heart rate averaged 499 ± 25 bpm after 8 wk of reperfusion. The heart rate values at 2 h and 8 wk of reperfusion were not significantly different from the heart rate recorded before occlusion of the LAD.
Figure 6 presents cardiac output (A), stroke volume (B), and heart rate (C) at rest before occlusion of the LAD (control), and each week for 8 wk of reperfusion (black bars) in 11 intact, conscious, and unrestrained mice, as well as one fully instrumented, sham-occluded (time control) mouse. Cardiac output was reduced at week 1 of reperfusion and remained reduced for 8 wk. Stroke volume was reduced at weeks 1, 5, 6, 7, and 8 of reperfusion. Heart rate was not significantly different during the 8-wk reperfusion period.
Figure 7 presents cardiac output (A), stroke volume (B), and heart rate (C) at rest and during the graded exercise test in 6 of the 11 mice, before occlusion of the LAD, and in the same 6 mice after 8 wk of reperfusion. Cardiac output increased during exercise both before and after 8 wk of reperfusion (significant exercise effect). Furthermore, the increase in cardiac output at each workload was higher before the ischemia, reperfusion, and infarction protocol compared with the cardiac output response after 8 wk of reperfusion (significant group effect and significant group × exercise interaction). Specifically, before the myocardial ischemia/reperfusion and infarction protocol, cardiac output increased at rest from 9.6 ± 0.6 ml/min to 18.9 ± 0.9 ml/min at the highest workload (97% increase from rest). In the same mice, following 8 wk of reperfusion, cardiac output increased at rest from 7.0 ± 1.2 ml/min to 11.7 ± 2.3 ml/min (67% increase from rest). The peak cardiac output decreased from 18.9 ± 0.9 ml/min before myocardial ischemia, reperfusion, and infarction to 11.7 ± 2.3 ml/min (38% decrease) after 8 wk of reperfusion.
Stroke volume (Fig. 7B) also increased during exercise both before and after 8 wk of reperfusion (significant exercise effect). Furthermore, stroke volume across workloads was higher before the ischemia, reperfusion, and infarction protocol compared with stroke volume after 8 wk of reperfusion (significant group effect without a significant group × exercise interaction).
Heart rate (Fig. 7C) also increased during exercise both before and after 8 wk of reperfusion (significant exercise effect). Heart rate significantly increased from 489 ± 18 bpm at rest to 798 ± 9 bpm at the highest workload before the myocardial ischemia/reperfusion and infarction protocol. After the myocardial ischemia/reperfusion and infarction protocol, in the same mice, heart rate significantly increased from 515 ± 29 bpm at rest to 764 ± 21 bpm at the highest workload. There were no differences in the heart rate response during exercise before and after 8 wk of reperfusion.
Fifty-six days (8 wk) after the ischemia, reperfusion, and infarction protocol, the hearts were harvested, sectioned, and stained with Masson trichrome to visualize the infarct scar (Fig. 8). Photomicrographs of sections from one noninfarcted mouse heart and one infarcted mouse heart following 8 wk of reperfusion are presented in Fig. 8. In the noninfarcted sections (left), the only collagen (i.e., blue stain) documents the placement of the unused occluder (arrows). Furthermore, note the small left ventricular chamber area and thick left ventricular wall. The right panel presents an infarcted heart following 8 wk of reperfusion. Note the intense blue staining indicating the abundance of collagen and the extremely large left ventricular chamber area and thin left ventricular wall.
Fig. 8.
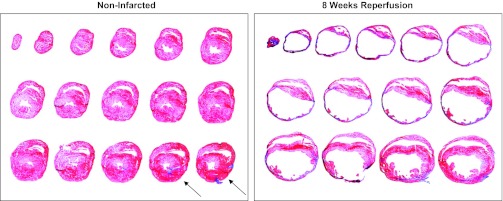
Photomicrographs of sections from one noninfarcted mouse heart and one infarcted mouse heart following 8 wk of reperfusion are presented. Sections were processed with Masson Trichrome stain. In the noninfarcted sections (left), the only collagen (i.e., blue stain) documents the placement of the unused occluder (arrows). Furthermore, note the small left ventricular chamber area and thick left ventricular wall. Right: infarcted heart following 8 wk of reperfusion. Note the intense blue staining indicating the abundance of collagen and the extremely large left ventricular chamber area and thin left ventricular wall. The sections were taken from the apex through the basal part of the left ventricle at 300-μm intervals.
In Fig. 9A, the average infarct size (closed circle) and individual infarct sizes (horizontal lines) following 8 wk of reperfusion in 11 mice are presented. Figure 9B presents the average (open circle) and individual (horizontal lines) left ventricular chamber sizes from the 5 noninfarcted hearts, as well as the 11 hearts following 8 wk of reperfusion (closed circle and horizontal lines). Left ventricular chamber size was significantly larger after 8 wk of reperfusion. Finally, Fig. 9C presents the average (open circle) and individual (horizontal lines) left ventricular wall thickness from the five noninfarcted hearts, as well as 11 hearts following 8 wk of reperfusion (closed circle and horizontal lines). Left ventricular wall thickness was significantly smaller after 8 wk of reperfusion.
DISCUSSION
Results from this study document within-animal changes in cardiac output, at rest and during exercise, before, during, and following myocardial ischemia, reperfusion, and infarction in chronically instrumented, intact, conscious, and unrestrained mice using a chronically implanted Doppler ultrasonic flow probe and coronary artery occluder. Furthermore, the results document profound cardiac structural remodeling following 8 wk of reperfusion (Fig. 8). This is the first murine study monitoring within-animal changes in cardiac function induced by myocardial ischemia, reperfusion and infarction in an intact, complex model, free of the confounding influences of anesthetics, surgical trauma, and restraint stress.
Cardiac function has been evaluated in mice using a variety of animal preparations ranging from the anesthetized open-chest condition to the conscious restrained state. Many of these preparations were limited by the use of anesthesia, the requirement for multiple blood sampling (dilution-based methods), an inability to record continuously (noninvasive imaging techniques), and the inability to perform exercise. However, one pioneering study, by Janssen et al. (20), used miniaturized transit time flow probes, as well as electromagnetic flow probes, to measure ascending aortic flow in adult conscious intact mice and mice with a myocardial infarction (permanent occlusion of the left descending coronary artery; MI). This pioneering study overcame the confounding influences of anesthesia and surgical trauma when recording cardiac output; however, anesthesia and surgical trauma during occlusion of the coronary artery, as well as permanent occlusion (without reperfusion), markedly alters the inflammatory, reparative, and recovery responses to coronary occlusion. For example, Nossuli et al. (36) demonstrated that the inflammation due to surgical trauma increases the background level of cytokine induction and accentuates the inflammatory response to myocardial reperfusion, causing significantly greater variability. Furthermore, many anesthetic agents confer protection against ischemia-reperfusion injury across species (8, 15, 44, 49), as well as in a variety of tissues, including heart (8, 9, 32, 39, 45), brain (28, 50, 54), kidney (27), lung (31), and liver (2). Anesthesia also significantly alters the autonomic nervous system (16), and disturbances in cardiac autonomic balance play a critical role in the determination of infarct size. Specifically, reductions in parasympathetic activity or increases in sympathetic activity increase the morbidity and mortality associated with myocardial infarction (3). Finally, most patients experiencing an acute myocardial infarction (AMI) receive reperfusion therapy. Furthermore, survival after AMI may improve even when reperfusion therapy is administered late, after irreversible necrosis has occurred. Thus, early or late reperfusion is recommended for individuals with myocardial infarction.
The original pioneering work recording cardiac output in conscious mice was also limited by the absence of serial measurements and within-animal changes in cardiac function induced by myocardial infarction, since the flow probes were implanted 3 wk after the MI. The procedures also prevented the study of cardiac output during activity or exercise. It is especially important to evaluate the cardiovascular system during exercise, since more can be learned about how a system operates when it is forced to perform than when it is at rest (40). Specifically, exertion may reveal cardiovascular phenotypes that are not apparent under resting conditions (12). Finally, the baseline cardiac output and heart rate values were high, and the reserve responses were low in the study by Janssen et al. (20).
Specifically, cardiac output averaged ∼20 ml/min in intact mice and 16 ml/min in MI mice (∼20% decrease in MI mice). Heart rate was more than 600 beats/min (bpm) for both intact and MI mice. In intact mice, volume loading increased cardiac output to ∼26 ml/min (30% increase). However, in MI mice, volume loading increased cardiac output to ∼20 ml/min (25% increase). These results were responsible, in part, for the spurious conclusion that mice have a limited cardiac output reserve. In contrast, in the current study, resting cardiac output averaged ∼9.5 ml/min before MI and 5.7 ml/min in the same mice after 8 wk of reperfusion (∼40% decrease in the same mice after 8 wk of reperfusion). Heart rate averaged ∼450 bpm before myocardial infarction and, in the same mice, heart rate averaged ∼500 bpm following 8 wk of reperfusion. Before MI, the graded exercise test increased cardiac output to ∼18.9 ml/min (97% increase from rest). Similarly, in the same mice, following 8 wk of reperfusion, the graded exercise test increased cardiac output to ∼11.7 ml/min (67% increase from rest). Heart rate increased from 450–500 bpm to ∼800 bpm in the same mice before myocardial infarction, as well as after 8 wk of reperfusion. Thus, although previous studies have documented very limited increases in cardiac output with interventions (20, 21), results from this study show nearly a 100% increase in cardiac output from rest to exercise.
The profound differences between results documented in the current study and the work by Janssen and colleagues (20) may be due to the fact that mice are very responsive to variations in ambient temperature and are often studied in rooms maintained between 21°C and 23°C, which is markedly below their thermoneutral zone of 30°C (46). Studies conducted in rooms with temperatures below thermoneutrality serve as a mild cold stress, and the mice respond with tachycardia due to a withdrawal of cardiac vagal tone and increased sympathetic tone (46). Accordingly, as emphasized by Swoap et al. (46), these findings support the need for carefully controlling ambient temperature during the assessment of cardiovascular function in mice. Thus, in the current study, examining responses in an environment within the thermoneutral zone for mice (46) likely accounts for the lower resting values. Thus, the limited cardiac output reserve reported in earlier investigations (20, 21) may be due, in part, to the high baseline values.
Advanced coronary care and early reperfusion strategies have dramatically improved survival rates for individuals experiencing an acute myocardial infarction (38). However, individuals who survive an acute myocardial infarction (AMI) are at risk of developing heart failure (30). The development of heart failure, following AMI, is associated with “ventricular remodeling”, i.e., changes in cardiac structure and function. Ventricular structural remodeling occurs in the area of the infarction (increased chamber dilation, wall thinning, and sphericity), as well as the noninfarcted regions of the ventricle (myocardial hypertrophy) and results in reduced cardiac function [Fig. 8; (7)]. The extent of ventricular remodeling, following AMI, is associated with the progression and prognosis of heart failure (51) and is dependent on the size of the infarct and on the process of cardiac repair (13).
In this context, although myocardial ischemia and reperfusion protocols are used in a variety of models to study the mechanisms of ischemia/reperfusion injury, as well to evaluate the therapeutic effects of potential interventions (6, 24, 53), with few exceptions (10, 11), most studies have investigated the short-term consequences of myocardial ischemia/reperfusion and infarction ranging from a few hours to a few days (5, 14, 18, 23–25, 37, 42, 52). Accordingly, little is known regarding the long-term consequences of myocardial ischemia/reperfusion and infarction; however, these data are critical for the extrapolation to the clinical situation. In this context, infarct sparing strategies that are successful shortly after reperfusion may be different when the evaluation takes place at later stages (11). For example, beneficial effects of interventions that seemed successful early after reperfusion did not alter infarct size at later time points after reperfusion (1, 35, 48). The chronic model described in this manuscript avoids these limitations.
Perspectives and Significance
Myocardial infarction in humans occurs in a complex setting of regulated systems, feedback controls, and redundant control mechanisms. Accordingly, investigations of myocardial infarction must be conducted in complex models with multiple systems and regulatory strategies to fully appreciate the physiological context. Currently, these investigations are mainly performed under conditions remote from the normal in vivo condition; thus, the extrapolation to the in vivo situation and the facilitation of translational aspect of the findings are limited. Furthermore, most myocardial infarctions in humans occur in the absence of anesthetic agents and surgical trauma, and the hearts are perfused with blood. In addition, most myocardial ischemia and reperfusion protocols cannot be conducted in humans. Thus, the study of relevant complex, conscious models are essential (29, 43), and animals must be considered. This article describes the application of existing technology, surgical techniques, and experimental protocols to achieve this goal. The methodology allows for the accurate documentation of within-animal changes in cardiac function, at rest and during exercise, induced by myocardial ischemia, reperfusion, and infarction in intact, conscious, complex, and unrestrained mice. Investigators may be encouraged to adopt these existing procedures to their investigations of myocardial infarction since highly reliable data can be obtained in mice under physiological conditions.
Our challenge, therefore, is to integrate molecular and cellular tools and techniques with physiological function in complex models with multiple systems and regulatory strategies (4, 22). Accordingly, applying the model described in this paper to gene-manipulated mouse models of cardiac pathology may be particularly important since it allows for detailed monitoring of within-animal changes in cardiac function induced by investigator interventions, and it allows for the differentiation between cardiac and peripheral causes of altered hemodynamic function (20).
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grant HL-88615.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: H.L.L. and S.E.D. conception and design of research; H.L.L. and S.E.D. performed experiments; H.L.L. and S.E.D. analyzed data; H.L.L. and S.E.D. interpreted results of experiments; H.L.L. and S.E.D. prepared figures; H.L.L. and S.E.D. drafted manuscript; H.L.L. and S.E.D. edited and revised manuscript; H.L.L. and S.E.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We wish to thank Hussein Janbaih, Richard Wang, and Amanda Blumberg for their expert technical assistance.
REFERENCES
- 1. Ausma J, Cleutjens J, Thone F, Flameng W, Ramaekers F, Borgers M. Chronic hibernating myocardium: interstitial changes. Mol Cell Biochem 147: 35–42, 1995 [DOI] [PubMed] [Google Scholar]
- 2. Beck-Schimmer B, Breitenstein S, Urech S, De CE, Wittlinger M, Puhan M, Jochum W, Spahn DR, Graf R, Clavien PA. A randomized controlled trial on pharmacological preconditioning in liver surgery using a volatile anesthetic. Ann Surg 248: 909–918, 2008 [DOI] [PubMed] [Google Scholar]
- 3. Billman GE. Cardiac autonomic neural remodeling and susceptibility to sudden cardiac death: effect of endurance exercise training. Am J Physiol Heart Circ Physiol 297: H1171–H1193, 2009 [DOI] [PubMed] [Google Scholar]
- 4. Billman GE. The grand challenge of physiology: to integrate function from molecules to man. Front Physiol 1: 5, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Briaud SA, Ding ZM, Michael LH, Entman ML, Daniel S, Ballantyne CM. Leukocyte trafficking and myocardial reperfusion injury in ICAM-1/P-selectin-knockout mice. Am J Physiol Heart Circ Physiol 280: H60–H67, 2001 [DOI] [PubMed] [Google Scholar]
- 6. Chen Z, Chua CC, Gao J, Hamdy RC, Chua BH. Protective effect of melatonin on myocardial infarction. Am J Physiol Heart Circ Physiol 284: H1618–H1624, 2003 [DOI] [PubMed] [Google Scholar]
- 7. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol 35: 569–582, 2000 [DOI] [PubMed] [Google Scholar]
- 8. Cope DK, Impastato WK, Cohen MV, Downey JM. Volatile anesthetics protect the ischemic rabbit myocardium from infarction. Anesthesiology 86: 699–709, 1997 [DOI] [PubMed] [Google Scholar]
- 9. De Hert SG, Turani F, Mathur S, Stowe DF. Cardioprotection with volatile anesthetics: mechanisms and clinical implications. Anesth Analg 100: 1584–1593, 2005 [DOI] [PubMed] [Google Scholar]
- 10. de Waard MC, Duncker DJ. Prior exercise improves survival, infarct healing, and left ventricular function after myocardial infarction. J Appl Physiol 107: 928–936, 2009 [DOI] [PubMed] [Google Scholar]
- 11. De CT, Cleutjens JP, Blankesteijn WM, Debets JJ, Smits JF, Janssen BJ. Long-term structural and functional consequences of cardiac ischaemia-reperfusion injury in vivo in mice. Exp Physiol 89: 605–615, 2004 [DOI] [PubMed] [Google Scholar]
- 12. Fitzgerald SM, Gan L, Wickman A, Bergstrom G. Cardiovascular and renal phenotyping of genetically modified mice: a challenge for traditional physiology. Clin Exp Pharmacol Physiol 30: 207–216, 2003 [DOI] [PubMed] [Google Scholar]
- 13. Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res 58: 88–111, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Girod WG, Jones SP, Sieber N, Aw TY, Lefer DJ. Effects of hypercholesterolemia on myocardial ischemia-reperfusion injury in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol 19: 2776–2781, 1999 [DOI] [PubMed] [Google Scholar]
- 15. Haessler R, Kuzume K, Chien GL, Wolff RA, Davis RF, Van Winkle DM. Anesthetics alter the magnitude of infarct limitation by ischemic preconditioning. Cardiovasc Res 28: 1574–1580, 1994 [DOI] [PubMed] [Google Scholar]
- 16. Halliwill JR, Billman GE. Effect of general anesthesia on cardiac vagal tone. Am J Physiol Heart Circ Physiol 262: H1719–H1724, 1992 [DOI] [PubMed] [Google Scholar]
- 17. Hartley CJ, Cole JS. An ultrasonic pulsed Doppler system for measuring blood flow in small vessels. J Appl Physiol 37: 626–629, 1974 [DOI] [PubMed] [Google Scholar]
- 18. Hoffmeyer MR, Scalia R, Ross CR, Jones SP, Lefer DJ. PR-39, a potent neutrophil inhibitor, attenuates myocardial ischemia-reperfusion injury in mice. Am J Physiol Heart Circ Physiol 279: H2824–H2828, 2000 [DOI] [PubMed] [Google Scholar]
- 19. Ishida T, Lewis RM, Hartley CJ, Entman ML, Field JB. Comparison of hepatic extraction of insulin and glucagon in conscious and anesthetized dogs. Endocrinology 112: 1098–1109, 1983 [DOI] [PubMed] [Google Scholar]
- 20. Janssen B, Debets J, Leenders P, Smits J. Chronic measurement of cardiac output in conscious mice. Am J Physiol Regul Integr Comp Physiol 282: R928–R935, 2002 [DOI] [PubMed] [Google Scholar]
- 21. Janssen BJ, Smits JF. Autonomic control of blood pressure in mice: basic physiology and effects of genetic modification. Am J Physiol Regul Integr Comp Physiol 282: R1545–R1564, 2002 [DOI] [PubMed] [Google Scholar]
- 22. Janssen PM. Challenges in cardiac muscle physiology. Front Physiol 1: 2, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jones SP, Girod WG, Granger DN, Palazzo AJ, Lefer DJ. Reperfusion injury is not affected by blockade of P-selectin in the diabetic mouse heart. Am J Physiol Heart Circ Physiol 277: H763–H769, 1999 [DOI] [PubMed] [Google Scholar]
- 24. Jones SP, Lefer DJ. Myocardial reperfusion injury: insights gained from gene-targeted mice. News Physiol Sci 15: 303–308, 2000 [DOI] [PubMed] [Google Scholar]
- 25. Jones SP, Trocha SD, Lefer DJ. Cardioprotective actions of endogenous IL-10 are independent of iNOS. Am J Physiol Heart Circ Physiol 281: H48–H52, 2001 [DOI] [PubMed] [Google Scholar]
- 26. Joyner MJ. Giant sucking sound: can physiology fill the intellectual void left by the reductionists? J Appl Physiol 111: 335–342, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Julier K, da SR, Garcia C, Bestmann L, Frascarolo P, Zollinger A, Chassot PG, Schmid ER, Turina MI, von Segesser LK, Pasch T, Spahn DR, Zaugg M. Preconditioning by sevoflurane decreases biochemical markers for myocardial and renal dysfunction in coronary artery bypass graft surgery: a double-blinded, placebo-controlled, multicenter study. Anesthesiology 98: 1315–1327, 2003 [DOI] [PubMed] [Google Scholar]
- 28. Kapinya KJ, Lowl D, Futterer C, Maurer M, Waschke KF, Isaev NK, Dirnagl U. Tolerance against ischemic neuronal injury can be induced by volatile anesthetics and is inducible NO synthase dependent. Stroke 33: 1889–1898, 2002 [DOI] [PubMed] [Google Scholar]
- 29. Lefer DJ, Bolli R. Development of an NIH consortium for preclinicAl AssESsment of CARdioprotective therapies (CAESAR): a paradigm shift in studies of infarct size limitation. J Cardiovasc Pharmacol Ther 16: 332–339, 2011 [DOI] [PubMed] [Google Scholar]
- 30. Lewis EF, Moye LA, Rouleau JL, Sacks FM, Arnold JM, Warnica JW, Flaker GC, Braunwald E, Pfeffer MA. Predictors of late development of heart failure in stable survivors of myocardial infarction: the CARE study. J Am Coll Cardiol 42: 1446–1453, 2003 [DOI] [PubMed] [Google Scholar]
- 31. Liu R, Ishibe Y, Ueda M. Isoflurane-sevoflurane adminstration before ischemia attenuates ischemia-reperfusion-induced injury in isolated rat lungs. Anesthesiology 92: 833–840, 2000 [DOI] [PubMed] [Google Scholar]
- 32. Lucchinetti E, Jamnicki M, Fischer G, Zaugg M. Preconditioning by isoflurane retains its protection against ischemia-reperfusion injury in postinfarct remodeled rat hearts. Anesth Analg 106: 17–23, table, 2008 [DOI] [PubMed] [Google Scholar]
- 33. Lujan HL, Janbaih H, Feng HZ, Jin JP, DiCarlo SE. Myocardial ischemia, reperfusion, and infarction in chronically instrumented, intact, conscious, and unrestrained mice. Am J Physiol Regul Integr Comp Physiol 302: R1384–R1400, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lujan HL, Palani G, Chen Y, Peduzzi-Nelson J, DiCarlo SE. Targeted ablation of cardiac sympathetic neurons reduces resting, reflex and exercise-induced sympathetic activation in conscious rats. Am J Physiol Heart Circ Physiol 296: H1305–H1311, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Michael LH, Ballantyne CM, Zachariah JP, Gould KE, Pocius JS, Taffet GE, Hartley CJ, Pham TT, Daniel SL, Funk E, Entman ML. Myocardial infarction and remodeling in mice: effect of reperfusion. Am J Physiol Heart Circ Physiol 277: H660–H668, 1999 [DOI] [PubMed] [Google Scholar]
- 36. Nossuli TO, Lakshminarayanan V, Baumgarten G, Taffet GE, Ballantyne CM, Michael LH, Entman ML. A chronic mouse model of myocardial ischemia-reperfusion: essential in cytokine studies. Am J Physiol Heart Circ Physiol 278: H1049–H1055, 2000 [DOI] [PubMed] [Google Scholar]
- 37. Palazzo AJ, Jones SP, Girod WG, Anderson DC, Granger DN, Lefer DJ. Myocardial ischemia-reperfusion injury in CD18- and ICAM-1-deficient mice. Am J Physiol Heart Circ Physiol 275: H2300–H2307, 1998 [DOI] [PubMed] [Google Scholar]
- 38. Parikh NI, Gona P, Larson MG, Fox CS, Benjamin EJ, Murabito JM, O'Donnell CJ, Vasan RS, Levy D. Long-term trends in myocardial infarction incidence and case fatality in the National Heart, Lung, and Blood Institute's Framingham Heart study. Circulation 119: 1203–1210, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pravdic D, Sedlic F, Mio Y, Vladic N, Bienengraeber M, Bosnjak ZJ. Anesthetic-induced preconditioning delays opening of mitochondrial permeability transition pore via protein kinase C-epsilon-mediated pathway. Anesthesiology 111: 267–274, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rowell LB. Human Circulation Regulation During Physical Stress. New York: Oxford University Press, 1986, p. 257–286 [Google Scholar]
- 41. Sanchez O, Arnau A, Pareja M, Poch E, Ramirez I, Soley M. Acute stress-induced tissue injury in mice: differences between emotional and social stress. Cell Stress Chaperones 7: 36–46, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scalia R, Gooszen ME, Jones SP, Hoffmeyer M, Rimmer DM, III, Trocha SD, Huang PL, Smith MB, Lefer AM, Lefer DJ. Simvastatin exerts both anti-inflammatory and cardioprotective effects in apolipoprotein E-deficient mice. Circulation 103: 2598–2603, 2001 [DOI] [PubMed] [Google Scholar]
- 43. Schwartz LL, Kloner RA, Arai AE, Baines CP, Bolli R, Braunwald E, Downey J, Gibbons RJ, Gottlieb RA, Heusch G, Jennings RB, Lefer DJ, Mentzer RM, Murphy E, Ovize M, Ping P, Przyklenk K, Sack MN, Vander Heide RS, Vinten-Johansen J, Yellon DM. New horizons in cardioprotection: recommendations from the 2010 National Heart, Lung, and Blood Institute Workshop. Circulation 124: 1172–1179, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sloan RC, Rosenbaum M, O'Rourke D, Oppelt K, Frasier CR, Waston CA, Allan AG, Brown DA. High doses of ketamine-xylazine anesthesia reduce cardiac ischemia-reperfusion injury in guinea pigs. J Am Assoc Lab Anim Sci 50: 349–354, 2011 [PMC free article] [PubMed] [Google Scholar]
- 45. Stadnicka A, Marinovic J, Ljubkovic M, Bienengraeber MW, Bosnjak ZJ. Volatile anesthetic-induced cardiac preconditioning. J Anesth 21: 212–219, 2007 [DOI] [PubMed] [Google Scholar]
- 46. Swoap SJ, Overton JM, Garber G. Effect of ambient temperature on cardiovascular parameters in rats and mice: a comparative approach. Am J Physiol Regul Integr Comp Physiol 287: R391–R396, 2004 [DOI] [PubMed] [Google Scholar]
- 47. Takagawa J, Zhang Y, Wong ML, Sievers RE, Kapasi NK, Wang Y, Yeghiazarians Y, Lee RJ, Grossman W, Springer ML. Myocardial infarct size measurement in the mouse chronic infarction model: comparison of area- and length-based approaches. J Appl Physiol 102: 2104–2111, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vanoverschelde JL, Melin JA. The pathophysiology of myocardial hibernation: current controversies and future directions. Prog Cardiovasc Dis 43: 387–398, 2001 [DOI] [PubMed] [Google Scholar]
- 49. Walsh RS, Tsuchida A, Daly JJ, Thornton JD, Cohen MV, Downey JM. Ketamine-xylazine anaesthesia permits a KATP channel antagonist to attenuate preconditioning in rabbit myocardium. Cardiovasc Res 28: 1337–1341, 1994 [DOI] [PubMed] [Google Scholar]
- 50. Wang C, Jin LJ, Jung HH, Zuo Z. Pretreatment with volatile anesthetics, but not with the nonimmobilizer 1,2-dichlorohexafluorocyclobutane, reduced cell injury in rat cerebellar slices after an in vitro simulated ischemia. Brain Res 1152: 201–208, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation 76: 44–51, 1987 [DOI] [PubMed] [Google Scholar]
- 52. Yang J, Jones SP, Suhara T, Greer JJ, Ware PD, Nguyen NP, Perlman H, Nelson DP, Lefer DJ, Walsh K. Endothelial cell overexpression of fas ligand attenuates ischemia-reperfusion injury in the heart. J Biol Chem 278: 15185–15191, 2003 [DOI] [PubMed] [Google Scholar]
- 53. Yet SF, Tian R, Layne MD, Wang ZY, Maemura K, Solovyeva M, Ith B, Melo LG, Zhang L, Ingwall JS, Dzau VJ, Lee ME, Perrella MA. Cardiac-specific expression of heme oxygenase-1 protects against ischemia and reperfusion injury in transgenic mice. Circ Res 89: 168–173, 2001 [DOI] [PubMed] [Google Scholar]
- 54. Zhao P, Peng L, Li L, Xu X, Zuo Z. Isoflurane preconditioning improves long-term neurologic outcome after hypoxic-ischemic brain injury in neonatal rats. Anesthesiology 107: 963–970, 2007 [DOI] [PubMed] [Google Scholar]