

Abstract
To examine whether hypomyelination in neonatal rats might be related to apoptosis of oligodendrocyte progenitors, we administered dexamethasone (0.5 mg/kg SC) to neonatal rats on postnatal (P) days 1 through 5. Immunofluorescent staining and Western blotting for myelin basic protein (MBP) were performed on P14. Morphologic changes associated with apoptotic death of oligodendrocyte progenitors were assessed by using immunofluorescent staining on P5 of surface markers present at different developmental stages of oligodendrocyte progenitors (O4 and O1) and by double-staining with terminal deoxynucleotidyl transferase-mediated digoxigenin–dUTP nick end-labeling (TUNEL) and O4 or O1. Administration of dexamethasone to neonatal rats reduced the expression of MBP in the white matter by P14. In addition, dexamethasone reduced the expression of O4-positive cells, presumably preoligodendrocytes, in the corpus callosum and induced degenerative changes, such as cytoplasmic condensation and fragmented, tortuous processes, in oligodendrocyte progenitors, and increased the number of TUNEL-positive pyknotic nuclei of oligodendrocyte progenitors. These findings suggest that the dexamethasone-induced decreased expression of MBP in the cerebral hemispheres of the neonatal rats is due to apoptotic degeneration of oligodendrocyte progenitors. Administration of dexamethasone during the critical period of brain development may increase the risk of apoptosis in oligodendrocyte progenitors, subsequently resulting in hypomyelination.
Abbreviations: MBP, myelin basic protein; TUNEL, terminal deoxynucleotidyl transferase-mediated digoxigenin–dUTP nick end-labeling
Corticosteroids have been widely used during the antenatal and postnatal periods to reduce the risk of respiratory distress syndrome and to prevent the development of bronchopulmonary dysplasia.8,10 However, the fetal and neonatal safety associated with perinatal corticosteroid therapy has not been established. There is growing evidence that antenatal steroids adversely affect the growth and development of the immature brain, and postnatal dexamethasone therapy is often associated with neurodevelopmental impairment and an increased risk of cerebral palsy.8,16,18,23,26 Animal studies have shown deleterious effects of repeated administration of corticosteroids, especially dexamethasone, on brain development.2,13,15,17,19,24,28,30 Repeated administration of dexamethasone adversely affects the myelination of the developing rat brain and disturbs myelin synthesis in fetal sheep.15,17 However, the precise mechanism of these adverse effects of corticosteroid on myelination in the developing brain is not well understood.
A recent study demonstrated that dexamethasone accelerated the apoptosis of oligodendrocytes, leading to a shift toward necrosis.29 Hypoxia–ischemia to the neonatal rat brain increases the number of pyknotic oligodendrocyte progenitors in ischemic cerebral hemispheres and triggers the death of late oligodendrocyte progenitors (preoligodendrocytes) by the apoptotic process.5 Therefore, we hypothesized in the current study that dexamethasone-induced hypomyelination in the neonatal rats might be related to the death of oligodendrocyte progenitors by apoptosis. To assess this hypothesis, we subcutaneously administered dexamethasone to neonatal rats for 5 consecutive days after birth.28 We used immunofluorescent staining of myelin basic protein (MBP) and surface markers present at different developmental stages of oligodendrocyte progenitors (that is, O4, O1) to study the morphology of developing oligodendrocyte progenitors and subsequent hypomyelination. In addition, Western blot analysis was performed for MBP, and the apoptotic death of oligodendrocyte progenitors was assessed by double-staining with terminal deoxynucleotidyl transferase-mediated digoxigenin–dUTP nick end-labeling (TUNEL) and O4 or O1.
Materials and Methods
Animals.
The animals used in this experiment were treated in accordance with the “Guide for the Care and Use of Laboratory Animals of Dankook University”. Timed-pregnant Sprague–Dawley rats (Cheil, Seoul, South Korea) were housed individually in breeding cages, kept on a 12:12-h light:dark cycle, and had free access to food and water. Littermates were randomly divided into 2 groups at birth for the administration of dexamethasone or saline, and pups remained with their dam until weaning. The dexamethasone group received daily injections of dexamethasone (0.5 mg/kg SC) at the dorsum of the neck on postnatal days (P) 1 through 5.28 The dose of 0.5 mg/kg was selected due to preliminary data from Western blotting for MBP (n = 2) and has been given to preterm infants to prevent or treat bronchopulmonary dysplasia.8,16 Pups in the saline group received subcutaneous normal saline according to the same regimen as for the dexamethasone group. After ether anesthesia, at 12 h after the last dexamethasone administration on P5 and on P14, pups were perfused with 4% paraformaldehyde. The brain were removed, kept in 30% sucrose in 0.1 M PBS for 72 h at 4 °C, and then frozen in OCT compound (Tissue-Tek, Sakura Finetek, Tokyo, Japan). Cryostat sections (thickness, 12 μm) were obtained and stored at –80 °C.
Immunofluorescent staining.
Primary antibodies for immunohistochemistry were antiO4 and antiO1 mouse monoclonal IgM antibodies (MAB345 and MAB344, Chemicon, Temecula, CA) and goat polyclonal antiMBP (SC13912, Santa Cruz Biotechnology, Santa Cruz, CA). Immunohistochemistry was performed as described previously.2 Briefly, frozen coronal sections (thickness, 12 μm) of the frontal lobe from the level of bregma were mounted on gelatinized slides and incubated overnight at 4 °C with antibodies against O4, O1, or MBP (dilution, 1:1000 in PBS). After 3 washes in PBS (10 min each), the sections were incubated with secondary antibodies (dilution, 1:200; Alexa Fluor 555–antimouse IgG, Molecular Probes, Invitrogen, Carlsbad, CA) at room temperature for 1.5 h. Sections from each group were stained together to eliminate the possible effects of different experimental conditions. Tissue sections stained with O4 or O1 antibodies were washed and processed for TUNEL staining (TUNEL Apoptosis Detection Kit, DNA Fragmentation/Fluorescence Staining, Upstate, Temecula, CA).
For TUNEL, tissue sections were incubated with proteinase K (20 μg/mL, Sigma-Aldrich, St Louis, MO) at room temperature for 15 min and then washed 3 times with TBS (10 mM Tris-HCl [pH 7.4], 150 mM NaCl) for 5 min each wash. The sections were exposed to equilibration buffer containing terminal deoxynucleotidyl transferase buffer (TDT, Invitrogen) and BSA (1 mg/mL) for 30 min. Next, each slide was incubated for 3 h at 37 °C in a solution containing 37 U TDT, 1 pmol digoxigenin-11-dUTP, TDT buffer, and BSA (1 mg/mL). The slides were washed in TBS and processed with antidigoxigenin antibody. Stained sections were evaluated under a fluorescence microscope (Olympus, Tokyo, Japan) and a confocal laser microscope (Carl Zeiss, Jena, Germany).
Quantification of immunofluorescent cells.
To evaluate the immunofluorescent staining data quantitatively, the numbers of fluorescent cells in each rat were obtained from 3 random, adjacent sections that included the genu corpus callosum, its overlying supracallosal radiation, caudoputamen, and external capsule within rostral 1mm from bregma. From each section, 3 fields extending laterally from the medial corpus callosum at the level of the sagittal sulcus were counted at 400× magnification (1 field = 0.196 mm2), and the density (cells/mm2) of cells per field and average cell density per rat were calculated.5 The densities of O4-and O1-labeled cells on P5 were compared between the dexamethasone (n = 6) and saline (n = 6) groups. In addition, the percentages of O4- or O1-labeled cells with TUNEL-positive nuclei on P5 were compared between the dexamethasone (n = 6) and saline (n = 6) groups.
Western blotting of MBP.
The cerebral hemispheres dissected from the pups on P14 were frozen in liquid nitrogen. The tissues were homogenized and centrifuged, and the supernatant was stored at –80 °C. Proteins (50 μg/lane) were separated on a 4%–20% gradient polyacrylamide gel, electrotransferred onto a microcellulose membrane, and incubated overnight at 4 °C with antiMBP antibody. Subsequently, the membranes were incubated for 4 h with horseradish-peroxidase–conjugated goat antirabbit antibodies (Santa Cruz). A chemiluminescent detection system (Amersham Bioscience, Valley Stream, NY) was used to detection labeled proteins. The results were analyzed by semiquantitative densitometry (Gel-Pro Analyzer, Mediacybernetics, Bethesda, MD). Relative densities were compared between the dexamethasone and saline groups (n = 5 per group, including the 2 preliminary blots run to select the dose of dexamethasone to be administered).
Statistical analysis.
The statistical analysis was performed by using SPSS version 12.0 (SPSS, Chicago, IL). Data are expressed as mean ± SEM. Values were compared between the dexamethasone and saline groups by using the Student t test and Mann–Whitney U test. P values less than 0.05 were considered to be statistically significant.
Results
Immunofluorescent expression of MBP on P14.
All rats that received dexamethasone on P1 through P5 were physically active; their body weights were reduced by 30.1% (that is, rats weighed 9.7 ± 0.71 g) on P5 and 34.1% (24.3 ± 0.9 g) on P14 compared with those of rats that received saline (13.9 ± 0.8 g and 36.8 ± 2.7 g, respectively).19 Five daily consecutive injections of dexamethasone between P1 and P5 reduced the immunofluorescent expression of MBP in the white matter of the cerebral hemispheres by P14. The expression of MBP was decreased in the corpus callosum, its overlying supracallosal radiation, the external capsule, and the caudoputamen of rats that received dexamethasone compared with saline (Figure 1 A through F). Western blot analysis to quantify changes in MBP in rat cerebral hemispheres collected on P14 revealed the accumulation of MBP isoforms including the 21.5-, 18.5-, 17-, and 14-kDa forms (Figure 2). The relative densities of the MBP bands decreased (P < 0.01) by 84.3% in dexamethasone-treated rats (n = 5) compared with saline-treated rats (n = 5) on P14.
Figure 1.
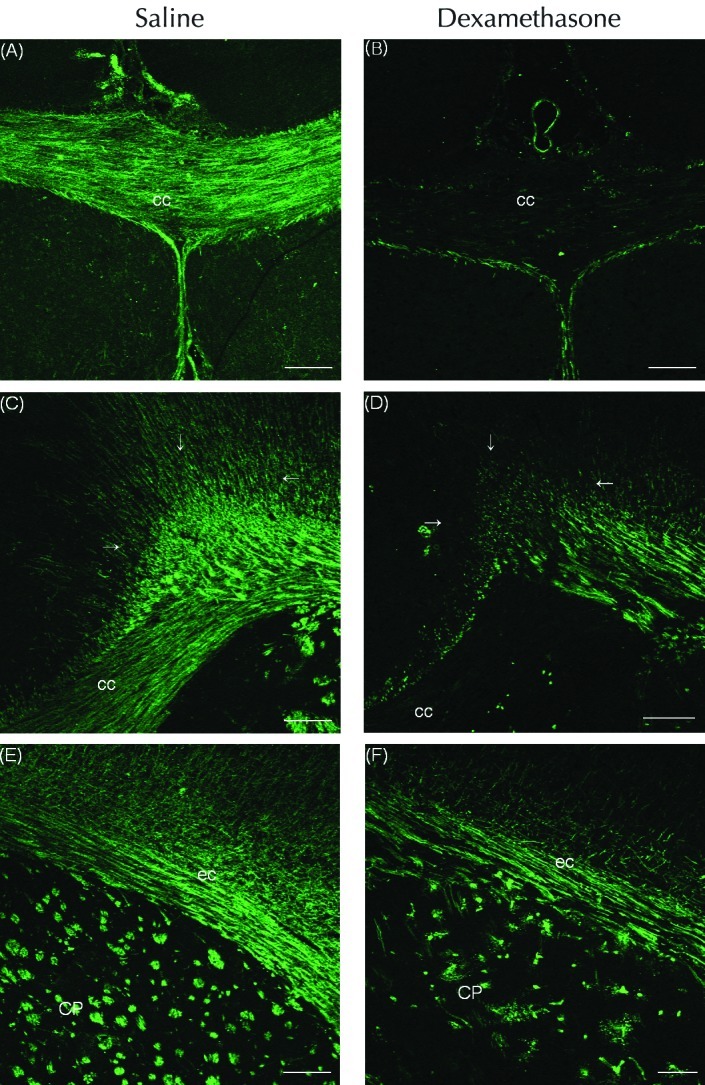
Immunofluorescent staining for myelin basic protein (MBP) in the cerebral hemispheres of (A, C, and E) saline- and (B, D, and F) dexamethasone-treated rats on P14. Compared with saline, repeated administration of dexamethasone (0.5 mg/kg) reduced the MBP-associated immunofluorescent signal in the corpus callosum (cc), external capsule (ec), caudoputamen (CP), and supracallosal radiation (outlined by arrows). Scale bar, 100 μm.
Figure 2.
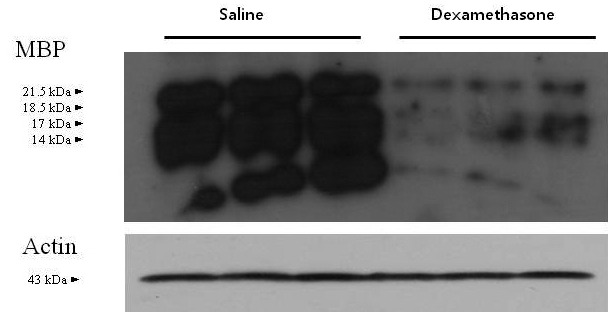
Western blotting of MBP (21.5-, 18.5-, 17-, and 14-kDa isoforms) in the cerebral hemispheres of saline- and dexamethasone-treated neonatal rats. Compared with saline, repeated administration of dexamethasone reduced the expression of MBP.
Immunofluorescent expression of O4 and O1 on P5.
The expression of cells labeled with O4 antibody (that is, O4-positive cells) was decreased in the corpus callosum and external capsule of the white matter in the cerebral hemispheres of the dexamethasone compared with saline group (Figure 3 A and B). At low power, O4-positive cells were more numerous in the corpus callosum of the saline rats than in the dexamethasone group (Figure 3 E and F). Confocal laser microscopy revealed that the pyknotic O4-positive cells in the dexamethasone rats displayed features of degeneration, including cell body condensation and few processes (Figure 3 J).5 Most O4-positive cells in saline-treated rats were simple multipolar cells with several processes (Figure 3 I), and those showing cell-body condensation had distinct, long, and well-branched and well-preserved processes (Figure 3 I). The expression of O1-positive cells did not differ significantly between the 2 groups (Figure 3 C and D). At low power, the number of O1-positive cells in the corpus callosum of the dexamethasone group was equivalent that in saline-treated rats (Figure 3 G and H). In the dexamethasone group, pyknotic O1 (+) cells showed condensation of the cell body, had fewer processes (which were arranged in a simple multipolar distribution), and were less mature than those of saline-treated rats (Figure 3 K and L).6 Many of the O1-positive cells in the saline group displayed a mature, complex, multipolar morphology characterized by a round soma with sharp, long, branched processes (Figure 3 K).6
Figure 3.

Immunofluorescent staining for (A, B, E, F, I, and J) O4 or (C, D, G, H, K, and L) O1 at P5 in rat pups injected with dexamethasone or saline. The expression of O4-positive cells was decreased in the corpus callosum (cc) and external capsule (ec) of the white matter in the cerebral hemispheres of rats that received (A) dexamethasone compared with (B) saline; (C and D) in contrast, expression of O4 did not differ between groups. At low power, cells labeled with O4 antibody were more numerous in the corpus callosum of rats that received (E) saline compared with (F) dexamethasone. In addition, the corpus callosum of dexamethasone-treated rats showed many pyknotic O4-positive cells with condensation of the cell body. However, at low power, O1-positive cells in the corpus callosum of the (H) dexamethasone group were equivalent in number to those in the (G) saline-treated rats. High-power confocal laser microscopy of (I) O4-positive cells of saline-treated rats revealed simple multipolar cells with several processes. In addition, the corpus callosum contained a few O4-positive cells with condensation of the cell body (arrowheads), but the processes of these were clear, long, and well-branched and -preserved (arrows). In contrast, the (J) pyknotic O4-positive cells of dexamethasone-treated rats displayed features of degeneration, including condensation of the cell body and few processes (arrowheads). The upper-right insert shows the cytoplasmic condensation in more detail; the bottom-left insert highlights the fragmented, tortuous o4-labeled processes. (K) Many O1-positive cells in the saline-treated rats displayed a mature, complex, multipolar morphology characterized by a round soma with clear, long, branched processes (arrows). (L) The corpus callosum of dexamethasone-treated rats contained O1-positive cells that were pyknotic and condensed and had few or bipolar (arrows) processes; the inset shows that these cells had simple multipolar processes and were less mature than those of saline-treated rats. Scale bars, 250 μm (A through D); 100 μm (E through H); and 25 μm (I through L).
Double-staining with TUNEL and O4 or O1 antibody on P5.
On P5, the corpus callosum showed numerous TUNEL-positive pyknotic (that is, apoptotic) nuclei (green) after the administration of dexamethasone or saline (Figure 4 A through H), and O4-positive cells (red) were decreased in dexamethasone- compared with saline-treated rats (Figure 4 A and C). O4-positive, but not O1-positive, cells with TUNEL-positive nuclei were increased markedly in the corpus callosum of the dexamethasone group (Figure 4). In the corpus callosum of the dexamethasone group, O4-positive cells TUNEL-positive pyknotic nuclei showed the loss of discrete processes, compared with the saline group. Dexamethasone treatment did not affect the processes of O1-positive cells, which remained similar in appearance to those of the saline-treated rats (Figure 4 F and H). By P5, the density (cells/mm2) of O4-positive cells in the corpus callosum was reduced (P < 0.05) by 50.9% in the dexamethasone-treated rats (n = 6) compared with the saline group (n = 6). In addition, the percentage of O4-positive cells with TUNEL-positive nuclei was increased (P < 0.05) by 51.6% in the dexamethasone group (n = 6) compared with the saline group (n = 6). However, neither the density of O1-positive cells nor the percentage of O1-positive cells with TUNEL-positive nuclei differed between the 2 groups.
Figure 4.

Confocal laser microscopy of the corpus callosum double-stained on P5 by using TUNEL and O4 or O1 antibody. Numerous TUNEL-positive nuclei (green) were visualized on P5 after administration of (A, B, E, and F) saline or (C, D, G, and H) dexamethasone. The expression of O4-positive cells (red) was increased in the corpus callosum of rats treated with (A) saline compared with (C) dexamethasone. (A through D) O4-positive cells with TUNEL-positive nuclei (suggesting apoptosis) were increased markedly in the corpus callosum of the dexamethasone compared with the saline group. However, administration of dexamethasone did not significantly reduce the expression of (E through H) O1-positive cells overall or those with with TUNEL-positive nuclei in the corpus callosum. High-power microscopy showed that in the corpus callosum, O4-positive cells with TUNEL-positive pyknotic nuclei showed the loss of discrete processes (arrows) in the (D) dexamethosone-treated compared with (B) saline-treated rats, which had a few O4-positive cells with TUNEL-positive nuclei. In contrast, dexamethasone did not alter the processes of O1-positive cells, which were (H) relatively well preserved and similar to those of (F) saline-treated rats. Several O1-positive cells with TUNEL-positive nuclei (arrowheads) were observed in the corpus callosums of both groups. Scale bars, 100 μm (A, C, E, and G) and 25 μm (B, D, F, and H).
Discussion
The involvement of corticosteroids in the myelin biosynthetic pathway is well known.15,30 Several studies have suggested that corticosteroids, especially dexamethasone, affect the maturation of oligodendrocytes and impair the formation of myelin.2,15,17,24,30 Other studies have reported that corticosteroids enhance the expression of genes related to the synthesis of MBP and exert trophic and protective effects in the nervous system.20,21,25,32 A study using the rat brain cell cultures showed that dexamethasone alters oligodendroglial differentiation and myelination depending on the developmental stage: early during myelination, dexamethasone has a stimulatory effect, whereas at later stages, it causes marked inhibition.1 In addition, corticosteroid administration during the critical period of brain development may impair neurogenesis and myelination.2,17,24 However, the mechanisms of dexamethasone-induced hypomyelination remain unclear, despite a recent study that demonstrated that dexamethasone accelerated the apoptosis of oligodendrocytes.29
In the current study, repeated administration of dexamethasone into neonatal rats between P1 and P5 markedly reduced the expression of MBP in the cerebral hemispheres by P14. In addition, the expression of O4-positive cells in the white matter was decreased on P5 in dexamethasone-treated rats, and pyknotic many O4-positive and several O1-positive cells were present in the corpus callosum of these rats. These pyknotic O4- and O1-positive cells showed morphologic changes suggestive of acute degeneration, including condensation of the cell body and loss and fragmentation of processes.5 Because O4 and O1 antibodies selectively bind to cell-surface glycoconjugates, the observed cytoplasmic labeling is consistent with a loss of plasma membrane integrity.5 In the current study, the pyknotic O4-positive cells of the dexamethasone-treated rats were morphologically similar to preoligodendrocytes, being characterized by a simple, multipolar arbor of processes.6 Many nonpyknotic O1-positive cells of the saline-treated rats displayed a morphology of immature oligodendrocytes, as complex multipolar cells with round somas and multiple distinct, long, branched processes. However, in dexamethasone-treated rats, O1-positive cells with pyknosis within the cell bodies seemed to have a less differentiated morphology with fewer processes than those of nonpyknotic O1-positive cells with a morphology of immature oligodendrocytes.6 These findings suggest that the O1-positive pyknotic cells were likely preoligodendrocytes rather than immature oligodendrocytes. Therefore, in the current study, the cells that showed degenerative changes in dexamethesone-treated rats were primarily preoligodendrocytes.
The 4 successive stages of oligodendrocyte development—early oligodendrocyte progenitors, late oligodendrocyte progenitors (preoligodendrocytes), immature oligodendrocytes, and mature oligodendrocytes—can be defined by the presence of various type-specific surface antigens.22 Preoligodendrocytes give rise to immature oligodendrocytes that are postmitotic myelinating cells, which in turn yield mature oligodendrocytes that express mature myelin markers, such as MBP.27 The O1 antibody labels immature oligodendrocytes, whereas the O4 antibody binds to both preoligodendrocytes and immature oligodendrocytes.9 O4-positive–O1-negative late oligodendrocyte progenitors (preoligodendrocytes) are highly susceptible to hypoxia–ischemia in the pathogenesis of periventricular leukomalacia.5,7 In vitro, preoligodendrocytes are markedly more susceptible to free-radical–mediated injury than are immature oligodendrocytes.4,12 Preoligodendrocytes are the primary oligodendrocyte-lineage stage that is destroyed by apoptosis after hypoxia–ischemia-induced white matter injury in neonatal rats.5,7 Periventricular leukomalacia is a major white matter disorder in premature infants.3,31 Various factors including hypoxia–ischemia and infection–inflammation contribute to the pathogenesis of periventricular leukomalacia,31 the risk for which is increased in infants that receive dexamethasone.16,18,26 Systemic administration of dexamethasone during brain development may be another risk factor associated with white matter injury in premature infants.13,18,26
To confirm that these degenerative morphologic changes in oligodendrocyte progenitors of dexamethasone-treated rats were related to apoptosis, double-staining with TUNEL and O4 or O1 was performed. Many of the TUNEL-positive nuclei in the corpus callosum on P5 originated from oligodendrocyte progenitors. This finding suggests that degenerative changes of oligodendrocyte progenitors may be related to apoptosis induced by dexamethasone. In the current study, the decreased MBP expression in the developing brain after dexamethasone treatment likely was due, at least in part, to apoptosis of oligodendrocyte progenitors during this specific developmental stage. The time period of P1 to P5 of rodents roughly corresponds to 20 to 32 wk after conception in human infants, when preoligodendrocytes comprise a major cell population in the white matter.6 The risk of the white matter damage is high during this well-defined period in human brain development.31 Because preoligodendrocytes is the predominant cell type in the white matter of neonatal rats between P1 and P5, they might be selectively targeted during white matter injury in our neonatal rats as in humans.6,31 However, O1-positive complex multipolar cells, presumably immature oligodendrocytes, were less susceptible to dexamethasone than were O4-positive cells, presumably preoligodendrocytes. Therefore, the death of O4-positive preoligodendrocytes might affect the subsequent differentiation of oligodendrocyte progenitors and hinder myelin formation. However, we were unable to document a significant decrease in the expression or density O1- compared with O4-positive cells in the corpus callosum by P5, despite the marked reduction of MBP expression by P14. Our data regarding changes in oligodendrocyte progenitors were limited to P5, and further study is needed to assess changes in oligodendrocyte progenitors after P5, especially regarding the possibility of the regeneration of oligodendrocyte progenitors after injury.5 Our findings suggest that the administration of dexamethasone during this critical period of brain development primarily affect oligodendrocyte progenitors, particularly preoligodendrocytes, with relatively less effect on immature oligodendrocytes. Therefore, preoligodendrocytes may be the principal target of dexamethasone-induced hypomyelination in neonatal rats, even though immature O1-positive cells might also be susceptible to dexamethasone-mediated injury.
Repeated administration of dexamethasone to our neonatal rats induced morphologic changes that were suggestive of apoptotic degeneration, including TUNEL-positive pyknotic nuclei, in oligodendrocyte progenitors. However, apoptotic changes in preoligodendrocytes were documented only after double-staining with TUNEL and O4 or O1. According to previous reports of studies using animal models of white matter disease after hypoxia–ischemia injury, caspase-3 plays a key role in the apoptosis of late oligodendrocyte progenitor cells.5,14 In addition, corticosteroids induce apoptosis in cells by changing corticosteroid receptor-mediated gene expression, which activates caspases, other proteases, and endonucleases.11 Additional studies are needed to confirm that dexamethasone-induced hypomyelination in the developing brain is due to the apoptotic death of oligodendrocyte progenitors. A previous study indicated that dexamethasone induced degeneration of dendrites but did not induce neuronal apoptosis in the hippocampus of postnatal rats.28 The reason underlying these different findings from the current and previous studies merits further attention.
Repeated administration of dexamethasone significantly reduced body weight in our neonatal rats. In another study, body and brain weights were decreased after repeated administration of dexamethasone, and corticosteroid-associated decreased neurogenesis was suggested as a possible cause of the decrease in brain weight.19 Dexamethasone may affect MBP synthesis by modulating gene expression or by acting on a glucocorticoid receptor.20,21,25,32 In the current study, dexamethasone may have interfered with protein synthesis, affecting brain growth and differentiation of oligodendrocyte progenitors and leading to hypomyelination. Therefore, impairment of protein synthesis by dexamethasone during the differentiation of oligodendrocyte progenitors may explain the decreased expression of MBP at P14 in the absence of significant decreases in the number of O1-positive cells.
In conclusion, we here demonstrated that repeated administration of dexamethasone to neonatal rats on P1 through P5 decreased MBP expression by P14. Through its effects on apoptosis, dexamethasone may induce degenerative changes in oligodendrocyte progenitors, mainly preoligodendrocytes, until P5. Together, the findings of the current and previous studies suggest that dexamethasone-induced hypomyelination is due, at least in part, to injury of preoligodendrocytes as well as to inhibition of myelin formation.2,11,14,17,20,28 Persistent, repeated exposure of immature brains to corticosteroids during critical periods for brain development may be harmful. However, the specific time of administration, the dose, and the circulating levels and types of corticosteroids associated with injury of oligodendrocyte progenitors and the impairment of myelin formation are not known. Additional studies are warranted to define and evaluate the administration time, dose, circulating levels, and types of corticosteroids that affect myelination in the developing brain.2,17,24
Acknowledgment
This study was supported by grants from the Institute of Medical Science, Dankook University Medical Center, in 2008.
References
- 1.Almazan G, Honegger P, Du Pasquier P, Matthieu JM. 1986. Dexamethasone stimulates the biochemical differentiation of fetal forebrain cells in reaggregating cultures. Dev Neurosci 8:14–23 [DOI] [PubMed] [Google Scholar]
- 2.Antonow-Schlorke I, Helgert A, Gey C, Coksaygan T, Schubert H, Nathaniels PW, Witte OW, Schwab M. 2009. Adverse effects of antenatal glucocorticoids on cerebral myelination in sheep. Obstet Gynecol 113:142–151 [DOI] [PubMed] [Google Scholar]
- 3.Back SA. 2006. Perinatal white matter injury: the changing spectrum of pathology and emerging insights into pathogenetic mechanisms. Ment Retard Dev Disabil Res Rev 12:129–140 [DOI] [PubMed] [Google Scholar]
- 4.Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. 1998. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci 18:6241–6253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. 2002. Selective vulnerability of late oligodendrocyte progenitors to hypoxia–ischemia. J Neurosci 22:455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. 2001. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci 21:1302–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Back SA, Riddle A, McClure MM. 2007. Maturation-dependent vulnerability of perinatal white matter in premature birth. Stroke 38:724–730 [DOI] [PubMed] [Google Scholar]
- 8.Committee on Fetus and Newborn 2002. Postnatal corticosteroids to treat or prevent chronic lung disease in preterm infants. Pediatrics 109:330–338 [DOI] [PubMed] [Google Scholar]
- 9.Craig A, Ling Luo N, Beardsley DJ, Wingate-Pearse N, Walker DW, Hohimer AR, Back SA. 2003. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp Neurol 181:231–240 [DOI] [PubMed] [Google Scholar]
- 10.Crowley PA. 1995. Antenatal corticosteroid therapy: a meta-analysis of the randomized trials, 1972 to 1994. Am J Obstet Gynecol 173:322–335 [DOI] [PubMed] [Google Scholar]
- 11.Distelhorst CW. 2002. Recent insights into the mechanism of glucocorticosteroid-induced apoptosis. Cell Death Differ 9:6–19 [DOI] [PubMed] [Google Scholar]
- 12.Fern R, Moller T. 2000. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate-release feedback loop. J Neurosci 20:34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.French NP, Hagan R, Evans SF, Godfrey M, Newnham JP. 1999. Repeated antenatal corticosteroids: size at birth and subsequent development. Am J Obstet Gynecol 180:114–121 [DOI] [PubMed] [Google Scholar]
- 14.Gu C, Casaccia-Bonnefil P, Srinivasan A, Chao MV. 1999. Oligodendrocyte apoptosis mediated by caspase activation. J Neurosci 19:3043–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gumbinas M, Oda M, Huttenlocher P. 1973. The effects of corticosteroids on myelination of the developing rat brain. Biol Neonate 22:355–366 [DOI] [PubMed] [Google Scholar]
- 16.Halliday HL, Ehrenkranz RA, Doyle LW. 2009. Late (>7 days) postnatal corticosteroids for chronic lung disease in preterm infants. Cochrane Database Syst Rev 21:CD001145. [DOI] [PubMed] [Google Scholar]
- 17.Huang WL, Harper CG, Evans SF, Newnham JP, Dunlop SA. 2001. Repeated prenatal corticosteroid administration delays myelination of the corpus callosum in fetal sheep. Int J Dev Neurosci 19:415–425 [DOI] [PubMed] [Google Scholar]
- 18.Jones RA. 2005. Randomized, controlled trial of dexamethasone in neonatal chronic lung disease: 13- to 17-year follow-up study: I. Neurologic, psychological, and educational outcomes. Pediatrics 116:370–378 [DOI] [PubMed] [Google Scholar]
- 19.Kanagawa T, Tomimatsu T, Hayashi S, Shioji M, Fukuda H, Shimoya K, Murata Y. 2006. The effects of repeated corticosteroid administration on the neurogenesis in the neonatal rat. Am J Obstet Gynecol 194:231–238 [DOI] [PubMed] [Google Scholar]
- 20.Melcangi RC, Magnaghi V, Cavarretta I, Riva MA, Martini L. 1997. Corticosteroid effects on gene expression of myelin basic protein in oligodendrocytes and of glial fibrillary acidic protein in type 1 astrocytes. J Neuroendocrinol 9:729–733 [DOI] [PubMed] [Google Scholar]
- 21.Neuberger TJ, Kalimi O, Regelson W, Kalimi M, De Vries GH. 1994. Glucocorticoids enhance the potency of Schwann cell mitogens. J Neurosci Res 38:300–313 [DOI] [PubMed] [Google Scholar]
- 22.Pfeiffer SE, Warrington AE, Bansal R. 1993. The oligodendrocyte and its many cellular processes. Trends Cell Biol 3:191–197 [DOI] [PubMed] [Google Scholar]
- 23.Powell K, Kerkering KW, Barker G, Rozycki HJ. 2006. Dexamethasone dosing, mechanical ventilation, and the risk of cerebral palsy. J Matern Fetal Neonatal Med 19:43–48 [DOI] [PubMed] [Google Scholar]
- 24.Raschke C, Schmidt S, Schwab M, Jirikowski G. 2008. Effects of betamethasone treatment on central myelination in fetal sheep: an electron-microscopical study. Anat Histol Embryol 37:95–100 [DOI] [PubMed] [Google Scholar]
- 25.Schumacher M, Akwa Y, Guennoun R, Robert F, Labombarda F, Desarnaud F, Robel P, De Nicola AF, Baulieu EE. 2000. Steroid synthesis and metabolism in the nervous system: trophic and protective effects. J Neurocytol 29:307–326 [DOI] [PubMed] [Google Scholar]
- 26.Shinwell ES, Karplus M, Reich D, Weintraub Z, Blazer S, Bader D, Yurman S, Dolfin T, Kogan A, Dollberg S, Arbel E, Goldberg M, Gur I, Naor N, Sirota L, Mogilner S, Zaritsky A, Barak M, Gottfried E. 2000. Early postnatal dexamethasone treatment and increased incidence of cerebral palsy. Arch Dis Child Fetal Neonatal Ed 83:F177–F181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skoff RP, Ghandour MS, Knapp PE. 1994. Postmitotic oligodendrocytes generated during postnatal cerebral development are derived from proliferation of immature oligodendrocytes. Glia 12:12–23 [DOI] [PubMed] [Google Scholar]
- 28.Tan CK, Yan J, Ananth C, Kaur C. 2002. Dexamethasone induces dendritic alteration but not apoptosis in the neurons of the hippocampus in postnatal rats. Neurosci Lett 326:206–210 [DOI] [PubMed] [Google Scholar]
- 29.Trousson A, Makoukji J, Petit PX, Bernard S, Slomianny C, Schumacher M, Massaad C. 2009. Cross-talk between oxysterols and glucocorticoids: differential regulation of secreted phopholipase A2 and impact on oligodendrocyte death. PLoS ONE 4:e8080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsuneishi S, Takada S, Motoike T, Ohashi T, Sano K, Nakamura H. 1991. Effects of dexamethasone on the expression of myelin basic protein, proteolipid protein, and glial fibrillary acidic protein genes in developing rat brain. Brain Res Dev Brain Res 61:117–123 [DOI] [PubMed] [Google Scholar]
- 31.Volpe JJ. 2001. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res 50:553–562 [DOI] [PubMed] [Google Scholar]
- 32.Zhu W, Wiggins RC, Konat GW. 1994. Glucocorticoid-induced upregulation of proteolipid protein and myelin-associated glycoprotein genes in C6 cells. J Neurosci Res 37:208–212 [DOI] [PubMed] [Google Scholar]