

Abstract
Circulating hormones influence multiple aspects of hypothalamic development and play a role in directing formation of neural circuits. Leptin is secreted by adipocytes and functions as a key developmental signal that promotes axon outgrowth from the arcuate nucleus (ARH) during a discrete developmental critical period. To determine the cellular mechanisms by which leptin impacts development of hypothalamic circuits, we examined roles for leptin receptor (LepRb) signals in neonatal mice. LepRb, ERK, and STAT3 signaling were required for leptin-stimulated neurite outgrowth from ARH explants in vitro. Neonatal mice with disrupted LepRb→ERK signaling displayed impaired ARH projections but were able to compensate by adulthood. LepRb→STAT3 signaling also plays a role in early circuit formation and controls the ultimate architecture of POMC, but not AgRP, projections. Thus, the developmental actions of leptin on feeding circuits are dependent on LepRb, and distinct signaling pathways are responsible for directing formation of NPY and POMC projections.
Introduction
Multiple regions of the hypothalamus interact to modulate energy homeostasis by integrating cues that signal metabolic state (Elmquist et al., 2005). The adipocyte-derived hormone leptin controls energy balance by activating the signaling form of the leptin receptor (LepRb) in the brain (Ahima and Flier, 2000), and the arcuate nucleus of the hypothalamus (ARH) represents a key site of leptin action (Morton et al., 2003; Coppari et al., 2005). In the ARH, leptin regulates the activity of two oppositely acting sets of LepRb-expressing neurons by stimulating the anorectic proopiomelanocortin (POMC)-containing neurons and inhibiting the orexigenic action of neurons that coexpress neuropeptide Y (NPY) and agouti-related protein (AgRP). Although these POMC and NPY/AgRP neurons project convergently to several hypothalamic regions, their direct inputs to the paraventricular nucleus of the hypothalamus (PVH) play a crucial role in regulating energy balance (Sawchenko, 1998).
Leptin binding to the extracellular domain of LepRb initiates signaling by activating the associated Jak2 tyrosine kinase, which phosphorylates specific residues on the intracellular domain of LepRb (Robertson et al., 2008). Each phosphorylation site recruits distinct intracellular signaling proteins, and LepRb Tyr1138 mediates the phosphorylation of signal transducer and activator of transcription-3 (STAT3). Mutation of LepRb Tyr1138 produces a dramatic hyperphagia and obesity in adult animals that rivals the phenotype of LepRb-null Leprdb/db (db/db) mice, suggesting that LepRb→STAT3 signaling represents a crucial mediator of leptin action on energy balance in vivo (Bates et al., 2003). LepRb Tyr985 promotes the activation of the extracellular regulated kinase (ERK) cascade, as well as binding the suppressor of cytokine signaling-3 to promote feedback inhibition of LepRb (Bjørbaek et al., 2001; Björnholm et al., 2007). Mutation of Tyr985 in vivo causes leanness in adult mice, consistent with the importance of Tyr985-mediated feedback inhibition in the control of energy balance (Björnholm et al., 2007). Leptin also activates phosphatidylinositol 3′-kinase (PI3K) and its downstream effectors (including Akt) in the hypothalamus, but this pathway appears to play a more modest role in long-term regulation of energy balance (Hill et al., 2008).
During development, axonal projections ascend from the ARH to reach the PVH during the second week of life (Bouret et al., 2004a). Leptin is required for the normal development of ARH projections: ARH projections to each of its targets are markedly reduced in leptin-deficient Lepob/ob (ob/ob) mice, and exogenous leptin can rescue these projections if administered during neonatal life (although not in adulthood) (Bouret et al., 2004b). The site of action for this neurotrophic action of leptin appears to be the ARH, because treatment of organotypic explants with leptin promotes neurite outgrowth from ARH neurons (Bouret et al., 2004b).
Although roles for LepRb signals in the control of energy homeostasis have been examined in adult animals, little is known about the signaling events that mediate the developmental actions of leptin. In the present study, we examined neurite outgrowth in vitro and used axonal labeling and immunohistochemistry in genetically altered mice in vivo to reveal distinct roles for individual LepRb signals in establishing patterns of hypothalamic connectivity.
Materials and Methods
Animals and genotyping.
Animal usage was in compliance with and approved by the Institutional Animal Care and Use Committee of the Saban Research Institute of Childrens Hospital of Los Angeles. All experimental animals were generated by intercrossing of heterozygous animals on the C57BL/6 background (n ≥ 6). Neonatal Leprdb/db, Leprtm1Mgmj/tm1Mgmj (also known as LeprS1138/S1138 or s/s) (Bates et al., 2003), Leprtm2Mgmj/tm2Mgmj (also known as LeprL985/L985 or l/l) (Björnholm et al., 2007), and wild-type (WT) littermate mice were identified by genotyping as described previously (Kowalski et al., 2001; Bates et al., 2003; Björnholm et al., 2007), because the various genotypes are visually indistinguishable until ∼3–4 weeks of age. Homozygous transgenic mice that selectively express the green fluorescent protein GFP and humanized renilla GFP (hrGFP) in POMC- and NPY-containing neurons, respectively, were kindly provided by Drs. M. Low (Oregon Health Science University, Portland, OR) and B. Lowell (Harvard Medical School, Boston, MA), respectively.
Phosphoproteins immunohistochemistry and analysis.
Male WT, POMC–eGFP, and NPY–hrGFP mice on P10 were injected intraperitoneally with leptin (10 mg/kg; Preprotech) or vehicle alone (5 mm Na citrate buffer) and were perfused 15 min (pAkt staining), 30 min (pERK staining), or 45 min (pSTAT3 staining) later with a solution of 2% (pSTAT3) or 4% (pERK and pAkt) paraformaldehyde. Frozen coronal sections were cut at 30 μm and then processed for immunofluorescence. For pSTAT3 immunostaining, sections were pretreated for 20 min in 0.5% NaOH and 0.5% H2O2 in potassium PBS, followed by immersion in 0.3% glycine for 10 min. Sections were then placed in 0.03% SDS for 10 min and placed in 4% normal serum plus 0.4% Triton X-100 plus 1% BSA (fraction V) for 20 min before incubation for 48 h with a rabbit anti-pSTAT3 antibody (1:1000; Cell Signaling Technology). For pERK and pAkt stainings, sections were incubated for 48 h in a rabbit anti-pERK (1:1000; Cell Signaling Technology) or a rabbit anti-pAkt (1:150; Cell Signaling Technology). The primary antibodies were localized with Alexa Fluor 568 goat anti-rabbit IgGs (1:200; Invitrogen). Sections were then counterstained using bis-benzamide (1:10,000; Invitrogen) to visualize cell nuclei and coverslipped with buffered glycerol, pH 8.5. Slides were then numerically coded to obscure the treatment group, and pSTAT3, pERK, and pAkt immunopositive cells were counted manually by using a Carl Zeiss Imager Z1 microscope and a 20× objective. GFP-positive cells were visualized using direct fluorescence. Only GFP-positive cells that had a corresponding bis-benzamide-stained nucleus were included from our counts. Immunoreactive (IR) cells were counted for each animals in three sections through the ARH. The mean number of immunopositive cells per hemisection was compared between groups by using ANOVA.
Measurement of Lepr-b mRNA.
Lepr-b mRNA levels were quantified from microdissected ARH with real-time PCR. Briefly, total RNA was isolated from microdissected ARH using TriReagent (Sigma). Lepr-b mRNA levels were quantified using TaqMan One-Step RT-PCR Master Mix (Applied Biosystems) with ABI Prism 7700 sequence detection system (Applied Biosystems). The RT-PCR conditions are 50°C, 2 min for reverse transcription, followed by 95°C for 10 min and then 40 cycles of 95°C, 15 s and 60°C, 1 min. The standard curve was calculated using a series of 10-, 100-, and 1000-fold dilutions of mRNA extracted from the ARH. The relative copy number of cyclophilin (primers sets; Applied Biosystems) was quantified, and the data were expressed as a percentage of maximum ratio of Lepr-b mRNA compared with cyclophilin mRNA. Primer and probe sets include the following: LepRb, 5′-GGAGACAGAGGCCCAGACATT-3′ (forward), 5′-AAACTTCCCTCCAGTTCCAAAAG-3′ (reverse), 5′-AGCAACCCACCATGATTTCACCACAAC-3′ (probe); 18S, 5′-CGGCTACCACATCCAAGGAA-3′ (forward), 5′-GGGCCTCGAAAGAGTCCTGT-3′ (reverse), 5′-CAGCAGGCGCGCAAATTACC-3′ (probe). Ct values ranged from 28 to 30 and from 27 to 29 for Lepr-b and cyclophilin, respectively. The data are shown as percentage of the ratio in mice on P2.
DiI implants.
Male mouse pups were anesthetized and perfused on P12, with 4% paraformaldehyde. The brains were removed, numerically coded to ensure unbiased processing and analysis, and crystals of DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate) were implanted as described previously (Bouret et al., 2004a) (Bouret et al., 2004b). Briefly, an insect pin was used to place a small crystal of DiI (Invitrogen) (20–40 μm in diameter) into the ARH of each brain under visual guidance. Although it was not possible to completely limit the tracer to the ARH in any of the cases, similar implants were achieved in animals of all genotypes. Successful implants were centered in the ARH and did not involve the ventromedial hypothalamus substantially. Cases with tracer implants that did not extend throughout the ARH or had aberrant patterns of tracer spread were discarded. After 6 weeks of incubation at 37°C, 80-μm-thick sections were collected through the hypothalamus from each brain and evaluated with both conventional fluorescence and confocal microscopy.
AgRP and aMSH immunohistochemistry.
Anesthetized adult male mice (P60–P80) were perfused transcardially with 4% paraformaldehyde, and the brains were frozen and sectioned at 20 μm and then processed for immunofluorescence as described previously (Bouret et al., 2004b). Briefly, sections were incubated for 72 h in a rabbit anti-AgRP (1:2000; Phoenix Pharmaceuticals) or a sheep anti-aMSH (1:40,000; Millipore Bioscience Research Reagents). The primary antibodies were localized with Alexa Fluor 488 goat anti-rabbit IgGs or Alexa Fluor 488 donkey anti-sheep IgGs (1:200; Invitrogen). Sections were counterstained using bis-benzamide (1:10,000; Invitrogen) to visualize cell nuclei and coverslipped with buffered glycerol, pH 8.5.
Quantitative analysis of fibers density.
For quantification, four to six animals per experiments were analyzed. A series of 20 adjacent optical sections were collected at 1 μm intervals through the PVH (two sections per animal) of animals of each genotype by using a Leica SP-confocal microscope equipped with a 20× objective (numerical aperture, 0.40; working distance, 360 μm). Image analysis was performed by using MetaMorph image analysis software (Universal Imaging) (Bouret et al., 2004a,b, 2008). Briefly, each image plane was binarized so as to isolate labeled fibers from background, as well as compensate for differences in fluorescence intensity, and was then skeletonized so that each fiber segment was 1 pixel thick. The integrated intensity was then calculated for each image, which reflects the total number of pixels in the skeletonized image and was proportional to the total length of labeled fibers in the image. To avoid over-counting fibers that were thicker than the distance between planes, each skeletonized image was compared with the precedent one, and pixels appearing in the previous image were rejected automatically. This procedure was performed on each image plane in the stack, and the values for all image planes in a stack were summed. The resulting value is an accurate index of fiber density in the volume sampled.
Isolated ARH explant cultures.
Brains were collected from P6 mice of each genotype and sectioned at 200 μm with a vibroslicer. The ARH was then carefully dissected out of each section under a stereomicroscope. Explants were cultured onto a rat tail collagen matrix (Millipore) in EOL-1 serum-free medium as described previously (Bouret et al., 2004b). Beginning on the first day in vitro, each explant was transferred to fresh EOL-1 medium containing either leptin (5 μg/ml; Peprotech) or vehicle alone (5 mm Na citrate buffer) (n = 5–7 cases per group). For the experiments using inhibitors, U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene] (10 μm; Cell Signaling Technology) or LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one] (5 μm[scap]; Calbiochem) was added 30 min before leptin. After 72 h, the explants were fixed in paraformaldehyde, and neurites extending from the explants were stained with β III tubulin (TuJ monoclonal antibody; Babco). The overall length and density of neurites was analyzed by collecting digital pictures of the explants that extend through their entire thickness and evaluated by using the MetaMorph software as described previously (Bouret et al., 2008). Briefly, each image was deconvolved, binarized according to threshold criteria to isolate labeled fibers from background, as well as to compensate for differences in fluorescence intensity, and then skeletonized so that each fiber segment was 1 pixel thick. The integrated intensity was calculated in four different regions of interest (100 × 100 μm) spaced at 100, 200, 300, 400, and 500 μm extending radially from the edge of the explants.
Statistical analysis.
Datasets were analyzed for statistical significance using Statview (SAS Institute) for a Kruskal–Wallis ANOVA, with a Fisher's least significant difference post hoc comparison.
Results
Functional LepRb signaling in neonatal ARH neurons
To assess the role for LepRb in mediating the developmental actions of leptin on ARH neurons, we first examined the expression of Lepr-b mRNA in the ARH of neonatal mice. The ARH contained relatively low levels of Lepr-b mRNA at P2 and P6, but levels increased markedly by P10 (Fig. 1A). This increase in Lepr-b mRNA coincides with the postnatal leptin surge (Ahima et al., 1998) and the period when ARH projections are developing (Bouret et al., 2004a).
Figure 1.
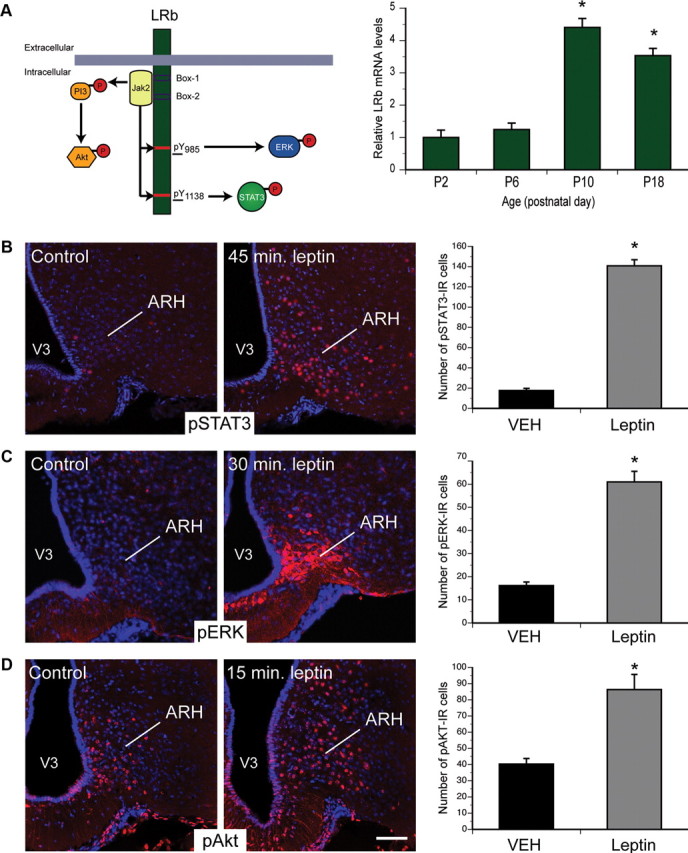
LepRb signaling in neonatal ARH neurons. A, Real-time PCR was used to compare levels of the long form of leptin receptor (LepRb) mRNA in the ARH of WT mice killed on P2, P6, P10, and P18 (n = 4 per group). B–D, Confocal images and quantitative comparisons of pSTAT3-IR, pERK-IR, and pAkt-IR cells (red fluorescence) 45 min (B), 30 min (C), and 15 min (D) after intraperitoneal administration of leptin (10 mg/kg) or vehicle alone (VEH) on P10 pups (n = 4 per group). V3, Third ventricle. The values shown are mean ± SEM. A, *p < 0.05 versus P2 and P6; B–D, *p < 0.05 versus vehicle. Scale bar, 25 μm.
Leptin acts via multiple intracellular signaling pathways, including STAT3, ERK, and PI3K→Akt, to modulate cellular and organismal physiology (Robertson et al., 2008) (Fig. 1A). We initially used the immunohistochemical detection of phosphorylated (activated) forms of STAT3 (pSTAT3), ERK (pERK), and Akt (pAkt) to examine the ability of leptin to activate these pathways in neonatal ARH neurons in postnatal mice. Peripheral injection of leptin in WT mice on P10 robustly induced pSTAT3 immunoreactivity after 45 min (Fig. 1B). Similarly, leptin increased the number of pERK-IR neurons after 30 min (Fig. 1C) and pAkt-IR neurons within 15 min (Fig. 1D). Thus, leptin activates these signaling pathways in the early postnatal ARH, as in adults.
Trophic action of leptin on ARH neurons requires specific LepRb signals
To understand whether LepRb and specific LepRb signaling pathways might participate in the neurotrophic actions of leptin, we performed a series of in vitro experiments using pharmacological and genetic manipulations of LepRb signaling. Previously, we demonstrated that leptin acts directly on ARH cells to promote neurite outgrowth from explanted neonatal ARH tissue in vitro (Bouret et al., 2004b). Addition of leptin (5 μg/ml) to the culture medium for 72 h increased the density of TuJ-stained neurites extending 100–400 μm from the edge of WT ARH explants relative to control explants maintained without leptin (Fig. 2A,B). In contrast, leptin failed to promote increased neurite outgrowth in explants derived from neonatal LepRb-null db/db mice (Fig. 2A,B), demonstrating the requirement for LepRb for this neurotrophic action of leptin.
Figure 2.
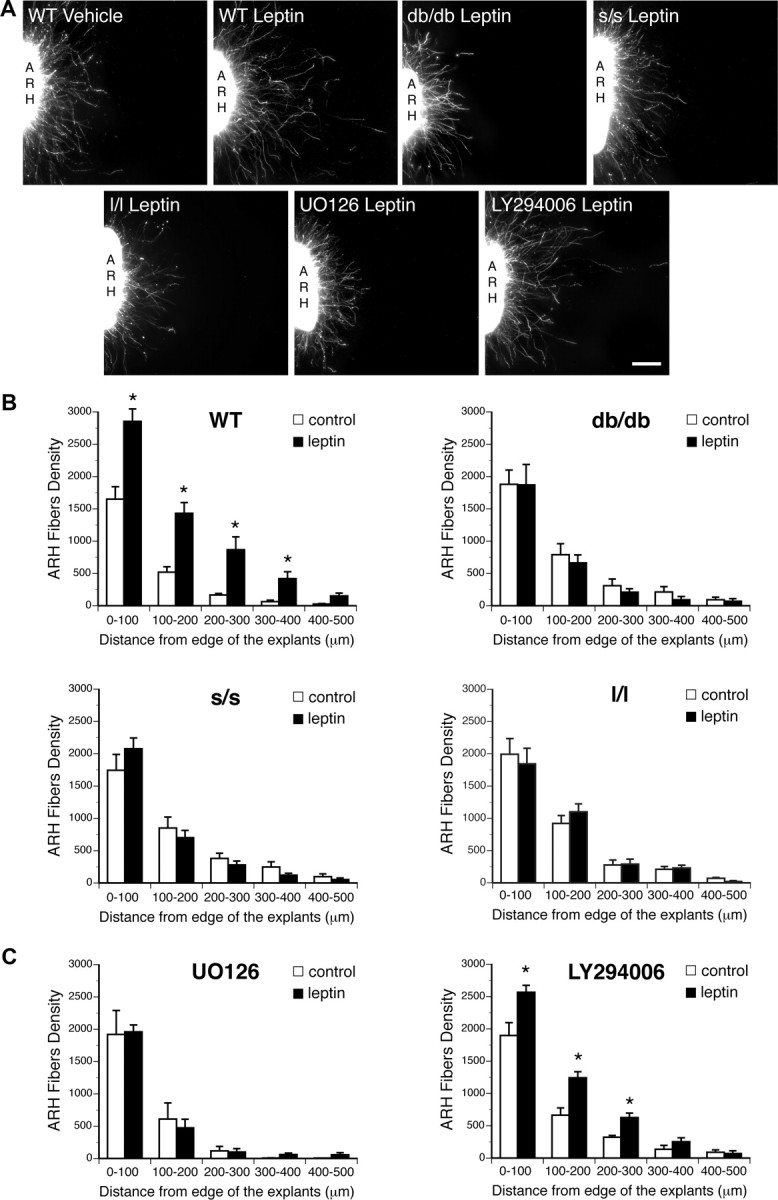
Effect of leptin signaling blockade on leptin-induced ARH neurite outgrowth in vitro. A, B, Isolated organotypic cultures of ARH from P6 WT, db/db, s/s, and l/l mice (n = 5–7 per case) were incubated for 72 h with serum-free medium containing leptin (5 μg/ml) or lacking leptin and then immunostained with β III tubulin, a marker of neurites. A, C, Isolated organotypic cultures of ARH from P6 WT mice (n = 5–7 per case) were incubated for 72 h with serum-free medium containing leptin (5 μg/ml) or lacking leptin in the presence or absence of U0126 (10 μm) or LY294002 (5 μm) and then immunostained with β III tubulin. The values shown are mean ± SEM. *p < 0.05 between control and leptin-treated explants. Scale bar, 90 μm.
To determine which LepRb-activated signals might be required for leptin-stimulated neurite outgrowth, we treated WT ARH explants with pharmacologic inhibitors of ERK and PI3K during leptin-stimulated neurite outgrowth assays. Exposure of ARH explants to U0126 (an inhibitor of MEKK1, the upstream activator of ERK) prevented the leptin-induced increase in neurite outgrowth observed in control cultures 72 h after leptin treatment (Fig. 2A,C). In contrast, treatment with LY294002 (which inhibits PI3K activity) failed to block leptin-stimulated ARH neurite outgrowth (Fig. 2A,C). Thus, leptin-stimulated neurite outgrowth in neonatal mouse ARH explants requires the activity of the ERK pathway, but not PI3K.
Given the lack of pharmacologic inhibitors specific for STAT3, we examined the importance of this pathway by determining the trophic effect of leptin in explants from LeprS1138/S1138 (s/s) mice, which contain a LepRb molecule mutant for Tyr1138 (and thus null for STAT3 signaling) (Bates et al., 2003). Leptin failed to enhance neurite outgrowth in ARH explants derived from s/s mice (Fig. 2A,B), demonstrating that LepRb→STAT3 signaling is required to transduce the growth promoting properties of leptin in vitro. Similarly, leptin failed to promote increased neurite outgrowth in explants derived from neonatal l/l mice (Fig. 2A,B). Together, these data suggest important roles for LepRb→ERK and LepRb→STAT3 signaling in initial axonal outgrowth during ARH circuit formation.
Development of ARH projections requires LepRb
To establish a role for LepRb in mediating the neurodevelopmental actions of leptin in vivo, we used DiI axonal labeling to evaluate projections from the ARH in db/db mice that lack LepRb. The density of DiI-labeled fibers in the PVH of db/db mice at P12 was half that observed in WT animals (Fig. 3). The distribution pattern of labeled fibers in the PVH was similar between db/db and WT mice, however, suggesting that LepRb deficiency alters the density but not the pattern of innervation. The disruption of ARH projections in db/db mice phenocopies that observed previously in leptin-deficient ob/ob mice (Bouret et al., 2004b), indicating that LepRb mediates the developmental actions of leptin on ARH projections.
Figure 3.

Development of ARH projections in mice mutant for LepRb signaling. Confocal images (A) and quantification (B) of ARH fibers, labeled with the anterograde tracer DiI, innervating the PVH of P12 WT, db/db, s/s, and l/l mice (n = 4–6 per group). The values shown are mean ± SEM. *p < 0.05 versus WT; #p < 0.05 versus db/db. Scale bar, 180 μm.
To determine the downstream LepRb signaling pathways mediating the developmental actions of leptin, we studied the postnatal development of ARH projections in s/s mice (Bates et al., 2003). The density of ARH-derived DiI-labeled fibers in the PVH of s/s mice was significantly reduced compared with that of WT mice, although the effect was less severe than that seen in the db/db animals (Fig. 3). In contrast, the density of ARH projections to the PVH in neonatal LeprL985/L985 mice (l/l; mutant for the ERK-activating Tyr985 of LepRb) (Björnholm et al., 2007) was similar to that observed in db/db mice (Fig. 3). Thus, much as the ERK and STAT3 signaling pathways are required for leptin-stimulated ARH neurite outgrowth in vitro, the LepRb residues that control these signaling pathways are important for the early development of ARH→PVH projections in vivo. The intermediate early developmental phenotype of mice mutant for LepRb→STAT3 signaling suggests the potential importance of STAT3 for the promotion of some, but not all, LepRb-regulated projections.
Distinct LepRb signaling pathways mediate formation of orexigenic versus anorexigenic ARH projections
To explore the potential cell-type specificity of the pathways effected by LepRb on ARH→PVH projections, we initially examined the ability of leptin to activate STAT3 and ERK in POMC and NPY/AgRP neurons of neonatal animals by determining the colocalization of leptin-stimulated pSTAT3 and pERK immunoreactivity with GFP in the ARH of transgenic mice expressing GFP in POMC- and NPY-containing neurons. This analysis revealed that leptin stimulated pSTAT3- and pERK immunoreactivity, respectively, in 35 and 20% of POMC neurons in the ARH of P10 mice (Fig. 4). Similarly, leptin stimulated pSTAT3 and pERK immunoreactivity, respectively, in 60 and 30% of NPY neurons in the ARH of P10 mice (Fig. 5). Thus, there exists no major divergence in signal activation by cell type, and such a mechanism cannot explain the differences in early ARH→PVH circuit development observed in the s/s compared with l/l and db/db lines.
Figure 4.

Leptin receptor signaling in ARH POMC neurons during postnatal development. Confocal images of pSTAT3 and pERK-IR cells (red fluorescence) 45 min (A) and 30 min (B) after intraperitoneal administration of leptin (10 mg/kg) or vehicle alone on P10 pups in which POMC-containing neurons have been labeled with the GFP (n = 4 per group). Arrows point to double-labeled neurons. Graphs represent the mean number of POMC neurons (gray bars) and mean number of double-labeled cells per hemisection after intraperitoneal administration of leptin (10 mg/kg, black bars) or vehicle alone (white bars). The values shown are mean ± SEM. *p < 0.05 versus vehicle. me, Median eminence; V3, third ventricle. Scale bar: A, 140 μm; B, 100 μm.
Figure 5.

Leptin receptor signaling in ARH NPY neurons during postnatal development. Confocal images of pSTAT3-IR and pERK-IR cells (red fluorescence) 45 min (A) and 30 min (B) after intraperitoneal administration of leptin (10 mg/kg) or vehicle alone on P10 pups in which NPY-containing neurons have been labeled with the hrGFP (n = 4 per group). Arrows point to double-labeled neurons. Graphs represent the mean number of NPY neurons (gray bars) and mean number of double-labeled cells per hemisection after intraperitoneal administration of leptin (10 mg/kg, black bars) or vehicle alone (white bars). The values shown are mean ± SEM. *p < 0.05 versus vehicle. me, Median eminence; V3, third ventricle. Scale bar: A, 110 μm; B, 130 μm.
To determine whether the defects in ARH projections observed in LepRb signaling mutant mice were permanent and/or affected formation of both the orexigenic AgRP and anorexigenic POMC neural pathways, we performed immunofluorescent labeling of AgRP and aMSH in adult (P80) db/db, s/s, and l/l mice. As for the axonal labeling study in neonates and similar to our previous observations in ob/ob mice, the density of AgRP- and aMSH-IR fibers was significantly reduced in the PVH of adult db/db mice relative to that of WT animals (Figs. 6, 7). In contrast to the marked perturbation in the density of ARH projections to the PVH observed in neonatal mice, in adult l/l mice, both AgRP and aMSH staining were similar to that of littermate control mice (Figs. 6, 7). Thus, these data suggest that, although the LepRb→ERK pathway is required for the early development of ARH→PVH projections, other ERK-activating pathways or signals compensate to normalize this pathway.
Figure 6.
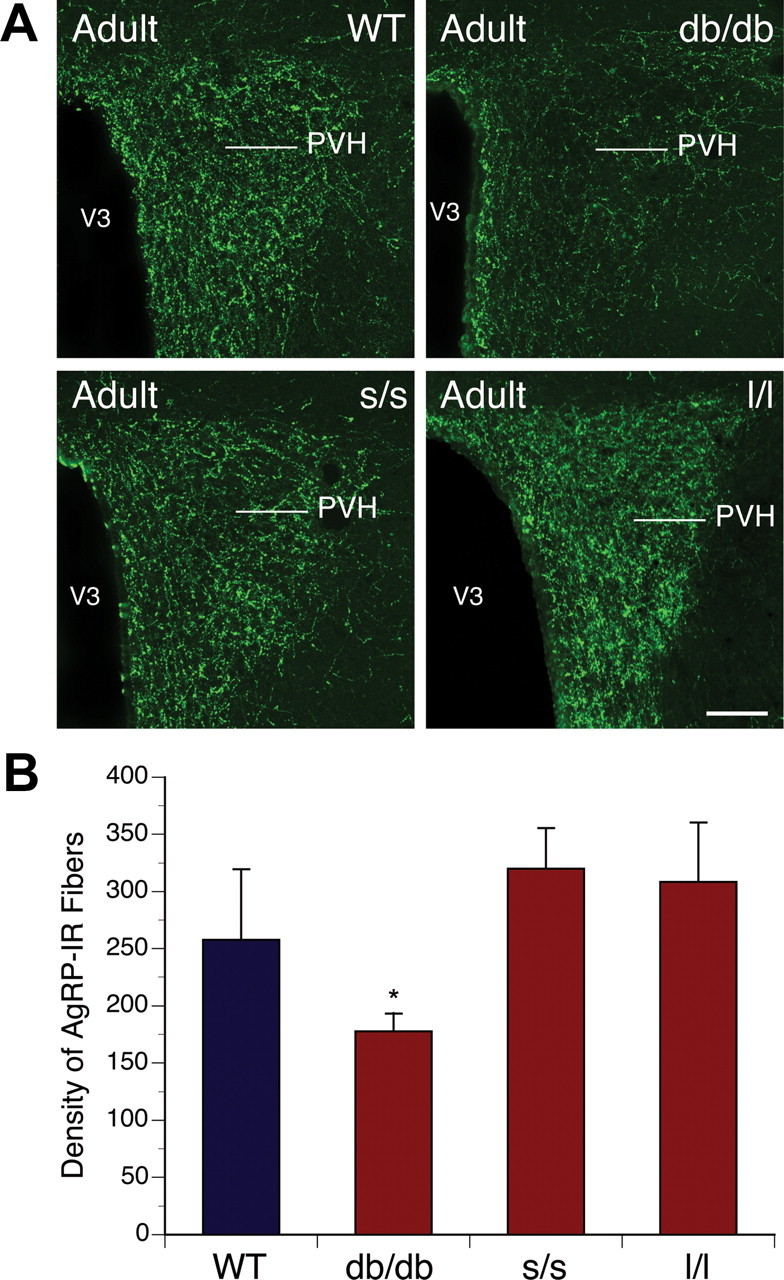
Arcuate AgRP neural projections in adult mice mutant for LepRb signaling. Confocal images (A) and quantification (B) of AgRP-IR fibers in the PVH of adult (P60) WT, db/db, s/s, and l/l mice (n = 4–6 per group). V3, Third ventricle. The values shown are mean ± SEM. *p < 0.05 versus db/db. Scale bar, 180 μm.
Figure 7.

aMSH neural projections in adult mice mutant for LepRb signaling. Confocal images (A) and quantification (B) of aMSH-IR fibers in the PVH of adult (P60) WT, db/db, s/s, and l/l mice (n = 4–6 per group). V3, Third ventricle. The values shown are mean ± SEM. *p < 0.05 versus db/db and s/s. Scale bar, 180 μm.
As in l/l mice, the density of AgRP immunostained fibers in adult s/s animals was comparable with that of WT littermates (Fig. 6). In contrast, however, the density of aMSH-IR fibers in the PVH remained significantly lower in the adult s/s mice (Fig. 7). Hence, disruption of LepRb→STAT3 signaling specifically perturbs the architecture of the ARH POMC circuitry but spares the AgRP circuit. This is consistent with the disruption of numerous other parameters of POMC neuron function and the sparing of AgRP neuron regulation in the obese s/s animals (Bates et al., 2003). Thus, LepRb→ERK and LepRb→STAT3 signaling play distinct roles in the development and architecture of the ARH→circuitry.
Discussion
It is well documented that STAT3-dependent signaling plays a major role in a number of leptin-driven hypothalamic functions in mature animals, including body weight regulation, feeding, lactation, and regulation of thyroid function (Bates et al., 2003; Gao et al., 2004). Evidence for activation of other signaling pathways is less clearly defined, but both PI3K-mediated activation of AKT and activation of MAPK signaling may also contribute to cellular regulation of energy balance by leptin (Robertson et al., 2008). Despite the lack of a clear regulatory effect of leptin on food intake in neonatal animals (Mistry et al., 1999), all three of these pathways appear functional in developing ARH neurons. Moreover, the results presented here indicate that distinct LepRb signaling pathways are required for normal development of hypothalamic neural projections that later convey the effects of leptin on energy balance. We found significant disruptions in ARH projections in both neonatal and adult db/db mice that were virtual phenocopies of those observed previously in leptin-deficient ob/ob mice (Bouret et al., 2004b). These data indicate that the LepRb, which is expressed and competent during the time of the postnatal leptin surge, mediates the developmental actions of leptin on ARH projections. Notably, both NPY- and POMC-containing projections appear affected by LepRb deficiency, and in vitro studies with organotypic explants cultures suggest that leptin acts via the LepRb directly on ARH neurons to promote neurite extension during postnatal life.
Alterations of ARH neuronal development appear to be coupled to at least two major LepRb signaling pathways. Despite the ability of leptin to promote STAT3, ERK, and Akt activation in neonatal ARH neurons, only STAT3 and ERK (but not PI3K) are required for normal development of ARH projections. The density of ARH projections to the PVH was clearly reduced in s/s mice compared with that of WT littermates, but the effect was not as dramatic as that seen for the receptor mutant (db/db). Remarkably, in adult s/s mice, the density of AgRP immunostained fibers was comparable with that of WT littermates, but the density of aMSH-IR fibers was clearly reduced in the PVH of s/s mice, suggesting that development of projections to the PVH from POMC neurons is more dependent on STAT3 signaling than is the development of projections from NPY neurons. This cell-type specificity of leptin during development may explain the sparing of many ARH projections in the s/s mice. Alternatively, STAT3 signaling may be coupled less efficiently to neurotrophic response of ARH neurons than it is to regulatory processes that impact food intake in adults. However, we cannot exclude that changes in immunostaining observed in the PVH of s/s mice are not attributable to alterations in peptide synthesis, transport, or release. Tract tracing analyses in adult mice using, for example, Phaseolus vulgaris leucoagglutinin injections in the ARH coupled with various peptide markers or genetically targeted axonal markers would be required to confirm that apparent defects in the densities of peptidergic axons in the PVH are attributable to true changes in the organization of hypothalamic circuits. Nevertheless, the observation that AgRP-containing projections appear relatively normal in mature s/s mice is consistent with previous data suggesting normal regulation of AgRP neurons in s/s animals (Bates et al., 2003). These findings also suggest that other LepRb signaling pathways, distinct from STAT3, are involved in the development of ARH AgRP/NPY projections.
Our in vitro data also demonstrate that leptin stimulates ARH neurite outgrowth independently of PI3K signaling, suggesting that this signaling pathway is unlikely to contribute to the establishment for ARH projections. Nevertheless, the LepRb→PI3K/Akt pathway appears crucial for the regulatory effect of leptin on feeding and energy balance in mature animals (Robertson et al., 2008), perhaps through activation of established ARH projections to the PVH. In addition, it remains possible that PI3K signaling is involved in other developmental events in hypothalamic circuitry, such as synapse formation and remodeling, and that leptin may influence neuronal excitability through the PI3K/Akt pathway in the developing hypothalamus as it does in adults (Hill et al., 2008).
MAPK signaling through ERKs is characteristic of neurotrophin action and leads to various and partially overlapping trophic effects on neurons, ranging from neuronal differentiation and plasticity to regulation of neuronal survival (Huber et al., 2003; Zhou and Snider, 2006). Here we provide in vitro evidence that MAPK signaling is important for the growth-promoting action of leptin on ARH neurites during development. Consistent with these findings, the ability of ARH axons to reach the PVH by P12 is severely disrupted in l/l mice, which lack the capacity to activate ERK phosphorylation via LepRb. In sharp contrast with the marked reduction in ARH axons that succeed in reaching the PVH in neonatal l/l mice, the density of both AgRP- and aMSH-labeled fibers in the PVH of adult l/l mice appears to be normal, suggesting that these peptidergic pathways retain considerable plasticity beyond P12. However, because aMSH-immunopositive fibers may originate from neurons located in the ARH or in the nucleus of the tractus solitarius (Sawchenko and Swanson, 1983; Broberger et al., 1998), it is also possible that compensatory changes may occur in brainstem POMC projections when arcuate projections are diminished and that these projections may be responsible for part of the normal pattern of aMSH innvervation observed in adult l/l mice.
Both ERK and STAT3 signaling pathways participate in the control of axon outgrowth in other neural systems (Doherty et al., 2000; Liu and Snider, 2001; Conway, 2006). However, molecular events that link these signals to cellular mechanisms controlling axon outgrowth and targeting remain unknown. Axonal outgrowth implies cytoskeletal rearrangements that contribute to growth cone extension, which often involves growth-associated proteins interacting with actin microfilaments or microtubules (Dent and Gertler, 2003). Notably, in cultured neurons, LepRb is concentrated at axonal growth cones in which it induces formation of dendritic filopodia (Shanley et al., 2002; O'Malley et al., 2007). Leptin has also been reported to influence growth cone morphogenesis through LepRb signaling in developing cortical neurons (Valerio et al., 2006). Moreover, the global impairment of ARH projection pathways observed in db/db mice supports the notion that defects in LepRb signaling affect the ability of arcuate neurons to extend axons to their targets rather than affecting target-dependent mechanisms. Consistent with this hypothesis, leptin induces neurite extension when coincubated with organotypic explants of the ARH, confirming that the neurotrophic effects of leptin have a direct site of action within the ARH (Bouret et al., 2004b). Also, neonatal leptin-induced LepRb signaling, as evidenced by pERK labeling, was primarily restricted to the ARH and was not observed in ARH terminal fields, such as the PVH and lateral septum (Bouret et al., 2004a), adding additional support to the importance of a direct site of action for leptin on ARH axonal growth. However, expression of LepRb does not seem sufficient for the developmental actions of leptin. For example, LepRb is expressed by neonatal dorsomedial hypothalamus (DMH) neurons (Caron et al., 2010), yet these projections appear to develop normally in ob/ob mice (Bouret et al., 2004b). It is also important to note that hypothalamic circuits appear to remain remarkably plastic during development and may have built-in redundancy. This property may explain the efficacy of leptin treatment in reducing body weight in adult mice, despite the permanent disruption in ARH projections, because leptin signaling in ob/ob mice may occur via different neural pathways, perhaps via intact LepRb–DMH projections.
In conclusion, these findings define crucial roles for distinct LepRb signals in the development and ultimate architecture of specific hypothalamic pathways involved in the regulation of energy balance. These results reveal that STAT3 and MAPK signaling mediates distinct aspects of development of peptidergic projections from the ARH to the PVH, which may contribute to the distinct metabolic phenotypes seen in the s/s and l/l mutants (Bates et al., 2003; Björnholm et al., 2007). Our data underline the importance of LepRb→STAT3 signaling in control of ARH POMC neural projections and indicate a novel role for LepRb→MAPK action in the development of ARH projections. In addition, the differential disruption of NPY/AgRP and POMC projections in mature mice provide evidence of cell-specific roles of individual LepRb signals in the development of hypothalamic neural projections, whereas the sustained defects of both peptidergic pathways in db/db mice suggest a convergent impact of multiple cellular signals on the architecture of ARH projections. A better understanding of the relationship between these cell-type-specific developmental signaling pathways and various genetic and environmental cues will be crucial as we seek to understand perinatal programming of the hypothalamus and obesity.
Footnotes
This work was supported by National Institutes of Health NIH Grants DK65900 (R.B.S.) and DK057768 (M.G.M., R.B.S.).
References
- Ahima RS, Flier JS. Leptin. Annu Rev Physiol. 2000;62:413–437. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- Ahima RS, Prabakaran D, Flier JS. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J Clin Invest. 1998;101:1020–1027. doi: 10.1172/JCI1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG., Jr STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–859. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- Bjørbaek C, Buchholz RM, Davis SM, Bates SH, Pierroz DD, Gu H, Neel BG, Myers MG, Jr, Flier JS. Divergent roles of SHP-2 in ERK activation by leptin receptors. J Biol Chem. 2001;276:4747–4755. doi: 10.1074/jbc.M007439200. [DOI] [PubMed] [Google Scholar]
- Björnholm M, Münzberg H, Leshan RL, Villanueva EC, Bates SH, Louis GW, Jones JC, Ishida-Takahashi R, Bjørbaek C, Myers MG., Jr Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J Clin Invest. 2007;117:1354–1360. doi: 10.1172/JCI30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret SG, Draper SJ, Simerly RB. Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice. J Neurosci. 2004a;24:2797–2805. doi: 10.1523/JNEUROSCI.5369-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004b;304:108–110. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- Bouret SG, Gorski JN, Patterson CM, Chen S, Levin BE, Simerly RB. Hypothalamic neural projections are permanently disrupted in diet-induced obese rats. Cell Metab. 2008;7:179–185. doi: 10.1016/j.cmet.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broberger C, Johansen J, Johansson C, Schalling M, Hökfelt T. The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci U S A. 1998;95:15043–15048. doi: 10.1073/pnas.95.25.15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron E, Sachot C, Prevot V, Bouret SG. Distribution of leptin-sensitive cells in the postnatal and adult mouse brain. J Comp Neurol. 2010;518:459–476. doi: 10.1002/cne.22219. [DOI] [PubMed] [Google Scholar]
- Conway G. STAT3-dependent pathfinding and control of axonal branching and target selection. Dev Biol. 2006;296:119–136. doi: 10.1016/j.ydbio.2006.04.444. [DOI] [PubMed] [Google Scholar]
- Coppari R, Ichinose M, Lee CE, Pullen AE, Kenny CD, McGovern RA, Tang V, Liu SM, Ludwig T, Chua SC, Jr, Lowell BB, Elmquist JK. The hypothalamic arcuate nucleus: a key site for mediating leptin's effects on glucose homeostasis and locomotor activity. Cell Metab. 2005;1:63–72. doi: 10.1016/j.cmet.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Dent EW, Gertler FB. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron. 2003;40:209–227. doi: 10.1016/s0896-6273(03)00633-0. [DOI] [PubMed] [Google Scholar]
- Doherty P, Williams G, Williams EJ. CAMs and axonal growth: a critical evaluation of the role of calcium and the MAPK cascade. Mol Cell Neurosci. 2000;16:283–295. doi: 10.1006/mcne.2000.0907. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Coppari R, Balthasar N, Ichinose M, Lowell BB. Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J Comp Neurol. 2005;493:63–71. doi: 10.1002/cne.20786. [DOI] [PubMed] [Google Scholar]
- Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, Fu XY. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A. 2004;101:4661–4666. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, Elmquist JK. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest. 2008;118:1796–1805. doi: 10.1172/JCI32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AB, Kolodkin AL, Ginty DD, Cloutier JF. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu Rev Neurosci. 2003;26:509–563. doi: 10.1146/annurev.neuro.26.010302.081139. [DOI] [PubMed] [Google Scholar]
- Kowalski TJ, Liu SM, Leibel RL, Chua SC., Jr Transgenic complementation of leptin-receptor deficiency. I. Rescue of the obesity/diabetes phenotype of LEPR-null mice expressing a LEPR-B transgene. Diabetes. 2001;50:425–435. doi: 10.2337/diabetes.50.2.425. [DOI] [PubMed] [Google Scholar]
- Liu RY, Snider WD. Different signaling pathways mediate regenerative versus developmental sensory axon growth. J Neurosci. 2001;21:RC164. doi: 10.1523/JNEUROSCI.21-17-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry AM, Swick A, Romsos DR. Leptin alters metabolic rates before acquisition of its anorectic effect in developing neonatal mice. Am J Physiol. 1999;277:R742–R747. doi: 10.1152/ajpregu.1999.277.3.R742. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Niswender KD, Rhodes CJ, Myers MG, Jr, Blevins JE, Baskin DG, Schwartz MW. Arcuate nucleus-specific leptin receptor gene therapy attenuates the obesity phenotype of Koletsky (fa(k)/fa(k)) rats. Endocrinology. 2003;144:2016–2024. doi: 10.1210/en.2002-0115. [DOI] [PubMed] [Google Scholar]
- O'Malley D, MacDonald N, Mizielinska S, Connolly CN, Irving AJ, Harvey J. Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Mol Cell Neurosci. 2007;35:559–572. doi: 10.1016/j.mcn.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson SA, Leinninger GM, Myers MG., Jr Molecular and neural mediators of leptin action. Physiol Behav. 2008;94:637–642. doi: 10.1016/j.physbeh.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawchenko PE. Toward a new neurobiology of energy balance, appetite, and obesity: the anatomists weigh in. J Comp Neurol. 1998;402:435–441. [PubMed] [Google Scholar]
- Sawchenko PE, Swanson LW. The organization of forebrain afferents to the paraventricular and supraoptic nuclei of the rat. J Comp Neurol. 1983;218:121–144. doi: 10.1002/cne.902180202. [DOI] [PubMed] [Google Scholar]
- Shanley LJ, O'Malley D, Irving AJ, Ashford ML, Harvey J. Leptin inhibits epileptiform-like activity in rat hippocampal neurones via PI 3-kinase-driven activation of BK channels. J Physiol. 2002;545:933–944. doi: 10.1113/jphysiol.2002.029488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerio A, Ghisi V, Dossena M, Tonello C, Giordano A, Frontini A, Ferrario M, Pizzi M, Spano P, Carruba MO, Nisoli E. Leptin increases axonal growth cone size in developing mouse cortical neurons by convergent signals inactivating glycogen synthase kinase-3beta. J Biol Chem. 2006;281:12950–12958. doi: 10.1074/jbc.M508691200. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Snider WD. Intracellular control of developmental and regenerative axon growth. Philos Trans R Soc Lond B Biol Sci. 2006;361:1575–1592. doi: 10.1098/rstb.2006.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]