

Abstract
Traumatic brain injury (TBI) causes many long-term neurological complications. Some of these conditions, such as posttraumatic epilepsy, are characterized by increased excitability that typically arises after a latent period lasting from months to years, suggesting that slow injury-induced processes are critical. We tested the hypothesis that trkB activation promotes delayed injury-induced hyperexcitability in part by promoting reactive axonal sprouting. We modeled penetrative TBI with transection of the Schaffer collateral pathway in knock-in mice having an introduced mutation in the trkB receptor (trkBF616A) that renders it susceptible to inhibition by the novel small molecule 1NMPP1. We observed that trkB activation was increased in area CA3 1 day after injury and that expression of a marker of axonal growth, GAP43, was increased 7 days after lesion. Extracellular field potentials in stratum pyramidale of area CA3 in acute slices from sham-operated and lesioned mice were normal in control saline. Abnormal bursts of population spikes were observed under conditions that were mildly proconvulsive but only in slices taken from mice lesioned 7–21 days earlier and not in slices from control mice. trkB activation, GAP43 upregulation, and hyperexcitability were diminished by systemic administration of 1NMPP1 for 7 days after the lesion. Synaptic transmission from area CA3 to area CA1 recovered 7 days after lesion in untreated mice but not in mice treated with 1NMPP1. We conclude that trkB receptor activation and reactive axonal sprouting are critical factors in injury-induced hyperexcitability and may contribute to the neurological complications of TBI.
Keywords: traumatic brain injury, hippocampus, lesion, plasticity, posttraumatic epilepsy
traumatic brain injury (TBI) causes devastating cognitive, sensory, emotional, and motor deficits in millions of patients. The consequences of TBI are as diverse as its causes, and include acute hemorrhaging, edema, and infection as well as many disabling, long-term neurological conditions. Many neurological complications of TBI, such as posttraumatic epilepsy and central pain syndromes, are characterized by increased excitability, typically arising after a latent period lasting from months to years (Agrawal et al. 2006; Annegers et al. 1998; da Silva et al. 1992; Tasker et al. 1991), suggesting that TBI triggers slow changes within the brain that promote increased excitability during this latent period. If the nature of these slow changes was understood, it might be possible to intervene and prevent some of the adverse complications of TBI prophylactically.
A common consequence of TBI is increased expression and signaling of neurotrophins and their receptors, in particular brain-derived neurotrophic factor (BDNF) and trkB receptors (see, e.g., Harris et al. 2010; Lindvall et al. 1994; Oyesiku et al. 1999; Royo et al. 2006; Wu et al. 2008). These changes are typically transient and observed shortly after the injury, suggesting that they then trigger other downstream effector processes. The nature of such trkB-triggered changes remains incompletely understood. During development, axonal outgrowth is promoted by neurotrophins (Huang and Reichardt 2003; Lu et al. 2005). One hypothesis is that trkB activation after injury promotes aberrant reactive axonal sprouting by pyramidal cells, which in turn promotes increased excitability.
Pyramidal cells can generate new local axon collaterals after injury in vivo (Greer et al. 2011; Nadler 2003; Perez et al. 1996). Irreparable damage to axons is a common feature of TBI and can result directly from the trauma of a penetrating injury or indirectly from the shearing and stretching of axons that occur with rapid acceleration and deceleration of the brain (Povlishock and Katz 2005). The cells whose axons are injured usually survive (Singleton et al. 2002), but the axons themselves may be damaged irreversibly and ultimately degenerate (Greer et al. 2011). In hippocampal slice cultures, axonal injury causes upregulation of BDNF and trkB, the growth of new axon collaterals by CA3 cells, and hyperexcitability. This reactive axonal sprouting results in a 50% increase in the probability that any two CA3 cells are connected by an excitatory synapse (McKinney et al. 1997). Upregulation of the growth-associated protein GAP43 after axonal injury is reduced in cultures from mice with decreased trkB receptor expression (Dinocourt et al. 2006). Inhibition of trkB receptors might therefore inhibit both injury-induced axonal sprouting and injury-induced increases in excitability. However, these mice had a constitutive decrease in trkB levels, and the process governing axonal sprouting in cultures might differ from those in intact tissue. Furthermore, there has been no physiological evidence of axonal regeneration after injury in vivo. Accordingly, the role of trkB after TBI is not yet well established.
As a more direct and physiologically relevant test of the hypothesis that TBI leads to hyperexcitability because of trkB receptor-dependent axonal sprouting, we performed transections of the Schaffer collateral pathway in knock-in mice expressing trkB receptors with a single point mutation (F616A) that allows for inhibition of autophosphorylation with the small molecule 1NMPP1 (Chen et al. 2005).
MATERIALS AND METHODS
All procedures involving animals were approved by the University of Maryland School of Medicine Institutional Animal Care and Use Committee.
Transgenic mice.
Chen et al. (2005) created a line of knock-in mice, called trkBF616A, in which all trkB receptors have a single amino acid substitution in the intracellular ATP binding domain that renders them sensitive to block by 1NMPP1 [1-naphthylmethyl 4-amino-1-tert-butyl-3-(p-methyl phenyl)pyrazolo[3,4-d]pyrimidine], a small molecule derived from the kinase inhibitor PP1 (Bishop et al. 2000), with high affinity in vitro and in living animals. The modified trkB receptors are fully functional in the absence of drug, and the mice have no detectable phenotype. We crossed trkBF616A mice with the thy1-GFPm line of mice (Feng et al. 2000), yielding mice with blockable trkB receptors and strong green fluorescent protein (GFP) expression in a subset of CA1 pyramidal cells.
Schaffer collateral transection in vivo.
Schaffer collateral (SC) transections were performed as described previously (Dinocourt et al. 2011). Briefly, 4- to 6-wk-old male mice were anesthetized with an intraperitoneal injection of a ketamine (40–80 mg/kg)-xylazine (5–10 mg/kg) mixture, immobilized in a stereotactic frame (David Kopf Instruments), and placed on a heating pad to maintain normal body temperature. An incision was made along the midline of the scalp, and the skin and fascia were retracted. Two holes (∼2 × 4 mm) were made in the skull over the dorsal surface of the brain. A microknife (Fine Science Tools) mounted on a stereotactic carrier was inserted into the brain to a depth of 3.5 mm from the cortical surface at a starting position 3 mm caudal from bregma and 3.25 mm lateral from the midline. The scalpel was left in place for 1 min before transection of the SC pathway along approximately two-thirds of the rostro-caudal extent of the hippocampus by moving the scalpel simultaneously 2 mm rostrally and 2.25 mm medially. The scalpel was then left in place for 1 min before being withdrawn slowly. The procedure was then repeated in the other hemisphere. Age-matched sham-operated control mice were treated in the same way, including the opening of the skull, but the dura was left intact and no lesion was made. It should be noted that this procedure also produces damage in the overlying neocortical tissue.
Osmotic pump implantation.
Osmotic minipumps (Alzet model 2001; 1.0 μl/h over 7 days) were used for in vivo drug delivery. Minipumps were filled with either sterile 1NMPP1 (0.14 nmol·g−1·h−1), using 2% DMSO-2% Tween 20 in 0.9% saline as a vehicle, or vehicle alone. Pumps were then incubated for priming in sterile 0.9% saline at 37°C for 4 h prior to implantation. Immediately after SC transection, a small incision was made just above the scapulae and a subcutaneous pocket was created to house the pump. Pumps were wiped with alcohol pads and inserted into the pocket. The incision was closed with 5.0 nylon sutures in an interrupted pattern. After 7 days of treatment the pump was removed, as hippocampal slices were being prepared for electrophysiology, and the volume of liquid remaining in the pump was measured to ensure pump efficacy.
Hippocampal slice preparation.
Age-matched sham-operated and lesioned mice were deeply anesthetized and decapitated, and the brain was removed and placed in ice-cold oxygenated artificial cerebrospinal fluid (ACSF) (Tian and Baker 2002) composed of (in mM) 125 NaCl, 2 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2, and 20 glucose and titrated to pH 7.4 by bubbling with 95% O2-5% CO2. The hippocampus was dissected free, and 400-μm-thick sections were cut with a vibratome according to standard procedures. Slices were then incubated at the air/gas interface for at least 1 h in a holding chamber at room temperature (RT) before being transferred to a recording chamber.
Electrophysiology.
Slices were continuously submerged in RT ACSF composed of (in mM) 125 NaCl, 5 KCl, 26 NaHCO3, 3 CaCl2, 1 MgCl2, and 20 glucose and saturated with 95% O2-5% CO2. Extracellular recordings were obtained with ACSF-filled glass micropipettes (tip resistance ∼1 MΩ) placed in stratum (str.) pyramidale for recording population spikes. Responses were evoked via a concentric metal stimulating electrode placed in str. radiatum at the border of areas CA3 and CA1. A constant-voltage stimulus (amplitude 1–20 V, duration 100 μs) was applied at 0.1 Hz. Voltage signals were amplified 100-fold and low-pass filtered at 1 kHz (npi Electronics) and digitized and analyzed with pCLAMP software (Molecular Devices).
To measure functional recovery across the lesion, stimuli were applied starting at a low voltage until a minimal response in area CA3 was elicited and thereafter increased in 0.2-V increments. These data were pooled, and a linear regression was applied (Pearson's correlation P < 0.01 for all experiments). To compare the response of CA3 cells to bicuculline application under different conditions, we calculated a modified coastline bursting index (CBI) (Korn et al. 1987). A 100-ms-long epoch of the evoked response in control ACSF and in ACSF containing a mildly proconvulsive concentration of bicuculline methylchloride (0.1 μM) was measured in the same slice. The CBI of the traces was then calculated as the sum of the point-to-point voltage differences during the length of the epoch after exporting the traces to Excel. The resulting CBI value is sensitive to changes in the number or amplitude of population spikes. It increases when the number of neurons in the vicinity of the recording electrode firing action potentials in response to the stimulus increases and when the number of cells firing multiple action potentials increases. The percent change in CBI in response to bicuculline application was then calculated. Mean values for sham-operated control animals were compared from all time points, and no difference was found (Student's t-test, P = 0.9) so they were pooled for quantitative analyses. For the number of population spikes, only responses >0.2 mV were counted. Population spike amplitude was calculated as the difference in voltage between the prestimulus baseline and the peak of the negative deflection.
Hippocampal slice cultures.
Organotypic slice cultures were prepared with the roller tube method (Gähwiler et al. 1998). Briefly, 400-μm-thick sections were prepared and mounted in droplets of chicken plasma supplemented with fibrin onto poly-l-lysine-coated glass coverslips. Coverslips were individually placed into flat-sided culture tubes and incubated in a roller drum at 37°C for 3–19 days in serum-containing medium. SC transections were made after 14 days in vitro. Cultures were placed in a petri dish and bathed for 10 min in ACSF containing 3 μM kynurenate. The SC pathway was transected by dragging a razor blade shard from the hippocampal fissure through to the edge of the culture. Sham-operated controls were bathed in cutting solution for the same period of time without transection of the SC pathway. Sham-operated and SC-lesioned cultures were placed back in roller tubes with either fresh medium or medium containing either 3 μM 1NMPP1 in 0.3% DMSO or DMSO alone. Cultures were incubated for 5 days and then removed at 19 days in vitro (DIV) and either fixed in 4% paraformaldehyde (PFA) or placed in lysis buffer for protein extraction.
Immunoprecipitation of trkB receptors.
Area CA3 was dissected out of hippocampal slices under ice-cold ACSF. The tissue was placed in lysis buffer, homogenized, and centrifuged. trkB protein was precipitated with protein A beads (KPL) and an anti-trkB antibody (BD Transduction no. 610101; 1:2,000). Resulting proteins were loaded onto a 4–12% Bis-Tris gel. After running in 1× NuPAGE MOPS SDS running buffer, the gel was transferred onto polyvinylidene difluoride (PVDF) membranes in 1× NuPAGE transfer buffer (in 10% methanol). The membrane was blocked with 5% nonfat dry milk in buffer containing 1 M Tris-buffered saline (TBS) and 0.05% Tween and probed with antibodies against phosphorylated trk receptors (Tyr490; Cell Signaling no. 9141; 1:1,000) at 4°C overnight. After rinses in TBS-Tween, the membranes were incubated for 1 h at RT in horseradish peroxidase-conjugated goat anti-rabbit IgG (Cell Signaling no. 7074; 1:1,000). The immunoblot was developed with SuperSignal West Femto Chemiluminescent Substrate (ThermoScientific). Membranes were then stripped, blocked, and reprobed with antibody against trkB (BD Transduction no. 610101; 1:2,000) that recognizes the full-length and truncated forms of trkB. Membranes were imaged on a Kodak Imaging Station 2000R for determination of protein levels with Kodak ID Image Analysis Software (Kodak Molecular Imaging Systems). Because only full-length trkB receptors can be phosphorylated, trkB activation was quantified as the ratio of phosphorylated trk to total full-length trkB.
Immunohistochemistry.
Hippocampal slices, prepared as described above and used for electrophysiological recording, were fixed overnight in 4% PFA. Slices were then embedded in 10% gelatin blocks and resectioned at 40 μm on a vibratome. All free-floating sections to be processed for GAP43 immunohistochemistry were first rinsed in 0.1 M PBS. Sections were incubated for 1 h at RT in PBS containing 0.3% Triton X-100 and 3% normal donkey serum (NDS) (Sigma). Slice cultures were fixed in 4% PFA overnight at 4°C, rinsed, and blocked in 1% bovine serum albumin and 1% NDS in 0.1 M PBS for 1 h at RT on a rocker. Tissue was incubated overnight at RT in a rabbit anti-GAP43 antibody (courtesy of Frank Margolis, University of Maryland, Baltimore) at 1:1,000. Sections were then rinsed and incubated for 1 h at RT in donkey anti-rabbit Cy3 secondary antibodies (JacksonImmuno no. 711-165-152; 1:500) in PBS containing 1% NDS. Tissue was again rinsed and then incubated in 1% DAPI (Invitrogen) in PBS for 30 min. Slices were then rinsed and positioned on 2% gelatin-coated slides and mounted with Fluoromount aqueous medium (Sigma).
Image analysis.
Stained sections were imaged under identical conditions on an Olympus FluoviewX confocal microscope. Images were taken with a ×20 oil immersion objective at 1-μm steps through the tissue, resulting in ∼40 optical sections/slice in two channels: 568-nm excitation for GAP43 staining and 488-nm excitation for DAPI. Images were analyzed for GAP43 fluorescence with ImageJ software (National Institutes of Health). Multichannel z-stacks were opened, and regions of interest in each layer (str. oriens, pyramidale, and radiatum) were chosen using only the DAPI staining to eliminate bias for GAP43 staining. Unmodified GAP43 images were then converted to 16-bit grayscale images, and background was subtracted by means of a thresholding procedure removing all signals below a user-selected cutoff, as determined in small regions adjacent to the tissue. The mean fluorescence intensity in arbitrary units (values as provided by ImageJ) was measured through 5 μm in the center of the stack. Data were then transferred to Microsoft Excel. For each slice, three regions of interest per layer were analyzed. Data were first averaged for each slice and then averaged across slices.
Western blot analysis.
Organotypic hippocampal cultures were removed from the coverslip and pooled (4 hippocampi/tube) in RIPA lysis buffer. For in vivo analysis, area CA3 was dissected from hippocampal slices and placed in ice-cold lysis buffer. All tissues were placed on ice and homogenized with a microtube pestle and a motorized holder for 1 min each. Homogenates were centrifuged for 10 min at 14 g and 4°C, and supernatant was removed to a new tube. Protein concentration was determined with the Bradford assay. Briefly, 20 μl of unknown sample or 20 μl of standard proteins was added to 150 μl of Coomassie reagent in a 96-well plate (3 per sample). Absorbance was read with a microplate reader and software. Protein concentrations were calculated based on the standard curve obtained with standard protein samples of known concentration. Protein concentrations were made equal by the addition of water to equal volumes, rendering 10 μg of unknown protein samples in 20 μl of volume. SDS-PAGE sample buffer (5 μl) was added to each tube, and the samples were incubated at 90°C for 5 min before being loaded onto a gel. Samples were separated out on a 4–12% Bis-Tris gel for 1 h at 150 V and then transferred to a PVDF membrane (Invitrogen) for 2 h at 30 V. The membrane was blocked in 5% nonfat milk in 0.1 M PBS for 1 h at RT on a rocker. Membranes were incubated in an antibody to rabbit GAP43 (courtesy of Frank Margolis; 1:1,000) in blocking solution overnight at RT. The membrane was washed and incubated for 1 h at RT in a horseradish peroxidase-conjugated anti-rabbit IgG antibody (Cell Signaling no. 7074; 1:1,000) diluted in 0.1 M PBS and then rinsed. Bands were imaged and analyzed as above. Membranes were stripped with stripping buffer and reprobed with a mouse anti-GAPDH antibody (Sigma no. G8795; 1:2,000) with a horseradish peroxidase-conjugated anti-mouse IgG antibody (Cell Signaling no. 7076; 1:1,000) secondary. Membranes were imaged and analyzed as described above. GAPDH was used as a loading control.
Statistics.
Statistical analyses were performed with the IBM SPSS Statistics 19 software package (IBM, Armonk, NY). Statistical significance for CBI, number of population spikes, Western blot analysis of phospho-trkB, and GAP43 and GAP43 immunofluorescence was determined by ANOVA and Tukey post hoc tests or a Fisher's least significant difference (LSD) (number of population spikes, Figs. 1C and 8C). P < 0.05 was accepted for statistical significance at α = 0.05. A Pearson's correlation (2 tailed) was used for analysis of CA3 versus CA1 population spikes in Figs. 3B and 7B, and all correlations were linear (P < 0.01). A Kolmogorov-Smirnov test was used to compare the distributions in Fig. 7C. For cultures with or without GAP43-immunoreactive axons (Fig. 5C), we used the Fisher exact test, using the GraphPad Software calculator (GraphPad Software, La Jolla, CA). All data are presented as means ± SE.
Fig. 1.
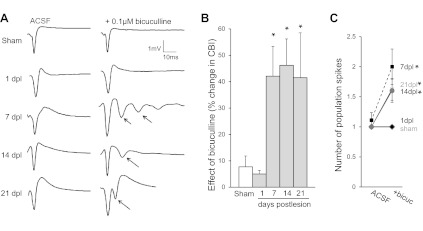
Schaffer collateral (SC) transection leads to a delayed and persistent increase in neuronal excitability. A: sample extracellular recordings of population spikes in slices from sham-operated control trkBF616A mice and trkBF616A mice that had SC lesions placed 1, 7, 14, and 21 days earlier. In all conditions, only a single population spike was recorded in control artificial cerebrospinal fluid (ACSF). Application of a low concentration of bicuculline (0.1 μM) had no significant effect on either the coastline burst index (CBI; B) or the number of population spikes in sham-operated control slices or slices taken at 1 day postlesion (dpl) (C) but increased both CBI and the number of population spikes significantly in slices taken at 7dpl, 14dpl, and 21dpl (*P < 0.05 compared with sham operated for all cases). Arrow, additional population spike.
Fig. 8.
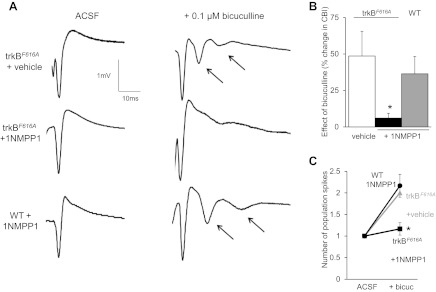
Blocking trkBF616A receptors with 1NMPP1 in vivo prevents lesion-induced hyperexcitability. A: sample traces of extracellular recordings in str. pyramidale of area CA3 in slices taken from mice lesioned 7 days earlier. Multiple population spikes were observed upon application of 0.1 μM bicuculline in slices from vehicle-treated trkBF616A mice and in WT mice treated with 1NMPP1, whereas inhibition of trkBF616A receptors with 1NMPP1 prevented the occurrence of multiple population spikes. Changes in the CBI (B) and number of population spikes (C) produced by bicuculline application were significantly less in slices from trkBF616A mice that received 1NMPP1 (n = 6) compared with 7dpl vehicle-treated trkBF616A mice (n = 4) and WT mice receiving 1NMPP1 (n = 6) (*P < 0.05 compared with vehicle treated, α = 0.05). Arrow, additional population spike.
Fig. 3.

Reestablishment of synaptic transmission across the lesion after 7 days. A: population spikes were recorded simultaneously in str. pyramidale of areas CA1 and CA3 in normal ACSF in response to stimuli delivered within str. radiatum of area CA3 in slices taken from sham-operated trkBF616A mice and trkBF616A mice in which a SC lesion was made 1 or 7 days earlier. One day after the lesion, no response could be elicited in area CA1 despite robust responses in area CA3 because of the transection of the SC axons. By 7 days after lesion, in contrast, robust responses were elicited in area CA1. B: responses were elicited with stimulation at various intensities in slices from sham-operated mice (n = 6 slices from 4 mice) or slices from mice lesioned 1 (n = 5 slices from 4 mice) or 7 (n = 5 slices from 5 mice) days earlier and are plotted together with a best-fit line (Pearson's correlation r values: sham operated r = 0.9; 1dpl r = 0.5; 7dpl r = 0.8; all correlations are significant at P = 0.01). The recovery of transmission from area CA3 to area CA1 indicates that CA3 cell axons had regenerated and regrown across the lesion after 7 days.
Fig. 7.
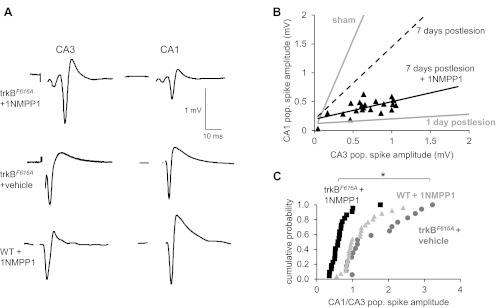
Inhibiting trkB receptors impaired the reestablishment of synaptic transmission across the lesion. A: population spikes were recorded simultaneously in str. pyramidale of areas CA1 and CA3 in normal ACSF in response to stimuli delivered within str. radiatum of area CA3 in slices taken from trkBF616A or WT mice treated with vehicle or 1NMPP1 for 7 days after SC lesions. After systemic administration of 1NMPP1, only very weak responses could be elicited in area CA1 despite the robust responses in area CA3. 1NMPP1 had no effect in WT mice. B: responses elicited with stimulation at various intensities in slices from trkBF616A mice treated with 1NMPP1 for 7 days after SC lesions (n = 7 slices from 5 mice) are plotted together with a best-fit line (Pearson's correlation r value; 7dpl+1NMPP1 r = 0.6; correlations are significant at P = 0.01). For comparison, the lines fit from the data for sham-operated control mice and mice tested at 1dpl and 7dpl shown in Fig. 3 are replotted. The poor recovery of transmission from area CA3 to area CA1 indicates that inhibition of trkB receptors impaired the regeneration and regrowth of axons across the lesion. C: plot of the cumulative probability distribution of the ratio of CA1 to CA3 population spike amplitudes obtained as in B. The distribution of responses in 1NMPP1-treated trkBF616A animals was significantly different from either 1NMPP1-treated WT animals or vehicle-treated trkBF616A animals (*P < 0.001, Kolmogorov-Smirnov test).
Fig. 5.
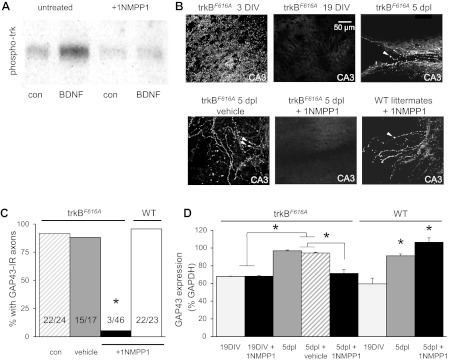
Injury-induced axonal sprouting was reduced by inhibiting trkBF616A receptors with 1NMPP1 in hippocampal slice cultures. A: brain-derived neurotrophic factor (BDNF; 50 ng/ml) triggered phosphorylation of trkB receptors in hippocampal slice cultures prepared from trkBF616A mice, as in wild-type (WT) animals. Administration of 3 μM 1NMPP1 to hippocampal slice cultures prepared from trkBF616A mice prevented BDNF-induced trkB autophosphorylation. B: confocal images showing numerous GAP43-immunoreactive puncta in slice cultures from trkBF616A mice after 3DIV, while axons are still growing, but not at 19DIV, when growth is complete. Five days after SC transection, GAP43-immunoreactive axons capped with growth cones (arrowheads) were apparent in the vicinity of the lesion in cultures treated with normal culture medium or medium containing DMSO. In contrast, no GAP43-immunoreactive axons were seen at 5dpl in cultures from trkBF616A mice in which 1NMPP1 was administered. GAP43-immunoreactive axons were apparent in cultures from WT mice treated with 1NMPP1, indicating that its action is specific for the mutated trkBF616A receptors. C: % of lesioned cultures displaying any GAP43-immunoreactive (IR) axons was calculated. 1NMPP1 produced a significant decrease in cultures from trkBF616A mice but not in cultures from WT mice (*Fisher's exact test, P < 0.0001, drug vs. medium; P = 1.0, medium vs. vehicle and medium vs. WT with drug, n from each condition shown in bars). D: GAP43 expression in slice cultures from trkBF616A and WT mice was quantified with Western blotting. GAP43 expression was significantly increased at 5dpl in untreated and vehicle-treated cultures (*P < 0.05, n = 7 replicates) but not after application of 1NMPP1 to cultures from trkBF616A mice. In cultures from WT mice, GAP43 expression increased significantly in the presence and absence of 1NMPP1 (*P < 0.05 compared with 19DIV; n = 6 replicates).
RESULTS
SC transection leads to delayed and persistent increase in excitability in ex vivo hippocampal slices.
To study the consequences of an axonal injury on intact tissue, we made lesions of the SC pathway in vivo in trkBF616A mice. Stereotactic coordinates were used to create a lesion of the SC pathway across approximately two-thirds of the length of the hippocampus that extended from the outer surface of the hippocampus to the hippocampal fissure (Dinocourt et al. 2011). To assay the consequences of these lesions on neuronal excitability, we prepared ex vivo hippocampal slices from animals lesioned 1, 7, 14, and 21 days earlier and from sham-operated control animals. Population spikes in str. pyramidale of area CA3 were recorded in response to local stimulation within str. radiatum. We observed single population spikes in slices from all animals at all times after lesion when recording in ACSF (Fig. 1, A and C), indicating that the lesioned tissue was not strongly hyperexcitable. Increased excitability in injured hippocampus can be revealed by mildly proconvulsive manipulations that slightly perturb the balance between excitation and inhibition (Dinocourt et al. 2011; McKinney et al. 1997). We therefore determined the effects of application of a very low concentration of the GABAA receptor antagonist bicuculline methylchloride (0.1 μM), a concentration 100-fold lower than normally used to elicit epileptiform discharge (Haas and Ryall 1980; Schwartzkroin and Prince 1980), on population spikes in slices from lesioned and sham-operated control mice. To quantify the effects of bicuculline on the number and amplitude of population spikes, we calculated the change in CBI (Korn et al. 1987), which detects changes in the number and/or amplitude of population spikes. This low concentration of bicuculline did not affect the CBI in slices obtained from control mice (increase in CBI = 7 ± 4%; n = 10 slices, 5 animals). With an ANOVA, there was a significant effect of the lesion [F(4,32) = 6.54, P = 0.001] on the change in CBI. Tukey post hoc analysis indicated that significant increases in CBI in slices in response to 0.1 μM bicuculline were observed from lesioned mice 7, 14, and 21 days earlier [increase in CBI = 42 ± 11% (n = 6 slices, 5 animals, P = 0.02), 46 ± 10% (n = 7 slices, 5 animals, P = 0.01), 42 ± 11% (n = 5 slices, 5 animals, P = 0.42) compared with sham-operated control animals; Fig. 1, A and B]. However, there was no significant change in CBI or number of population spikes in slices from mice lesioned 1 day earlier [increase in CBI = 5 ± 1% (n = 5 slices, 5 animals, P = 0.9) compared with sham-operated control animals; Fig. 1]. The increase in CBI in lesioned slices was due largely to significant increases in the number of population spikes [F(4,38) = 1.62, P = 0.001; Fig. 1, A and C]. We conclude that axonal injury produces a delayed increase in the excitability of the population of injured cells revealed under mildly proconvulsive conditions that is persistent for at least 21 days.
Regrowth of axonal connections restores transmission across the lesion.
The growth-associated protein of 43 kDa (GAP43) is expressed at high levels in growing axons and has been used to selectively label newly sprouted or regenerating axons (Bendotti et al. 1997; Benowitz et al. 1990; Dinocourt et al. 2006; Harris et al. 2010; McKinney et al. 1997; Oestreicher et al. 1997). We measured a significant difference in GAP43 protein levels [F(7,37) = 3.20, P = 0.01] in area CA3 of hippocampal tissue with Western blotting and detected significant increases at 7 days after lesion compared with time-matched 7-day sham-operated control [201 ± 24% and 106 ± 5%, respectively (P = 0.01, n = 7 animals); Fig. 2], suggesting that a period of transient reactive axonal sprouting had occurred with a delay after the injury.
Fig. 2.

Expression of GAP43 protein was increased after injury. A: Western blot analysis of GAP43 protein at 1, 7, 14, and 21 days after SC lesions in tissue taken from area CA3 in slices from sham-operated trkBF616A mice (left) and trkBF616A mice with a SC lesion (right). GAPDH was used as a loading control. B: GAP43 expression was quantified by normalizing to the GAPDH loading control for sham-operated and lesioned mice at each time point. GAP43 protein levels were significantly increased at 7 days after lesion (*P < 0.05 compared with 7 day sham operated).
If GAP43 is indicative of growing axons, are they capable of restoring synaptic transmission across the lesion? We recorded population spikes simultaneously in areas CA1 and CA3 in response to stimuli delivered in area CA3 over a range of intensities (Fig. 3). In slices from unlesioned sham-operated mice, there was a large increase in the amplitude of the responses in both regions as stimulus strength was increased. In slices taken from mice 1 day after lesion, in contrast, responses were increased with increasing stimulation strength in area CA3 but not in area CA1, as expected if SC axons were transected. Because stimulation in area CA3 elicited robust responses in both regions by 7 days after lesion, however, we infer that transmission had been restored across the lesion by newly grown axons.
trkB activation is increased after injury.
BDNF-trkB signaling promotes axonal growth during CNS development and injury-induced axonal sprouting in hippocampal slice cultures (Dinocourt et al. 2006). We assayed BDNF-trkB signaling after injury in vivo in hippocampal tissue from area CA3 taken 24 h, 48 h, and 7 days after SC transection. trkB was first immunoprecipitated, and then phospho-trk and total trkB levels were analyzed by Western blotting. We observed a significant peak in phospho-trk levels at 24 h after lesion {82 ± 6% [F(6,27) = 5.90, P = 0.001 for ANOVA, P < 0.01 for Tukey post hoc analysis, n = 4 animals]; Fig. 4} compared with all other time points and time-matched, sham-operated control animals. Since BDNF binding to trkB initiates the autophosphorylation of the receptor, increases in phospho-trk levels indicate that BDNF-trkB signaling has occurred at a time likely to correspond to the initiation of GAP43 induction and axonal sprouting.
Fig. 4.

Activation of trkB receptors after SC transection. A: trkB receptors in area CA3 in slices from intact trkBF616A mice and trkBF616A mice in which a SC lesion was placed 24 h, 48 h, and 7 days earlier were immunoprecipitated with protein A beads bound to trkB antibodies. The resulting protein was separated by SDS-PAGE, and membranes were probed with a phospho-trk antibody, then stripped and reprobed with a phosphorylation-independent anti-trkB antibody. B: phosphorylation of trkB receptors, quantified after normalizing to total trkB, was significantly increased 24 h after lesion (*P < 0.05 compared with 24 h sham operated; n = 4 mice in each condition). Single lanes with representative bands for each condition are shown for clarity of the figure.
Inhibiting trkBF616A receptors with 1NMPP1 prevents axonal sprouting and functional recovery in vitro and in vivo.
Is activation of trkB receptors required for injury-induced axonal sprouting? We used knock-in trkBF616A mice (Chen et al. 2005) that allow pharmacological inhibition of trkB signaling in vivo with 1NMPP1 to address this question. We first tested the effectiveness of trkB inhibition in hippocampal slice cultures prepared from these mice. Cultures were treated with BDNF (50 ng/ml) in control culture medium or medium containing 3 μM 1NMPP1 (in 0.3% DMSO) and then subjected to immunoprecipitation of trkB receptors. Immunoblotting with antibodies to phosphorylated trk receptors revealed that BDNF triggered strong activation of trkB in control medium but not in medium containing 1NMPP1 (Fig. 5A). We thus confirmed that BDNF activates the trkBF616A receptors effectively in the absence of 1NMPP1 and that 1NMPP1 is an effective inhibitor of the trkBF616A receptors in vitro.
Next, sham-operated and SC-lesioned cultures (McKinney et al. 1997) were maintained for 5 days in medium containing either 3 μM 1NMPP1 or vehicle (0.3% DMSO). Axonal sprouting was assessed by immunocytochemistry (Fig. 5, B and C) and Western blotting for GAP43 (Fig. 5D). Immature cultures showed high levels of GAP43 immunoreactivity at 3DIV; however, GAP43 levels were undetectable by histological methods by 14DIV (not shown) and 19DIV, when the cultures have matured, as described in wild-type mice and rats (Dinocourt et al. 2006; McKinney et al. 1997). By 5 days after a SC transection, GAP43-immunoreactive axons, many capped with growth cones, were apparent in 92% (22 of 24) and 88% (15 of 17) of lesioned cultures maintained in normal culture medium or medium with vehicle, respectively, and these were not significantly different (Fisher's exact test, P = 1.0, medium vs. vehicle). A peak in GAP43 protein levels [F(7,45) = 23.32, P = 0.001 for ANOVA] at 5 days after lesion was also seen in cultures in control medium (97 ± 1%) and medium with vehicle (95 ± 1%) and was significantly different (P < 0.05 for Tukey post hoc analysis) from that of GAP43 protein at 19DIV with and without drug (68 ± 1% and 68 ± 1%, respectively). We conclude that the regulation of GAP43 expression is similar in hippocampal tissue from trkBF616A receptor-expressing and wild-type mice, and that the vehicle has no effect on GAP43 expression.
In contrast to these controls, only 5% (3 of 46) of cultures prepared from trkBF616A mice and treated with the inhibitor 1NMPP1 for 5 days after the lesion contained visible GAP43-immunoreactive axons, with the majority showing none, significantly different from controls (Fisher's exact test, P < 0.0001). In wild-type cultures, treatment with 1NMPP1 had no effect on the percentage of lesioned cultures that displayed GAP43-immunoreactive axons [22 of 23 (96%), Fisher's exact test, P = 1.0, medium vs. 1NMPP1] or on the increase in GAP43 protein expression, indicating that the effect of 1NMPP1 is specific to the mutated trkB receptor. The lack of GAP43-positive axons and the prevention of increased GAP43 protein levels after SC transection supports use of this mouse model for determining whether trkB receptors play a role in hyperexcitability and injury-induced axonal sprouting in vivo.
1NMPP1 is able to cross the blood-brain barrier (Chen et al. 2005). We therefore tested the effects of systemic 1NMPP1 administration in adult trkBF616A mice via osmotic minipumps. Pumps containing 1NMPP1 (0.14 nmol·g−1·h−1 in 2% Tween 20/2% DMSO vehicle) or vehicle alone were implanted at the time the SC lesions were made. To determine how effective 1NMPP1 administered in this manner was at inhibiting trkB receptors, we harvested cortical tissue 7 days later and placed it in a solution of 50 ng/ml BDNF for 5 min before homogenizing the tissue and analyzing trkB autophosphorylation as described above. We observed that phospho-trkB levels were ∼50% lower in tissue from lesioned mice treated with 1NMPP1 than in tissue from untreated sham-operated or wild-type lesioned mice treated with 1NMPP1 [F(2,20) = 8.67, P = 0.002 for ANOVA; 15 ± 3% (n = 5 animals), 32 ± 3% (n = 5 animals), and 25 ± 5% (n = 5 animals) respectively, P < 0.05; Fig. 6, A and B].
Fig. 6.
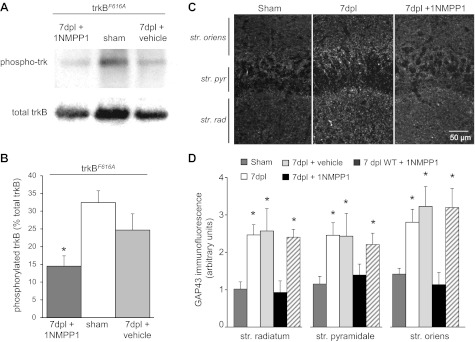
trkB activation and injury-induced GAP43 upregulation were decreased by inhibiting trkBF616A receptors with 1NMPP1 in vivo. A: BDNF-induced phosphorylation of trkB receptors was reduced significantly in cortical tissue prepared from trkBF616A mice administered 1NMPP1 systemically in vivo for 7dpl compared with tissue from vehicle-treated and sham-operated control trkBF616A mice. B: BDNF-induced phosphorylation of trkB receptors, quantified after normalizing to total trkB, was significantly decreased in 1NMPP1-treated mice compared with sham-operated and vehicle-treated control (*P < 0.05). C: treatment of trkBF616A mice with 1NMPP1 in vivo for 7 days reduced immunostaining for GAP43. D: quantification of fluorescence intensity of GAP43 immunostaining in str. radiatum, pyramidale, and oriens was significantly increased at 7dpl in all layers (*P < 0.05 compared with sham operated; n = 6) and in slices from WT mice treated with 1NMPP1 but was similar to controls in slices from trkBF616A mice treated with 1NMPP1.
Hippocampal tissue harvested from these same mice was stained with antibodies against GAP43. Confocal images were taken in area CA3 adjacent to the lesion site (Fig. 6C), and the intensity of GAP43 immunofluorescence was quantified. Inhibiting trkF616A receptors with 1NMPP1 significantly decreased the amount of GAP43 immunoreactivity in tissue subjected to SC transection compared with tissue from untreated mice taken 7 days after lesion and had no effect on tissue from vehicle control (Fig. 6D). We conclude that inhibiting trkB receptors inhibits injury-induced axonal sprouting.
We next asked whether inhibiting trkB signaling with 1NMPP1 would impair functional recovery of synaptic transmission across the lesion. At 7 days after lesion, stimulation in area CA3 triggered responses in area CA1 in wild-type mice treated with 1NMPP1 (n = 6 slices from 5 mice) and in trkBF616A mice treated with vehicle (n = 4 slices from 4 mice) but not in trkBF616A mice treated with 1NMPP1 (n = 7 slices from 5 mice) (Fig. 7). The lack of effect in wild-type mice indicates that the ability of 1NMPP1 to inhibit recovery is due solely to its action at the mutant trkBF616A receptors. We conclude that trkB receptor activation is necessary for injury-induced axonal sprouting and that these newly sprouted axons can mediate recovery of transmission across the lesion.
Blocking trkBF616A receptors with 1NMPP1 prevents injury-induced hyperexcitability.
To determine whether axonal sprouting contributes to the hyperexcitability after injury, we tested the effects of 0.1 μM bicuculline in slices from trkBF616A and wild-type mice treated with 1NMPP1 at 7 days after lesion. Stimulation in area CA3 resulted in single population spikes in all slices in control saline (Fig. 8, A and C). Bicuculline produced a significant increase in the number of population spikes and in the CBI [F(4,31) = 5.12, P = 0.003 for ANOVA] in slices from wild-type animals but not in slices from trkBF616A mice treated with 1NMPP1 (increase in CBI 36 ± 12% and 6 ± 3% respectively, P < 0.05 for Tukey post hoc analysis; Fig. 8). We conclude that trkB receptor activation promotes the development of hyperexcitability after TBI by promoting axonal sprouting.
DISCUSSION
Axonal injury induces delayed and persistent hyperexcitability.
To study the consequences of a relatively selective injury to the axons of brain cells in isolation, we developed a surgical approach to transect the SC pathway and thereby damage the axons of large numbers of CA3 pyramidal cells (Dinocourt et al. 2011). We have previously shown that this procedure results in very low amounts of cell death (Dinocourt et al. 2011). We have also reported that SC transection results in a delayed and persistent increase in the excitability of the denervated CA1 cell population (Dinocourt et al. 2011). We now report that this injury also triggers a delayed increase in the excitability of the CA3 cell population, as observed in ex vivo hippocampal slices from lesioned animals. Under control conditions, single stimuli delivered in str. radiatum within CA3, presumably triggering a mix of antidromic and orthodromic activation of CA3 cells, produced single population spikes in extracellular recordings made in str. pyramidale in slices from unlesioned animals and from animals lesioned 1–21 days earlier. In slices from unlesioned animals, application of a mildly proconvulsive concentration of bicuculline (0.1 μM), a concentration 100-fold lower than that normally used to induce epileptiform discharge (see, e.g., Haas and Ryall 1980; Schwartzkroin and Prince 1980), had no significant effect on the number of population spikes or on the CBI, a measure of neuronal excitability. This concentration of bicuculline is thus subthreshold for producing a significant imbalance of excitation and inhibition in intact, normal tissue. In slices from animals lesioned 7–21 days earlier, in contrast, multiple population spikes and a significant increase in the CBI were elicited in the presence of 0.1 μM bicuculline. These effects resemble the epileptiform discharge produced by high concentrations of GABAA receptor antagonists in healthy tissue (Haas and Ryall 1980; Schwartzkroin and Prince 1980). They were not seen in slices from animals 1 day after the lesion.
The increased effect of modest disinhibition on the response of the CA3 cell population to local stimulation provides evidence that some change has taken place in area CA3 after the lesion that results in an increase in the excitability of CA3 cell population. This increase in excitability was delayed by more than 1 day with respect to the lesion, and it persisted for at least 3 wk afterwards. Our finding of a delayed and persistent increase in excitability of a subtle nature is a confirmation under more physiological conditions of our previous observations in neonatal hippocampal slice cultures (McKinney et al. 1997), in area CA1 after SC transection in vivo (Dinocourt et al. 2011), and in the undercut neocortex (Prince and Tseng 1993).
It is interesting that there was no detectable difference in the responses in control saline at any time point examined. Hippocampal excitability is normally regulated within narrow limits by a balance of synaptic excitation and inhibition (Traub and Miles 1991). The lack of abnormal responses in control saline, despite the abnormal responses observed in 0.1 μM bicuculline, suggests that the CA3 cell population had compensated homeostatically for the lesion-induced increase in excitability by effectively increasing GABAergic inhibition so as to maintain a balance. This compensatory process could be due to increased excitation of interneurons, an increase in potency at GABAergic synapses, or sprouting of the axons of interneurons (Jin et al. 2011). The lack of overt hyperexcitability under normal conditions, but its release by mild disinhibition, resembles the phenotype of most forms of human epilepsy, which is latent most of the time until some as yet poorly understood condition triggers a seizure.
Axonal injury induces trkB-induced axonal sprouting.
The injury-induced increase in excitability of the CA3 cell population was preceded by activation of trkB receptors, as detected by increased trkB autophosphorylation in tissue harvested from the lesioned area 24 h after the transection. Our findings are consistent with the previously observed increase in BDNF and trkB protein levels in area CA3 after SC transection in hippocampal slice cultures (Dinocourt et al. 2006). In addition to the rapid activation of trkB receptors after injury in vivo (Binder et al. 1999; Hu et al. 2004), rapid, transient increases in trkB mRNA are also observed (Hicks et al. 1998, 1999).
There are multiple means by which increased BDNF-trkB signaling could promote increased excitability. Neurotrophins promote axonal outgrowth during development and in the adult CNS (Huang and Reichardt 2003; Lu et al. 2005). Neurotrophins also act pre- and postsynaptically to facilitate excitatory synaptic transmission in the hippocampus (Kang and Schuman 1995; Leßmann et al. 1994; Levine et al. 1995; Tyler and Pozzo-Miller 2001).
Five days after the increase in trkB activation, we observed an increase in the expression and immunoreactivity of the growth-associated protein GAP43. GAP43 is expressed at low levels in the adult brain but is expressed at high levels early in development while axons are actively growing (Benowitz and Routtenberg 1997; Skene 1989). Increased GAP43 after injury is often interpreted as evidence of axonal sprouting (Benowitz et al. 1990; Harris et al. 2010; McKinney et al. 1997). Indeed, we observed that many GAP43-immunoreactive processes had growth cone-like structures at their tips. Furthermore, GAP43 immunoreactivity was present in the axons of CA3 pyramidal cells in injured slice cultures (Dinocourt et al. 2006). Most convincingly, however, we observed that stimulation in area CA3 failed to elicit a response in area CA1 1 day after the SC transection, whereas CA1 cell responses of normal amplitude were readily obtained 7 days after the lesion. The reestablishment of synaptic transmission across the lesion provides strong evidence that new, functional CA3 cell axon collaterals do indeed sprout after transection and can restore damaged synaptic connections.
Sprouting of new axonal collaterals, GAP43 upregulation, and reorganization of excitatory synaptic circuitry occur in many models of brain injury, including controlled cortical impact (Hunt et al. 2010; Norris and Scheff 2009) and fluid percussion injury (Emery et al. 2000; Golarai et al. 2001; Greer et al. 2011; Santhakumar et al. 2001), and are not unique to our model of axonal injury. Axonal sprouting after injury has also been demonstrated in the rodent neocortex after subcortical undercuts that are likely to transect large numbers of pyramidal cell axons (Salin et al. 1995). Sprouting of CA3 (Nadler et al. 1980) and CA1 (Bausch and McNamara 2004; Cavazos et al. 2004; Esclapez et al. 1999; Perez et al. 1996) pyramidal cell axons has also been observed after excitotoxic injury in vivo and in vitro. It is now clear that reactive axonal sprouting is a much more common response of forebrain neurons to injury than previously considered.
Inhibiting trkB receptors prevents injury-induced axonal sprouting and hyperexcitability.
Our previous study suggested that neurotrophins promote axonal sprouting after SC transection in vitro (Dinocourt et al. 2006). We have now taken advantage of the availability of transgenic mice with pharmacologically blockable trkBF616A receptors (Chen et al. 2005) to demonstrate that the injury-induced increases in GAP43 expression and in the number of GAP43-immunoreactive axons are dependent upon trkB signaling. SC transection failed to increase GAP43 expression and immunoreactivity in tissue from mice in which trkBF616A receptors were inhibited with 1NMPP1 administration. Furthermore, there was virtually no recovery of excitatory synaptic transmission from area CA3 to area CA1 after inhibition of trkB signaling, providing functional evidence of a critical role of trkB activation as a trigger of reactive axonal sprouting. These results are further supported by data from He et al. (2004) showing that kindling-induced epileptogenesis is eliminated in TrkB−/− mice, making the receptor an attractive target for therapeutic intervention. The upregulation of GAP43 and recovery of transmission after SC transection in wild-type mice treated with 1NMPP1 provide evidence that its actions are mediated specifically by inhibition of the trkBF616A receptor.
Is injury-induced axonal sprouting required for the increased excitability of area CA3 after axonal damage? We observed that hyperexcitability was induced by 0.1 μM bicuculline in area CA3 7 days after SC transection in untreated trkBF616A mice but not in tissue from mice treated with 1NMPP1. Injury-induced hyperexcitability thus requires activation of trkB receptors in this model. Because axonal sprouting promotes increased excitability and also requires trkB receptor activation in this model, we suggest that it is one critical cause of injury-induced hyperexcitability. Aberrant axonal sprouting in injured hippocampal slice cultures results in an increase in the probability that pyramidal cells are connected by excitatory synapses, and thereby mediates the rapid spread of activity through the CA3 cell population. Our findings do not rule out a contribution of other trkB receptor-dependent processes as well.
The results presented in this report make several significant advances on our previous work. After axonal transection, regeneration readily occurred in slice cultures made from neonatal tissue. Because the microenvironment of the intact nervous system is known to impede axonal regeneration, particularly in adults, it is significant that we have demonstrated extensive axonal regeneration and reformation of functional synapses after axonal injury in vivo. Our previous hypomorphic trkB model made it more difficult to distinguish the role of trkB at the time of injury from developmental alterations resulting from decreased trkB expression. Using the superior trkBF616A model, we now provide a more definitive answer for the role of trkB receptors in axonal regeneration and hyperexcitability.
Implications for genesis of posttraumatic epilepsy.
Epilepsy is a debilitating consequence of TBI. The nature of the changes in the brain that lead to a permanently epileptic state after a latent period lasting from months to years (Agrawal et al. 2006) is not known. The hyperexcitability produced in our study of penetrating brain injury only occurs with a delay of several days and persists for at least several weeks after the acute consequences of the injury have passed. We have demonstrated that pyramidal cells in vivo can undergo significant axonal regeneration after axonal injury and that trkB activation is a key factor in the delayed development of network hyperexcitability. It would be of interest to next determine whether transient inhibition of trkB signaling leads to a lasting prevention of injury-induced hyperexcitability. If so, then transient local inhibition of neurotrophin receptors may offer a potential benefit as a prophylactic, therapeutic treatment to reduce the risk of development of posttraumatic epilepsy and other consequences of TBI.
GRANTS
This work was supported by grants from the National Institute of Neurological Disorders and Stroke (R01 NS-40338 to S. M. Thompson, F31 NS-071981-01A1 to S. Aungst), the Epilepsy Foundation of America Predoctoral Fellowship to S. Aungst, and the Sandler Foundation and Whitehall Foundation to P. M. England.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.A. and S.M.T. conception and design of research; S.A. performed experiments; S.A. analyzed data; S.A. and S.M.T. interpreted results of experiments; P.M.E. contributed reagents; S.A. prepared figures; S.A. drafted manuscript; S.A. and S.M.T. edited and revised manuscript; S.A., P.M.E., and S.M.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Frank Margolis for the gift of the GAP43 antibody; Dr. A. Puche for the osmotic pumps, technical advice, and use of the confocal microscope; Dr. B. Krueger for help with immunoprecipitation; and Drs. B. Alger and E. Powell for comments on the manuscript.
This work was submitted by S. Aungst in partial fulfillment of the requirements for a PhD in Neuroscience.
REFERENCES
- Agrawal A, Timothy J, Pandit L, Manju M. Post-traumatic epilepsy: an overview. Clin Neurol Neurosurg 108: 433–439, 2006 [DOI] [PubMed] [Google Scholar]
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med 338: 20–24, 1998 [DOI] [PubMed] [Google Scholar]
- Bausch SB, McNamara JO. Contributions of mossy fiber and CA1 pyramidal cell sprouting to dentate granule cell hyperexcitability in kainic acid-treated hippocampal slice cultures. J Neurophysiol 92: 3582–3595, 2004 [DOI] [PubMed] [Google Scholar]
- Bendotti C, Baldessari S, Pende M, Southgate T, Guglielmetti F, Samanin R. Relationship between GAP-43 expression in the dentate gyrus and synaptic reorganization of hippocampal mossy fibres in rats treated with kainic acid. Eur J Neurosci 9: 93–101, 1997 [DOI] [PubMed] [Google Scholar]
- Benowitz LI, Rodriguez WR, Neve RL. The pattern of GAP-43 immunostaining changes in the rat hippocampal formation during reactive synaptogenesis. Brain Res Mol Brain Res 8: 17–23, 1990 [DOI] [PubMed] [Google Scholar]
- Benowitz LI, Routtenberg A. GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci 20: 84–91, 1997 [DOI] [PubMed] [Google Scholar]
- Binder DK, Routbort MJ, McNamara JO. Immunohistochemical evidence of seizure-induced activation of trk receptors in the mossy fiber pathway of adult rat hippocampus. J Neurosci 19: 4616–4626, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407: 395–401, 2000 [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Jones SM, Cross DJ. Sprouting and synaptic reorganization in the subiculum and CA1 region of the hippocampus in acute and chronic models of partial-onset epilepsy. Neuroscience 126: 677–688, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos KW, Zhang C, Johnson NM, England PM, Shokat KM, Ginty DD. A chemical-genetic approach to studying neurotrophin signaling. Neuron 46: 13–21, 2005 [DOI] [PubMed] [Google Scholar]
- da Silva AM, Nunes B, Vaz AR, Mendonca D. Posttraumatic epilepsy in civilians: clinical and electroencephalographic studies. Acta Neurochir Suppl (Wien) 55: 56–63, 1992 [DOI] [PubMed] [Google Scholar]
- Dinocourt C, Aungst S, Yang K, Thompson SM. Homeostatic increase in excitability in area CA1 after Schaffer collateral transection in vivo. Epilepsia 52: 1656–1665, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinocourt C, Gallagher SE, Thompson SM. Injury-induced axonal sprouting in the hippocampus is initiated by activation of trkB receptors. Eur J Neurosci 24: 1857–1866, 2006 [DOI] [PubMed] [Google Scholar]
- Emery DL, Raghupathi R, Saatman KE, Fischer I, Grady MS, McIntosh TK. Bilateral growth-related protein expression suggests a transient increase in regenerative potential following brain trauma. J Comp Neurol 424: 521–531, 2000 [PubMed] [Google Scholar]
- Esclapez M, Hirsch JC, Ben-Ari Y, Bernard C. Newly formed excitatory pathways provide a substrate for hyperexcitability in experimental temporal lobe epilepsy. J Comp Neurol 408: 449–460, 1999 [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28: 41–51, 2000 [DOI] [PubMed] [Google Scholar]
- Gähwiler BH, Thompson SM, McKinney RA, Debanne D, Robertson RT. Organotypic slice cultures of neural tissue. In: Culturing Nerve Cells (2nd ed.), edited by Banker G, Goslin K. Cambridge, MA: MIT Press, 1998, p. 461–498 [Google Scholar]
- Golarai G, Greenwood AC, Feeney DM, Connor JA. Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J Neurosci 21: 8523–8537, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer JE, McGinn MJ, Povlishock JT. Diffuse traumatic axonal injury in the mouse induces atrophy, c-Jun activation, and axonal outgrowth in the axotomized neuronal population. J Neurosci 31: 5089–5105, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas HL, Ryall RW. Is excitation by enkephalins of hippocampal neurones in the rat due to presynaptic facilitation or to disinhibition? J Physiol 308: 315–330, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris NG, Mironova YA, Hovda DA, Sutton RL. Pericontusion axon sprouting is spatially and temporally consistent with a growth-permissive environment after traumatic brain injury. J Neuropathol Exp Neurol 69: 139–154, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XP, Kotloski R, Nef S, Lulkart BW, Prada LF, McNamara JO. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron 43: 31–42, 2004 [DOI] [PubMed] [Google Scholar]
- Hicks RR, Li C, Zhang L, Dhillon HS, Prasad MR, Seroogy KB. Alterations in BDNF and trkB mRNA levels in the cerebral cortex following experimental brain trauma in rats. J Neurotrauma 16: 501–510, 1999 [DOI] [PubMed] [Google Scholar]
- Hicks RR, Zhang L, Dhillon HS, Prasad MR, Seroogy KB. Expression of trkB mRNA is altered in rat hippocampus after experimental brain trauma. Brain Res Mol Brain Res 59: 264–268, 1998 [DOI] [PubMed] [Google Scholar]
- Hu B, Liu C, Bramlett H, Sick TJ, Alonso OF, Chen S, Dietrich WD. Changes in trkB-ERK1/2-CREB/Elk-1 pathways in hippocampal mossy fiber organization after traumatic brain injury. J Cereb Blood Flow Metab 24: 934–943, 2004 [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72: 609–642, 2003 [DOI] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN. Regionally localized recurrent excitation in the dentate gyrus of a cortical contusion model of posttraumatic epilepsy. J Neurophysiol 103: 1490–1500, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Reorganization of inhibitory synaptic circuits in rodent chronically injured epileptogenic neocortex. Cereb Cortex 21: 1094–1104, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267: 1658–1662, 1995 [DOI] [PubMed] [Google Scholar]
- Korn SJ, Giacchino JL, Chamberlin NL, Dingledine R. Epileptiform burst activity induced by potassium in the hippocampus and its regulation by GABA-mediated inhibition. J Neurophysiol 57: 325–341, 1987 [DOI] [PubMed] [Google Scholar]
- Leßmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. Neuroreport 6: 21–25, 1994 [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA 92: 8074–8077, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindvall O, Kokaia Z, Bengzon J, Elmer E, Kokaia M. Neurotrophins and brain insults. Trends Neurosci 17: 490–496, 1994 [DOI] [PubMed] [Google Scholar]
- Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci 6: 603–614, 2005 [DOI] [PubMed] [Google Scholar]
- McKinney RA, Debanne D, Gähwiler BH, Thompson SM. Lesion-induced axonal sprouting and hyperexcitability in the hippocampus in vitro: implications for the genesis of posttraumatic epilepsy. Nat Med 3: 990–996, 1997 [DOI] [PubMed] [Google Scholar]
- Nadler JV. The recurrent mossy fiber pathway of the epileptic brain. Neurochem Res 28: 1649–1658, 2003 [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Cotman CW. Selective reinnervation of hippocampal area CA1 and the fascia dentata after destruction of CA3-CA4 afferents with kainic acid. Brain Res 182: 1–9, 1980 [DOI] [PubMed] [Google Scholar]
- Norris CM, Scheff SW. Recovery of afferent function and synaptic strength in hippocampal CA1 following traumatic brain injury. J Neurotrauma 26: 2269–2278, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestreicher AB, de Graan PN, Gispen WH, Verhaagen J, Schrama LH. B-50, the growth associated protein-43: modulation of cell morphology and communication in the nervous system. Prog Neurobiol 53: 627–686, 1997 [DOI] [PubMed] [Google Scholar]
- Oyesiku NM, Evans CO, Houston S, Darrell RS, Smith JS, Fulop ZL, Dixon CE, Stein DG. Regional changes in the expression of neurotrophic factors and their receptors following acute traumatic brain injury in the adult rat brain. Brain Res 833: 161–172, 1999 [DOI] [PubMed] [Google Scholar]
- Perez Y, Morin F, Beaulieu C, Lacaille JC. Axonal sprouting of CA1 pyramidal cells in hyperexcitable hippocampal slices of kainate-treated rats. Eur J Neurosci 8: 736–748, 1996 [DOI] [PubMed] [Google Scholar]
- Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil 20: 76–94, 2005 [DOI] [PubMed] [Google Scholar]
- Prince DA, Tseng GF. Epileptogenesis in chronically injured cortex: in vitro studies. J Neurophysiol 69: 1276–1291, 1993 [DOI] [PubMed] [Google Scholar]
- Royo NC, Conte V, Saatman KE, Shimizu S, Belfield CM, Soltesz KM, Davis JE, Fujimoto ST, McIntosh TK. Hippocampal vulnerability following traumatic brain injury: a potential role for neurotrophin-4/5 in pyramidal cell neuroprotection. Eur J Neurosci 23: 1089–1102, 2006 [DOI] [PubMed] [Google Scholar]
- Salin P, Tseng GF, Hoffman S, Parada I, Prince DA. Axonal sprouting in layer V pyramidal neurons of chronically injured cerebral cortex. J Neurosci 15: 8234–8245, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhakumar V, Ratzliff AD, Jeng J, Toth Z, Soltesz I. Long-term hyperexcitability in the hippocampus after experimental head trauma. Ann Neurol 50: 708–717, 2001 [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Prince DA. Changes in excitatory and inhibitory synaptic potentials leading to epileptogenic activity. Brain Res 183: 61–76, 1980 [DOI] [PubMed] [Google Scholar]
- Singleton RH, Zhu J, Stone JR, Povlishock JT. Traumatically induced axotomy adjacent to the soma does not result in acute neuronal death. J Neurosci 22: 791–802, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene JH. Axonal growth-associated proteins. Annu Rev Neurosci 12: 127–156, 1989 [DOI] [PubMed] [Google Scholar]
- Tasker RR, DeCarvalho G, Dostrovsky JO. The history of central pain syndromes, with observations concerning pathophysiology and treatment. In: Pain and Central Nervous System Disease: the Central Pain Syndromes, edited by Casey KL. New York: Raven, 1991, p. 31–58 [Google Scholar]
- Tian GF, Baker AJ. Protective effect of high glucose against ischemia-induced synaptic transmission damage in rat hippocampal slices. J Neurophysiol 88: 236–248, 2002 [DOI] [PubMed] [Google Scholar]
- Traub RD, Miles R. Neuronal Networks of the Hippocampus. New York: Cambridge Univ. Press, 1991 [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci 21: 4249–4258,2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Lu D, Jiang H, Xiong Y, Qu C, Li B, Mahmood A, Zhou D, Chopp M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma 25: 130–139, 2008 [DOI] [PubMed] [Google Scholar]