

Abstract
In the mammalian CNS, excessive release of glutamate and overactivation of glutamate receptors are responsible for the secondary (delayed) neuronal death following neuronal injury, including ischemia, traumatic brain injury (TBI), and epilepsy. The coupling of neurons by gap junctions (electrical synapses) increases during neuronal injury. We report here that the ischemic increase in neuronal gap junction coupling is regulated by glutamate via group II metabotropic glutamate receptors (mGluRs). Specifically, using electrotonic coupling, Western blots, and siRNA in the mouse somatosensory cortex in vivo and in vitro, we demonstrate that activation of group II mGluRs increases background levels of neuronal gap junction coupling and expression of connexin 36 (Cx36) (neuronal gap junction protein), and inactivation of group II mGluRs prevents the ischemia-mediated increases in the coupling and Cx36 expression. We also show that the regulation is via cAMP/PKA (cAMP-dependent protein kinase)-dependent signaling and posttranscriptional control of Cx36 expression and that other glutamate receptors are not involved in these regulatory mechanisms. Furthermore, using the analysis of neuronal death, we show that inactivation of group II mGluRs or genetic elimination of Cx36 both dramatically reduce ischemia-mediated neuronal death in vitro and in vivo. Similar results are obtained using in vitro models of TBI and epilepsy. Our results indicate that neuronal gap junction coupling is a critical component of glutamate-dependent neuronal death. They also suggest that causal link among group II mGluR function, neuronal gap junction coupling, and neuronal death has a universal character and operates in different types of neuronal injuries.
Introduction
Gap junctions (which are morphological correlates of electrical synapses) are intercellular channels between two neighboring cells that allow direct diffusion of ions and small molecules (Bennett and Zukin, 2004). The channels are made of proteins known as connexins, which are encoded in mammals by a family of 21 connexin genes (Söhl et al., 2005), and connexin 36 (Cx36) is the main neuronal connexin (Belluardo et al., 2000; Rash et al., 2000). In the developing mammalian CNS, coupling of neurons by gap junctions and the expression of Cx36 transiently increase (usually during the first 2 postnatal weeks) (Park et al., 2011) and play a role in a number of developmental events, including the regulation of neuronal death (Cusato et al., 2003), neuronal migration (Lo Turco and Kriegstein, 1991), neuronal differentiation (Bani-Yaghoub et al., 1999), synaptogenesis, and neural circuit formation (Peinado et al., 1993; Kandler and Katz, 1995; Personius et al., 2001). The coupling and expression of Cx36 then decrease (usually during postnatal weeks 3–4) (Connors et al., 1983; Kandler and Katz, 1995; Arumugam et al., 2005). However, Cx36 expression and coupling increase in the adult CNS following neuronal injury such as ischemia (Oguro et al., 2001; de Pina-Benabou et al., 2005), spinal cord and traumatic brain injury (TBI) (Chang et al., 2000; Frantseva et al., 2002), epilepsy (Gajda et al., 2003; Samoilova et al., 2003), and inflammation (Garrett and Durham, 2008).
It has been suggested previously that excessive release of glutamate and overactivation of glutamate receptors [mainly NMDA receptors (NMDARs)] are primarily responsible for the secondary (delayed) neuronal death that occurs during neuronal injuries (Choi, 1988; Arundine and Tymianski, 2004; Hazell, 2007). Recently, we demonstrated that pharmacological blockade and genetic elimination of neuronal gap junctions both dramatically reduce NMDAR-mediated neuronal death in developing and mature CNS neurons (de Rivero Vaccari et al., 2007; Wang et al., 2010; Park et al., 2011). In addition, studying the rat and mouse hypothalamus and cortex, we established a role for group II metabotropic glutamate receptors (mGluRs) in mechanisms for the developmental increase in neuronal gap junction coupling and showed that these mechanisms also directly control the death/survival mechanisms in developing neurons (Park et al., 2011). Specifically, we showed that chronic activation of group II mGluRs augments the developmental increase in neuronal gap junction coupling in vivo and in vitro and makes neurons significantly more susceptible to NMDAR-mediated excitotoxicity. In contrast, chronic inactivation of group II mGluRs prevents the developmental increase in neuronal gap junction coupling and also prevents NMDAR-mediated neuronal death. However, the mechanisms for increase in neuronal gap junction coupling during neuronal injury are not known. Furthermore, whether gap junctions contribute to death or survival in injured neurons is controversial (Perez Velazquez et al., 2003; Decrock et al., 2009). Given that there is excessive release of glutamate during neuronal injury, in the present study we tested the hypothesis that, as in development, group II mGluRs control the injury-mediated increase in neuronal gap junction coupling and, via regulation of gap junctions, they influence injury-induced neuronal death.
Materials and Methods
Animal care.
The use of animal subjects in these experiments was approved by the University of Kansas Medical Center Animal Care and Use Committee. The experiments were conducted in accordance with the National Institutes of Health guidelines. We used wild-type mice (C57BL/6) and Cx36 knock-out mice (C57BL/6 background strain). The Cx36 knock-out was originally created by Dr. David Paul (Harvard Medical School, Cambridge, MA). Mice were genotyped as described previously (de Rivero Vaccari et al., 2007).
Culture preparation, induction of injuries, and culture treatments.
Most of the experiments were conducted in neuronal cultures and some in purified glial cultures. Neuronal cultures were prepared as reported previously (Park et al., 2011) from the somatosensory cortex obtained from day 16 embryos. Pregnant mice were anesthetized with isoflurane before embryos were removed. After disaggregation using papain, neurons were plated on glass coverslips and raised in Neurobasal medium (Invitrogen; catalog #21103), in which the percentage of neurons reaches ∼95%. The medium was supplemented with B-27 (Invitrogen; catalog #17504) and 0.5 mm l-glutamine. The culture medium was changed twice a week. Most of the experiments were conducted in cultures on day in vitro 25 ± 1 d (i.e., at the time when the developmental uncoupling of neuronal gap junctions and Cx36 downregulation already have occurred) (see Fig. 1A) (Arumugam et al., 2005); only small interfering RNA (siRNA) transfections were done on day in vitro 7, and neurons were taken to the experiments on day in vitro 9, which was done because the transfections induced neurodegeneration in mature cultures. Purified glial cortical cultures were prepared and maintained as reported (Park et al., 2011).
Figure 1.

Changes in Cx36 protein expression during development and following ischemia in vitro. Data from Western blot experiments in neuronal somatosensory cortical cultures are presented. A, Developmental changes in Cx36 expression. B, OGD-induced changes in Cx36 expression in mature cultures [day in vitro 25 (DIV25)]. Duration of OGD was 30 min. A representative blot (above) and statistical data (below) are shown. In both graphs, optical density signals are normalized relative to tubulin and compared with the control (day in vitro 1 in A; pre-OGD condition in B; set at 1.0). Statistical analysis was as follows: ANOVA with post hoc Tukey relative to the control; n = 6 (A) and 5–11 (B) per group; data are shown as mean ± SEM. NS, Nonsignificant.
To induce oxygen–glucose deprivation (OGD), the cultures were transferred for 30 min from normal conditions (95% air plus 5% CO2; 10 mm glucose) to OGD conditions (95% N2 plus 5% CO2; no glucose). To induce hypoosmotic shock, the osmolarity of the culture medium was reduced for 60 min by 30% (from 290 to 203 mOsm). To induce hydrostatic pressure injury, cultures were placed in a modular chamber, where an increased pressure (103 mmHg) was generated by filling the chamber with 95% air plus 5% CO2 for 60 min. To induce epileptic discharges, 4-aminopyridine (voltage-dependent K+-channel blocker; 100 μm) was added to the culture medium for 60 min and then washed out. Shams for all injuries included change in the culture medium (with or without drugs in it), but no injury.
In cultures, the following drugs and concentrations were used: (RS)-1-aminoindan-1,5-dicarboxylic acid (AIDA) (100 μm), group I mGluR antagonist; AMPA (25 μm), AMPA receptor agonist; d,l-2-amino-5-phosphonovalerate (AP5) (100 μm), NMDAR antagonist; (±)-β-(aminomethyl)-4-chlorobenzenepropanoic acid (baclofen) (20 μm), GABAB receptor (GABABR) agonist; bicuculline methiodide (20 μm), GABAA receptor (GABAAR) antagonist; biphenyl-indanone A (BINA) (3 μm), positive allosteric modulator of mGluR2; 8-bromo-cAMP, sodium salt (8-Br-cAMP) (100 μm), a cell-permeable analog of cAMP that activates cAMP-dependent protein kinase (PKA); carbenoxolone (25 μm), nonspecific blocker of gap junctions; 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (10 μm), AMPA receptor antagonist; (S)-3,5-dihydroxyphenylglycine (DHPG) (10 μm), group I mGluR agonist; glutamate (2 μm), glutamate receptor agonist; 3-isobutyl-1-methylxanthine (IBMX) (50 μm), nonspecific phosphodiesterase inhibitor that increases intracellular cAMP levels; (9S,10S,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo{1,2,3-fg:3′,2′,1′-kl}pyrrolo{3,4-i}{1,6}benzodiazocine-10-carboxylic acid hexyl ester (KT5720) (0.5 μm), blocker of the activity of PKA; (2S)-2-amino-2-{(1S,2S)-2-carboxycycloprop-1-yl}-3-(xanth-9-yl)propanoic acid (LY341495) (2 μm), group II mGluR antagonist; (1R,4R,5S,6R)-4-amino-2-oxabicyclo{3.1.0}hexane-4,6-dicarboxylic acid (LY379268) (2 μm), group II mGluR agonist; (RS)-α-methylserine-O-phosphate (MSOP) (100 μm), group III mGluR antagonist; muscimol (25 μm), GABAAR agonist; nimodipine (20 μm), blocker of L-type voltage-gated Ca2+ channels; NMDA (50 μm), NMDAR agonist; 3-amino-2-(4-chlorophenyl)propanephosphonic acid (phaclofen) (100 μm), GABABR antagonist; (RS)-α-cyclopropyl-4-phosphonophenyl glycine (PPG) (10 μm), group III mGluR agonist; tetrodotoxin (TTX) (2 μm), blocker of voltage-gated sodium channels. All drugs were obtained from Sigma-Aldrich or Tocris. In Western blot and electrotonic coupling experiments in cultures, all agents (e.g., LY379268, LY341495, etc.) were present in the culture medium for 10 min before, during (30 or 60 min), and 10 min after injury or sham injury and then washed out.
Photothrombotic focal cerebral ischemia.
Ischemia was induced as described previously (Wang et al., 2010). Adult (2- to 2.5-month-old) mice were anesthetized with isoflurane and placed in a stereotaxic frame. The skull was exposed by incision of the skin. Rose Bengal (1 mg in 100 μl of sterile saline) was injected intraperitoneally. A fiber optic bundle of a cold light source (Zeiss 1500; 1.5 mm aperture; 3000 K light intensity) was placed in the right hemisphere at anteroposterior −1.7 mm and lateral to midline +2.0 mm from bregma. To activate Rose Bengal, 15 min after its injection, the brain was illuminated through the intact skull for the following 30 min. Sterile saline or drugs (dissolved in saline) were injected to animals intraperitoneally in doses as described below in the paper. In all drug treatment and ischemic experiments, an analgesic was used to reduce possible pain reactions (buprenex; 2 mg/kg).
Electrotonic coupling.
Experiments were conducted as described in detail previously (Park et al., 2011). Briefly, the pipette solution contained the following (in mm): 145 KMeSO4, 10 HEPES, 2 MgCl2, 0.1 CaCl2, 1.1 EGTA, 2 Na-ATP, and 0.3 Na-GTP, pH 7.2, 3–7 MΩ electrode resistance. Cells were patched using Multiclamp 700-B amplifier and pCLAMP10 software (Molecular Devices). To determine electrotonic coupling, dual whole-cell current-clamp recordings were conducted in cultures from pairs of neurons. Because in cell cultures different cell types are morphologically indistinguishable and electrophysiological characterization of cell types is not elaborated, neurons were chosen randomly. Test current steps (500 ms, −100 pA) were applied to cell 2 (injected cell) and electrotonic responses were detected in cell 1 (noninjected cell). Recordings were done at a holding potential of −65 mV. Data were analyzed off-line with Clampfit 10 (Molecular Devices). The coupling coefficient was calculated as the response amplitude in the noninjected cell (cell 1) divided by the amplitude in the injected cell (cell 2). Incidence of electrotonic coupling was calculated as percentage of neuronal pairs that demonstrated the coupling. Cells were considered as coupled if the coupling coefficient was >1.6%. Only one pair of neurons per coverslip was tested.
Western blots.
Experiments were performed as reported in detail previously (Arumugam et al., 2005; Park et al., 2011). Briefly, the primary somatosensory cortex (obtained from the right ischemic region, right sham region, or left contralateral region) or cortical cultures were homogenized in a lysis buffer, and total protein was determined using the Bio-Rad DC protein assay method. Fifty micrograms of protein was loaded in each lane, transferred to 0.45 μm polyvinylidene difluoride membrane, and processed with a blocking solution and antibodies. Rabbit anti-Cx36 (0.5 μg/ml; Zymed; catalog #51-6300), rabbit anti-connexin 43 (Cx43) (0.2 μg/ml; Zymed; catalog #71-0700), rabbit anti-mGluR2 (0.5 μg/ml; Millipore; catalog #AB9209), rabbit anti-mGluR3 (0.5 μg/ml; Sigma-Aldrich; catalog #G1545), and mouse anti-tubulin (1:10,000; Sigma-Aldrich; catalog #T6793) were used as the primary antibodies, and they were visualized with horseradish peroxidase-conjugated anti-rabbit antibody (1:10,000; Zymed; catalog #G21234) or anti-mouse IgG antibody (1:10,000; Sigma-Aldrich; catalog #M5899). Signals were enhanced using ECL detection reagents (GE Healthcare). Band optical density was determined by using Quantity One quantification analysis software 4.5.2 (Bio-Rad). All optical density signals were normalized relative to tubulin and experimental samples were compared with controls (set at 1.0). Tubulin levels per unit of total protein did not vary significantly among samples used in this study.
siRNA.
Experiments were conducted as described in detail previously (Park et al., 2011). The mGluR2 and mGluR3 siRNAs were purchased from Dharmacon RNAi Technology (catalog #M-080176-00 and L-094437-01, respectively). Each siRNA consisted of four pooled 19 nt duplexes and was used in a final concentration of 50 nm. Cells were transfected using Lipofectamine 2000 (Invitrogen) 48 h before the induction of OGD and were processed for Western blot analysis 2 h after OGD. Both transfections effectively reduced protein levels. Scrambled siRNAs were used as controls and were ineffective.
Reverse transcription–quantitative real-time PCR.
Experiments were performed as reported previously (Park et al., 2011). Total RNA was isolated from cultures using Trizol method (Invitrogen). Total RNA (1 μg) was reverse transcribed with oligo-dT primers and the SuperScript II kit (Invitrogen) according to the manufacturer's instructions. One microliter of the reverse transcription reaction material was used as template for reverse transcription–quantitative real-time PCR (RT-qPCR) using a Bio-Rad iCycler in a total volume of 20 μl with SYBR Green PCR Master Mix (Applied Biosystems) and amplified for 40 cycles for 15 s at 95°C and for 20 s at 60°C. The following primer pairs were used: mouse Cx36, 5′-CCAGTAAGGAGACAGAACCAGAT-3′ (sense) and 5′-GATGATGTAGAAGCGGGAGATAC-3′ (antisense); mouse β-actin, 5′-GATGGTGGGAATGGGTCAGAA-3′ (sense) and 5′-CTCATTGTAGAAGGTGTGGTGC-3′ (antisense). In all experiments, Cx36 mRNA signals were normalized to β-actin mRNA signals, and normalized values were compared with controls (set at 1.0). β-Actin levels did not vary significantly among samples used in this study.
Methyl thiazolyl tetrazolium assay.
Neuronal viability in cultures was quantitatively evaluated by methyl thiazolyl tetrazolium (MTT) assay as reported previously (Park et al., 2011). Cultures were raised in 24-well plates. In all tests, LY341495 and carbenoxolone were added to the culture medium before induction of injury and remained in the medium during and for 24 h after the induction of injury, after which the cells were taken for analysis. Neurons were incubated with MTT (MTT Cell Viability Assay kit; Biotium; 40 μm, 400 μl per well) at 37°C for 4 h. Then the medium was carefully aspirated and 400 μl of DMSO per well was added to dissolve the blue formazan product. To measure the absorbance, 200 μl of the medium from each well in 24-well plate were transferred into an independent well in a 96-well plate. The values of absorbance at 570 nm were measured using a microplate reader (μQuant; Bio-Tek). Furthermore, as indicated above, we used the cultures that were mostly neuronal (up to 95%). However, to control specifically for neuronal cell death, a separate group of cultures was subjected to a high concentration of glutamate (500 μm) for 24 h that killed all neurons, but did not affect glial cell survival. The absorbance in these purified glial cultures was measured, averaged, and the result was subtracted from the individual absorbance data in neuronal culture groups so that the final result would represent only neuronal death/survival. Finally, the absorbance results in experimental groups were normalized to control groups.
Fluoro-Jade B staining.
This staining is selective and highly sensitive for identification of degenerating neurons (for details, see Wang et al., 2010). Staining was conducted as reported previously (Wang et al., 2010). Animals were anesthetized and transcardially perfused with PBS (0.01 mol/L; 100 ml) followed by a perfusion with 4% paraformaldehyde (dissolved in 50 ml of PBS; pH 7.4). The brains were removed. Fifty-micrometer-thick coronal sections were cut with a Leica VT1000S vibratome and mounted on gelatin-coated slides. The sections were hydrated in alcohol, incubated with potassium permanganate (0.006%; 10 min), incubated with Fluoro-Jade B (0.001%; 30 min), rinsed, and air-dried. The staining was visualized using a Nikon Eclipse 80i fluorescent microscope, FITC filter, and Photometrics ES2 digital camera. The ischemic cortical region was outlined and the total number of stained pixels was measured in the outlined region using a densitometric thresholding technique implemented with NIH ImageJ software. The threshold was set at a level just above that which would have counted background and nonspecific staining in areas outside the outlined region. The analysis was done in five cortical sections (including −1.5, −1.6, −1.7, −1.8, and −1.9 mm from bregma), and data were averaged for five sections. The staining analysis was done by a person blinded to experimental groups. Since Fluoro-Jade B has an excellent sensitivity and selectivity for identification of degenerating neurons, no additional costaining with neuronal markers was necessary.
Statistical analysis.
Data were analyzed using the two-tailed Student's t test (paired, when possible), ANOVA with post hoc Tukey's or Fisher's exact probability test, and InStat software (GraphPad Software). The number of samples indicated for in vitro experiments (including Western blot, siRNA, RT-qPCR, and MTT) represents the number of plate wells/coverslips with cells obtained at least from three independent cell culture preparations. The number of samples indicated for experiments in animals represents the number of animals. Data are reported as mean ± SEM.
Results
Regulation of neuronal gap junction coupling during ischemia in vitro
To determine changes during neuronal injury in neuronal gap junction coupling and expression of Cx36, we used OGD (see Materials and Methods), which is considered an in vitro model of ischemia (Lobner and Choi, 1994). Tests were done in wild-type mouse neuronal somatosensory cortical cultures after their maturation (Fig. 1A). We used Western blots to examine Cx36 protein expression and electrotonic coupling to analyze the coupling coefficient and incidence of electrotonic coupling (see Materials and Methods). As expected, OGD increased the levels of Cx36 protein (maximum at 2 h after OGD), the coupling coefficient, and the incidence of electrotonic coupling (Figs. 1B, 2A–C), suggesting increased neuronal gap junction coupling.
Figure 2.

Ischemic increase in neuronal gap junction coupling in vitro is regulated by glutamate via group II mGluRs and is action potential-dependent. Data from experiments in wild-type mouse mature neuronal somatosensory cortical cultures are shown. All pharmacological agents were present in the culture medium for 10 min before, during, and 10 min after OGD or sham OGD and then washed out. The analysis was done 2 h after OGD or sham OGD. A, B, Electrotonic coupling. Statistical data and representative traces (a1, b1) are shown. Presented are the incidence of electrotonic coupling and the coupling coefficient. Statistical analysis was as follows: Fisher's exact probability test (A; see Table 1 for the number of samples) and ANOVA with post hoc Tukey (B; responses from all of the tested pairs are included in the analysis). C–E, Western blots: Cx36 expression. Statistical data and representative blots (c1–e2) are shown. Optical density signals are normalized relative to tubulin and compared with the control (set at 1.0). Statistical analysis was as follows: ANOVA; n = 6–15 per group. In all graphs, statistical significance is shown relative to control (i) and nontreated plus OGD (ii). In B–E, the data are shown as mean ± SEM. Glu, Glutamate.
We tested the role of chemical neurotransmitter receptors in the ischemic increase in neuronal gap junction coupling. In general, if a receptor contributes to the ischemia-mediated increase in coupling, the following events must occur: release of the corresponding endogenous neurotransmitter by ischemic cells, increase in background levels of coupling and Cx36 expression by activation of the receptor, and prevention of the ischemia-mediated increases in coupling and Cx36 expression by receptor blockade. Depending on the amount of endogenous neurotransmitter released and the amount of receptors occupied by the neurotransmitter during ischemia, administration of the receptor agonist may or may not augment the ischemic increases in coupling and Cx36 expression. Excessive and rapid (10–45 min) release of glutamate has been found in neuronal cortical cultures during and after OGD (Lobner and Choi, 1994) and in the rat brain after focal cerebral ischemia (De Cristóbal et al., 2001; Yoshioka et al., 2009). We have previously shown that group II mGluRs regulate the transient increase in neuronal gap junction coupling during development and that activating these receptors leads to increased Cx36 expression in mature neurons (Park et al., 2011). Therefore, we tested whether group II mGluRs regulate the increase in neuronal gap junction coupling following ischemia.
In cortical cultures, activation of group II mGluRs (with LY379268; 2 μm) increased background levels of Cx36 and neuronal gap junction coupling (Fig. 2A–C, Table 1). Inactivation of these receptors (with LY341495; 2 μm) did not affect background levels of Cx36 and gap junction coupling (which were already low in mature cultures), but prevented the OGD-mediated increase (Fig. 2A–C, Table 1). Administration of glutamate (2 μm; an endogenous agonist for all glutamate receptors) also increased background Cx36 levels, and this effect was prevented by inactivation of group II mGluRs (Fig. 2D). Furthermore, the blockade of action potentials with TTX (2 μm; a voltage-gated sodium channel blocker) did not preclude LY379268 from increasing the background levels of Cx36, but prevented the OGD-induced Cx36 upregulation (Fig. 2E). From these data, we conclude that action potential-dependent synaptic release of glutamate regulates the OGD-mediated increases in neuronal gap junction coupling and Cx36 expression, the regulation is via activation of group II mGluRs, and these receptors likely are postsynaptic (as suggested by the TTX data).
Table 1.
Incidence of electrotonic coupling in mature neuronal somatosensory cortical cultures under different experimental conditions
| Conditions | No. of tested neuronal pairs | No. of coupled neuronal pairs |
|---|---|---|
| WT cultures | ||
| Sham OGD | ||
| Control | 26 | 0 |
| LY379268 (2 μm) | 23 | 4 (i) p = 0.042 |
| LY341495 (2 μm) | 24 | 0 (i) NS |
| OGD | ||
| Nontreated | 32 | 7 (i) p = 0.013 |
| LY379268 (2 μm) | 25 | 10 (i) p = 0.0003, (ii) NS |
| LY341495 (2 μm) | 40 | 1 (i) NS, (ii) p = 0.019 |
| Cx36 knock-out cultures | ||
| Sham OGD | ||
| Control | 15 | 0 |
| LY379268 (2 μm) | 18 | 0 |
| OGD | ||
| Nontreated | 18 | 0 |
Statistical analysis was as follows: Fisher's exact probability test, shown relative to control (i) and nontreated plus OGD (ii) in wild-type cultures. NS, Nonsignificant.
A number of other neurotransmitter receptors may regulate neuronal gap junction coupling (Hatton, 1998; Arumugam et al., 2005). However, neither background levels of Cx36 were affected by activation of NMDARs, group I mGluRs, group III mGluRs or GABABRs (with 50 μm NMDA, 10 μm DHPG, 10 μm PPG, and 20 μm baclofen, respectively) (Fig. 3A) nor the OGD-induced increases in Cx36 expression were prevented by inactivation of these receptors (with 100 μm AP5, 100 μm AIDA, 100 μm MSOP, and 100 μm phaclofen, respectively) (Fig. 3B). This suggests that these receptors are not involved in the OGD-dependent regulation of Cx36.
Figure 3.

Group I and group III metabotropic glutamate, AMPA, NMDA, GABAA, and GABAB receptors are not involved in the ischemic regulation of Cx36 expression. Pharmacological treatments were performed in wild-type mouse mature neuronal cortical cultures that were subjected to OGD or sham OGD followed by assessment of Cx36 protein expression 2 h after OGD or sham OGD (see text for details on the used drug concentrations; all agents were present in the culture medium for the time as described for Fig. 2). A–E, Analysis of the role of group I and III mGluRs, NMDARs, and GABABRs (A, B), AMPA receptors (C), and GABAARs (D, E). Statistical data and representative blots (a1–e2) are shown. Optical density signals are normalized relative to tubulin, and normalized values are compared with the control (set at 1.0; not shown in B). In all graphs, statistical analysis was as follows: ANOVA with post hoc Tukey; statistical significance is shown relative to control (i) and nontreated plus OGD (ii); n = 5–13 per group; the data are shown as mean ± SEM. Bic, Bicuculline; Musc, muscimol; Nimod, nimodipine.
Inactivation of AMPA receptors (with CNQX; 10 μm) prevented the OGD-mediated increase in Cx36 expression (Fig. 3C). However, their activation (with AMPA; 25 μm) did not affect background Cx36 levels (Fig. 3C). Therefore, we believe that AMPA receptors do not play a role in the ischemic regulation of Cx36, and the effect of CNQX in decreasing Cx36 expression is most likely through a decrease in the general level of electrical activity in cultures (i.e., similar to the effect of TTX) (Fig. 2E).
Our previous studies in developing neurons showed (Park et al., 2011) that GABAARs negatively regulate the transient developmental increase in neuronal gap junction coupling via GABAAR-mediated excitation and influx of Ca2+ through L-type voltage-gated Ca2+ channels. In the present study in mature neurons, inactivation of GABAARs (with bicuculline; 20 μm) increased background levels of Cx36 (Fig. 3D). However, the increase was prevented by blockade of group II mGluRs, indicating that this was caused by disinhibition-mediated release of glutamate from neurons and activation of group II mGluRs rather than via direct effect. In addition, activation of GABAARs (with muscimol; 25 μm) prevented the OGD-mediated increase in Cx36 expression (Fig. 3E). However, this effect was not altered by blockade of L-type voltage-gated Ca2+ channels (with nimodipine; 20 μm), suggesting that the muscimol effect is not due to GABAAR-mediated excitation, but rather is due to inhibition of the general level of electrical activity (again, similar to the effect of TTX) (Fig. 2E). Considering these findings, we conclude that GABAARs are not involved directly in the ischemic regulation of neuronal gap junctions.
Regulation of neuronal gap junction coupling during ischemia in vivo
To determine whether group II mGluRs also regulate neuronal gap junction coupling during ischemia in vivo, we used photothrombotic focal cerebral ischemia in adult wild-type mice (see Materials and Methods) and measured Cx36 protein levels in the ischemic region (right primary somatosensory cortex) and the corresponding unaffected contralateral region. The expression of Cx36 was increased in the ischemic region 2 h after ischemia followed by a decrease (Fig. 4A). In nonischemic mice, an intraperitoneal injection of group II mGluR agonist (LY379268) elevated background levels of Cx36 2 h after injection (Fig. 4B). Injection of an antagonist (LY341495) did not have an effect (Fig. 4B). However, the antagonist completely abolished the ischemic increase in Cx36 expression (Fig. 4C). These data support the in vitro findings (see above) that the ischemic increase in neuronal gap junction coupling is regulated via activation of group II mGluRs.
Figure 4.

Increase in Cx36 expression after photothrombotic focal cerebral ischemia is regulated by group II mGluRs. Data from Western blot experiments in the primary somatosensory cortex of adult wild-type mice are presented. A–C, Statistical data and representative blots (a1–c2) are shown. In A, “C” indicates contralateral cortex (left hemisphere), and “I” indicates ipsilateral cortex (right hemisphere), which also is ischemic. In B and C, all data are from the ipsilateral cortex: nonischemic (B) and ischemic (C). In all experiments, optical density signals are normalized relative to tubulin and compared with the control: contralateral cortex (A), saline ipsilateral (B), and saline contralateral (C; not shown); set at 1.0. Statistical analysis was as follows: Student's t test; shown relative to the contralateral cortex (A) and saline ipsilateral (B, C); n = 6–10 per group; data are shown as mean ± SEM. Injections were as follows: saline (100 μl), LY379268 (10 mg/kg; in 100 μl of saline), LY341495 (5 mg/kg; in 100 μl of saline); in B, each injection was single; in C, two injections were done (immediately before and immediately after the induction of ischemia).
Cellular mechanisms for ischemic increase in neuronal gap junction coupling
It has been established previously (Conn et al., 2005) that group II mGluRs include mGluR2 and mGluR3 and that activation of these receptors suppresses cAMP/PKA-dependent signaling. In mature neuronal cortical cultures, we used administration of 8-Br-cAMP plus IBMX (100 μm plus 50 μm; a cell-permeable analog of cAMP plus a nonspecific phosphodiesterase inhibitor) and KT5720 (0.5 μm; a blocker of the activity of PKA) to increase and decrease, respectively, the activity of cAMP/PKA-dependent signaling. We also used Western blot analysis to confirm that the ischemic regulation of neuronal gap junctions is via cAMP/PKA. The effect of activation of group II mGluRs (i.e., increased background Cx36 levels) was mimicked by blockade of PKA and was prevented by elevation in cytoplasmic cAMP (Fig. 5A). Furthermore, the effect of inactivation of group II mGluRs (i.e., reduction of the OGD-mediated Cx36 upregulation) was mimicked by elevation in cytoplasmic cAMP levels and prevented by blockade of the activity of PKA (Fig. 5B). This confirmed that the regulation by group II mGluRs is via cAMP/PKA-dependent signaling.
Figure 5.

The ischemic regulation of Cx36 by group II mGluRs is through cAMP/PKA-dependent signaling. A, B, Pharmacological treatments were performed in wild-type mouse mature neuronal cortical cultures that were subjected to sham OGD (A) or OGD (B) followed by assessment of Cx36 protein expression 2 h after sham OGD or OGD. Statistical data and representative blots are shown. Optical density signals are normalized relative to tubulin, and normalized values are compared with the control (set at 1.0; not shown in B). Statistical analysis was as follows: ANOVA with post hoc Tukey; statistical significance is shown relative to control (i), nontreated plus OGD (ii), LY379268 alone (iii), and LY341495 alone (iv); n = 7–11 per group; data are shown as mean ± SEM. Treatments were as follows: PKA(+), activation of the cAMP/PKA-dependent signaling by 8-Br-cAMP plus IBMX (100 μm plus 50 μm); PKA(−), blockade of the activity of PKA with KT5720 (0.5 μm); LY379268, 2 μm; LY341495, 2 μm; all agents were present in the culture medium for the time as described for Figure 2.
We also tested the importance of mGluR2 versus mGluR3. First, we used an siRNA approach in neuronal cortical cultures. Because siRNA transfections induced neuronal death in mature cultures, day in vitro 7–9 cultures were used (see Materials and Methods). The experiment showed that genetic suppression of both mGluR2 and mGluR3 prevented the OGD-mediated increase in Cx36 expression (Fig. 6A,B). Second, to determine whether there are developmental changes, between days in vitro 7 and 25, in the expression of mGluR2 and mGluR3, we used Western blots and observed no changes and decreases in the levels of mGluR2 and mGluR3, respectively (Fig. 6C). Finally, selective agonists and antagonists for mGluR2 and mGluR3 and positive allosteric modulators for mGluR3 are not available, but we tested a positive allosteric modulator of mGluR2, BINA (3 μm), to see whether it potentiates the effect of OGD on Cx36 expression. Five-minute-long OGD or BINA treatment (50 min) alone did not have an effect on Cx36 levels (as measured using Western blots 2 h after OGD or BINA administration); however, the expression of Cx36 was increased when 5 min OGD was given in the beginning of a 50 min BINA treatment (Fig. 6D). Together, these data suggest that the mGluR2 is important for the ischemic control of Cx36 expression. Given the developmental decrease in mGluR3 expression, the role of this receptor in Cx36 regulation during ischemia seems less likely than that of mGluR2; however, it cannot be completely ruled out.
Figure 6.

The role of mGluR2 versus mGluR3 in the ischemic control of Cx36 expression. Data from siRNA (A, B) and Western blot (A–D) experiments in wild-type mouse neuronal cortical cultures are shown. A, B, Transfections were done on day in vitro 7 (DIV7), followed by OGD or sham OGD on DIV9 and Western blot analysis 2 h after OGD/sham OGD. In sham OGD conditions (control), siRNA suppression of mGluR2 (A) and mGluR3 (B) decreases the receptor protein levels but does not significantly affect background levels of Cx36. Following OGD, the cells transfected with scrambled siRNA (SCR siRNA) demonstrate increased Cx36 expression, but this increase does not occur in cells transfected with active siRNA (either mGluR2 or mGluR3). C, Developmental changes in the expression of mGluR2 and mGluR3. D, In mature cultures, BINA (3 μm; 50 min) potentiates the effect of a 5-min-long OGD on Cx36 expression. In all figures, statistical data and representative blots (a1–d1) are shown. Optical density signals are normalized relative to tubulin, and normalized values are compared with the scrambled siRNA in control conditions (A, B), corresponding DIV7 (C), and control (D), set at 1.0. Statistical analysis was as follows: ANOVA with post hoc Tukey (A, B, D); Student's t test (C); *p < 0.05, **p < 0.01, ***p < 0.001; n = 5–10 (A, B), 4 (C), 3 (D) per group; data are shown as mean ± SEM. Stainings were done sequentially on one membrane.
Specificity of the mechanisms for increase in neuronal gap junction coupling
We examined whether regulation by group II mGluRs is specific for neuronal Cx36-containing gap junctions. First, electrotonic coupling was measured in neuronal cortical cultures prepared from Cx36 knock-out mice. As expected, the coupling was not observed in Cx36-deficient neurons, and remained absent following OGD or treatment with group II mGluR agonist (Fig. 7A,B; Table 1), both of which increase the coupling in wild-type neurons (Fig. 2A,B). Second, using purified glial cortical cultures and the primary somatosensory cortex (both obtained from wild-type mice), we performed Western blots to examine the expression of Cx43, a presumptive astrocytic connexin (Rash et al., 2000). Cx43 levels were increased 2 h after ischemia both in vitro and in vivo (Fig. 7C,D). However, background levels of Cx43 and ischemia-mediated increases were not altered by, respectively, activation and inactivation of group II mGluRs. Furthermore, background Cx43 levels were not altered by administration of glutamate in vitro (Fig. 7E). These data are in agreement with previous studies indicating that the expression of Cx43 also increases following ischemia (Danesh-Meyer et al., 2008). However, our data show that the ischemic regulation of glial Cx43 is not via glutamate-dependent mechanisms, and group II mGluRs particularly. Moreover, because no increase in the coupling occurs in Cx36-deficient neurons (Fig. 7A,B), these mechanisms are exclusive for Cx36.
Figure 7.

The regulation by group II mGluRs is specific for Cx36-containing gap junctions. A, B, Electrotonic coupling experiments in Cx36 knock-out (Cx36 KO) mouse mature neuronal somatosensory cortical cultures reveal no coupling between the neurons. Statistical data for the incidence of electrotonic coupling and the coupling coefficient (A, B, respectively) are presented (see Table 1 for the number of samples). For comparison, data from the OGD group in wild-type (WT) neuronal cultures from Figure 2, A and B, are included in the boxes. C–E, Cx43 expression. Statistical data and representative blots (c1–e1) from Western blot experiments in the primary somatosensory cortex obtained from adult wild-type mice (C) and in wild-type mouse mature purified glial cortical cultures (D, E) are shown. Statistical analysis was as follows: ANOVA with post hoc Tukey; n = 8 (C), 8–13 (D), and 7–10 (E) per group; data are shown as mean ± SEM. Optical density signals are normalized relative to tubulin, and normalized values are compared with the nonischemic saline (C) and control (D, E), set at 1.0. Experimental conditions and drug concentrations are the same as in Cx36 experiments (see Figs. 2, 4). In C, all measurements of Cx43 expression were done in the right (ipsilateral) cortex.
Changes in Cx36 mRNA levels following ischemia
During development, the increase in neuronal gap junction coupling is regulated by group II mGluRs via transcriptional regulation of the Cx36 gene (Park et al., 2011). To determine whether the same regulation occurs during ischemia, we measured Cx36 mRNA levels with RT-qPCR. Surprisingly, the levels of Cx36 mRNA were not elevated 2 h after ischemia both in vitro and in vivo (Fig. 8A,B) (i.e., at the time when protein levels are increased) (Figs. 2C, 4A). In addition, activation of group II mGluRs did not affect background Cx36 mRNA levels also both in vitro and in vivo (Fig. 8A,B). This suggests that the ischemic increase of Cx36 likely is controlled at the posttranscriptional level.
Figure 8.

Cx36 mRNA levels are unchanged 2 h following ischemia or activation of group II mGluRs. Statistical data from RT-qPCR experiments in wild-type mouse mature neuronal somatosensory cortical cultures (A) and in the primary somatosensory cortex of adult wild-type mice (B) are shown. Experimental conditions and drug concentrations are the same as in Western blot experiments in vitro (Fig. 2) and in vivo (Fig. 4). In both graphs, relative Cx36 transcript levels are normalized to β-actin, and normalized values are compared with the control (set to 1.0). Statistical analysis was as follows: ANOVA with post hoc Tukey (A, B, left panels) and Student's t test (A, B, right panels); shown relative to control (A), contralateral cortex (“C”) (B, left panel), and ipsilateral cortex (“I”) 2 h after saline injection (“saline”) (B, right panel); n = 5–9 (A) and 4–7 (B) per group; data are shown as mean ± SEM.
Functional implications
Previously, we (Wang et al., 2010) demonstrated that pharmacological blockade of neuronal gap junctions and Cx36 knock-out both dramatically reduce NMDAR-mediated excitotoxicity and neuronal death caused by focal cerebral ischemia in adult mice. Furthermore, we (Park et al., 2011) showed that chronic inactivation of group II mGluRs, which prevents transient developmental increase in neuronal gap junction coupling, also prevents NMDAR-mediated neuronal death. In the present study, we tested whether mechanisms regulating ischemic increase in neuronal gap junction coupling also regulate ischemic neuronal death. The amount of neuronal death was measured using MTT assay in cultures and Fluoro-Jade B staining in brain sections. In wild-type mouse cortical cultures, there was substantial amount of neuronal death 24 h after OGD (Fig. 9A) that was dramatically reduced both by blockade of gap junctions with a nonspecific gap junction blocker, CBX (carbenoxolone) (25 μm), and by inactivation of group II mGluRs (with LY341495; 2 μm) (i.e., the treatment that also prevents the OGD-mediated upregulation of neuronal gap junctions) (Fig. 2A–C). Strikingly, in Cx36 knock-out cultures, the amount of OGD-mediated neuronal death was statistically insignificant and pharmacological blockade of gap junctions or group II mGluRs did not have any additional neuroprotective effects (Fig. 9B). Furthermore, in the primary somatosensory cortex of adult wild-type mice, dramatic neuronal death was detected 48 h after ischemia (Fig. 9C–F), but was substantially reduced by inactivation of group II mGluRs (Fig. 9C,G). In Cx36 knock-out mice, the level of ischemic neuronal death was significantly lower than in wild-type animals, and inactivation of group II mGluRs did not have any additional effects (Fig. 9C,H). From these observations, we conclude that the ischemic increase in neuronal gap junction coupling is a critical component of neuronal death mechanisms during ischemia.
Figure 9.

Inactivation of group II mGluRs and neuronal gap junctions both reduce ischemic neuronal death. A, B, Statistical data from MTT experiments in wild-type (A) and Cx36 knock-out (B) mature neuronal cortical cultures are shown. Statistical analysis was as follows: ANOVA with post hoc Tukey; shown relative to nontreated plus OGD group; ***p < 0.001; n = 6–12 per group. C–H, Statistical data (C) and representative images (D–H) from Fluoro-Jade B staining experiments in adult mice are shown. All images are taken at bregma −1.7 mm (i.e., at the center of ischemic injury). Images in D–F are taken from the same brain section and the regions that are boxed in D and E are shown at a higher magnification as E and F, respectively. Saline and LY341495 injections were done in triplicate: immediately before, immediately after, and 24 h after the induction of ischemia (in doses as in Fig. 4). In C, the number of stained pixels in the ischemic cortical region is analyzed: ANOVA; statistical difference is shown relative to the saline-treated WT mice (***p < 0.001) and between saline- and LY341495-treated Cx36 knock-out mice (NS, nonsignificant); n = 4–7 mice per group; data are shown as mean ± SEM. Hipp, Hippocampus; SSp, primary somatosensory cortex; Thal, thalamus.
Other types of neuronal injury
TBI, spinal cord injury, and epilepsy also demonstrate excessive release of glutamate and glutamate-dependent excitotoxicity (Choi, 1988; Arundine and Tymianski, 2004) as well as increased neuronal gap junction coupling and Cx36 expression (Perez-Velazquez et al., 1994; Chang et al., 2000; Frantseva et al., 2002; Gajda et al., 2003; Samoilova et al., 2003). We tested whether group II mGluRs control the expression of Cx36 and neuronal death during other types of neuronal injuries too. We used three in vitro injury models: hypoosmotic shock as a model of cytotoxic and osmotic edemas that occur during stroke and TBI (Unterberg et al., 2004); hydrostatic pressure injury, which represents mechanical aspects of TBI (Morrison et al., 1998); and administration of 4-aminopyridine as a model of epileptic seizures (Wong and Yamada, 2001). The injuries were induced as described in Materials and Methods. Tests were conducted in mature neuronal cortical cultures using Western blots, RT-qPCR, and MTT assay to determine the expression of Cx36 protein and mRNA and the amount of injury-induced neuronal death. As with ischemia, each of the three injuries induced Cx36 protein increases 2 h after injury, but did not alter Cx36 mRNA levels (Figs. 10A,C, 11A,C, 12A,C). The injury-mediated increases in Cx36 protein were prevented by inactivation of group II mGluRs (Figs. 10B, 11B, 12B). Furthermore, neuronal death was detected 24 h following each of the three injuries and was dramatically reduced by inactivation of group II mGluRs or pharmacological blockade of gap junctions (Figs. 10D, 11D, 12D). Calcium imaging tests confirmed that administration of 4-aminopyridine causes seizure-like activity (Fig. 12E).
Figure 10.
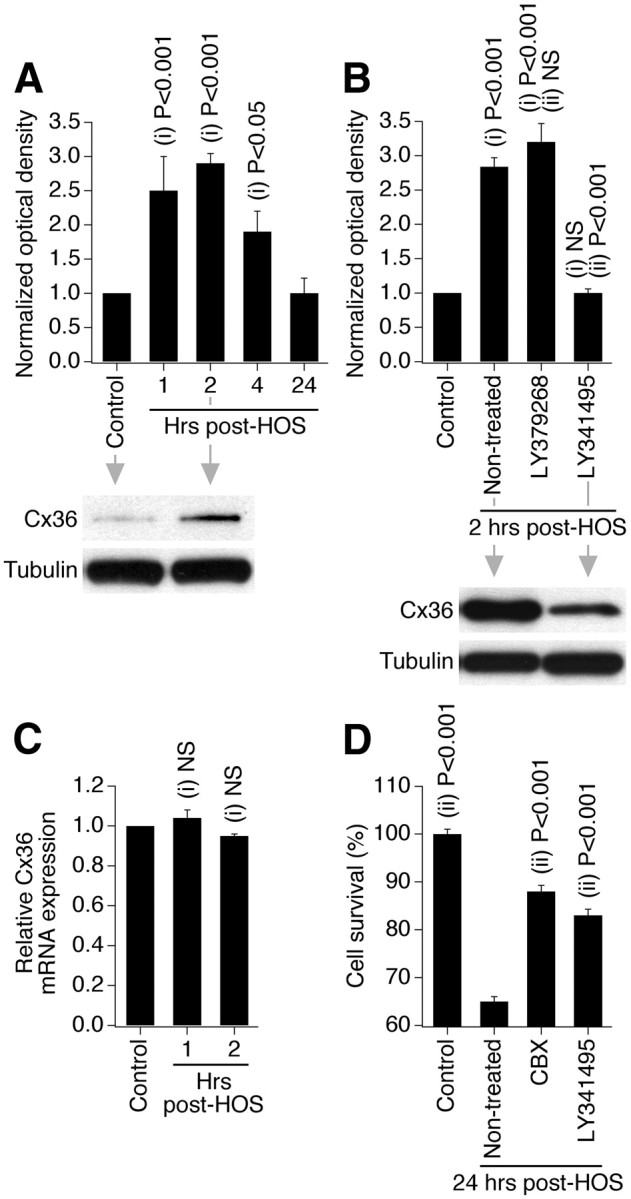
Group II mGluRs regulate increase in Cx36 expression and neuronal death during different types of neuronal injury: hypoosmotic shock (HOS). Data from Western blot (A, B), RT-qPCR (C), and MTT (D) experiments in wild-type mouse mature neuronal somatosensory cortical cultures are presented. Experimental conditions, drug concentrations, and analyses are the same as in OGD experiments shown in Figures 1B, 2C, 8A, and 9A; however, because cultures were subjected to HOS for 60 min (see Materials and Methods), the drug treatments were 30 min longer than during OGD. Statistical analysis was as follows: ANOVA with post hoc Tukey; shown relative to control (i) and nontreated plus HOS (ii); n = 5–10 (A), 4–7 (B), 5 (C), and 10 (D) per group; mean ± SEM.
Figure 11.
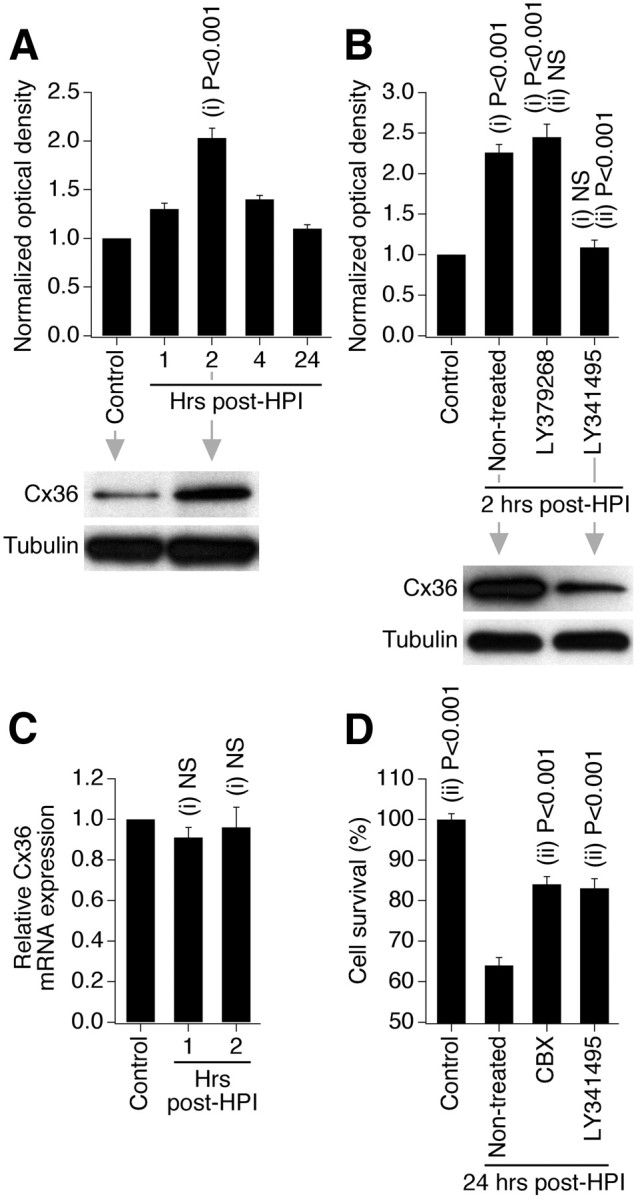
Group II mGluRs regulate increase in Cx36 expression and neuronal death during different types of neuronal injury: hydrostatic pressure injury (HPI). Data from Western blot (A, B), RT-qPCR (C), and MTT (D) experiments in wild-type mouse mature neuronal somatosensory cortical cultures are presented. Experimental conditions, drug concentrations, and analyses are the same as in ischemic experiments shown in Figures 1B, 2C, 8A, and 9A; however, because cultures were subjected to HPI for 60 min (see Materials and Methods), the drug treatments were 30 min longer than during OGD. Statistical analysis: ANOVA with post hoc Tukey; shown relative to control (i) and nontreated plus HPI (ii); n = 4–12 (A), 5–7 (B), 5 (C), and 6–8 (D) per group; data are shown as mean ± SEM.
Figure 12.

Group II mGluRs regulate increase in Cx36 expression and neuronal death during different types of neuronal injury: epileptic seizures. Data from Western blot (A, B), RT-qPCR (C), MTT (D), and fura-2 Ca2+ imaging (E) experiments in wild-type mouse mature neuronal somatosensory cortical cultures are presented. Experimental conditions, drug concentrations, and analyses are the same as in ischemic experiments shown in Figures 1B, 2C, 8A, and 9A; however, because cultures were subjected to 4-aminopyridine (4AP) for 60 min (see Materials and Methods), the drug treatments were 30 min longer than during OGD. Statistical analysis was as follows: ANOVA with post hoc Tukey; shown relative to control (i) and nontreated plus 4AP (ii); n = 4–12 (A), 5–7 (B), 5 (C), and 6–8 (D) per group; data are shown as mean ± SEM. E, Administration of 4AP induces neuronal intracellular Ca2+ increases that represent seizure-like activity. The activity recordings of these three neurons are done simultaneously from one microscope field (data are representative for 76 neurons). Administration of 4AP is indicated by a bar above the recordings.
Discussion
We (Park et al., 2011) previously showed that group II mGluRs control the developmental increase in neuronal gap junction coupling, which normally occurs during the first 2 weeks of postnatal development, and that these regulatory mechanisms also influence death/survival mechanisms in developing neurons. In the present study, under ischemic conditions in the somatosensory cortex of adult mice and in mature neuronal cortical cultures, we found that group II mGluRs also mediate increased expression of Cx36 and the concomitant neuronal gap junction coupling that occurs during the first 2 h after ischemia. Based on previous studies on postischemic glutamate release (Lobner and Choi, 1994; De Cristóbal et al., 2001; Yoshioka et al., 2009) and on our data shown in Figures 2, D and E, and 3D, we suggest that the regulation likely is via action potential-dependent synaptic release of glutamate and activation of the receptors that are postsynaptic. We also conclude that the increased neuronal gap junction coupling, mediated by group II mGluRs, is a critical determinant of the extent and spread of ischemia-mediated neuronal death. Experiments using other injury models in vitro (which represent some pathological aspects of TBI and epilepsy) produced essentially identical results. This suggests that the causal link among group II mGluR function, neuronal gap junction coupling, and neuronal death is universal, and operates in different types of neuronal injuries.
The data indicate that there are mechanistic similarities underlying the regulation of Cx36 expression during development (Park et al., 2011) and neuronal injury (present study). For example, as in development, the regulation of Cx36 by group II mGluRs in mature injured neurons is via cAMP/PKA-dependent signaling. Furthermore, in both conditions (development and injury), other glutamate receptors and GABABRs do not participate in the increase in Cx36 expression. In addition, the regulation by group II mGluRs is specific for neuronal gap junctions in both development and injury, as activation of the receptors does not induce gap junction coupling in Cx36-deficient neurons and does not affect the expression of glial Cx43.
However, there are important differences between developmental and injury-dependent regulatory mechanisms. For example, while there is a direct negative contribution of GABAARs in regulation of the developmental increase in Cx36 expression (via the GABAAR-dependent excitation and control of the Cx36 gene expression) (Park et al., 2011), GABAARs contribute only indirectly during neuronal injury (likely via GABAAR-dependent inhibition of the general level of electrical activity and the synaptic release of glutamate) (Fig. 3D,E). Furthermore, molecular mechanisms for the group II mGluR-dependent increase in Cx36 expression are different: they include transcriptional regulation via a neuron-restrictive silencer element in the Cx36 gene promoter in developing neurons (Park et al., 2011) but involve posttranscriptional regulation during neuronal injury (Fig. 8); the latter may be via mechanisms that include translational regulation or protein stability, both of which we are currently assessing. In addition, both mGluR2 and mGluR3 play a role in the developmental increase in Cx36 expression (Park et al., 2011), but the role of mGluR2 in the ischemic regulation of Cx36 in mature neurons seems more important than that of mGluR3 (Fig. 6).
Our previous work (Wang et al., 2010) showed that NMDAR-mediated neuronal death in the brain of adult mice was dramatically reduced by pharmacological inactivation of Cx36-containing gap junctions (with a relatively selective blocker mefloquine) or genetic knock-out of Cx36. In that study, we also demonstrated that the reduced level of neuronal death in knock-out animals was not due to the reduced expression of NMDARs, activity of NMDARs, or permeability of the blood–brain barrier to NMDA. In addition, NMDAR-mediated neuronal death was substantially reduced (and, in some cases, prevented) in developing neuronal cultures by Cx36 knock-out, pharmacological blockade of gap junctions, or chronic inactivation of group II mGluRs (i.e., the treatment that also prevents normal developmental increase in neuronal gap junction coupling) (de Rivero Vaccari et al., 2007; Park et al., 2011). In the present study, using adult animals and mature cultures, we show that inactivation of group II mGluRs and genetic knock-out of Cx36 both dramatically reduce neuronal death caused by neuronal injury (Figs. 9–12). Furthermore, in Cx36-deficient animals/cultures, inactivation of group II mGluRs does not result in an additional reduction in neuronal death. Together, the results strongly suggest that neuroprotection is specifically due to blockade of Cx36-containing gap junctions or inhibition of their regulatory mechanisms. Given that, in the mature CNS, Cx36-containing gap junctions are restricted largely to GABAergic interneurons and presumably are not expressed in pyramidal cells (Deans et al., 2001; Hormuzdi et al., 2001), but the coupling between pyramidal neurons increases within 1 h following neuronal injury (Frantseva et al., 2002), it is possible that both these neuronal types are subject to neuronal death because of expression and/or upregulation of neuronal gap junctions. Future studies will clarify this issue.
In the present work, we did not test the effects of group II mGluR agonists on death/survival of injured neurons, as this was done in a number of previous studies by others (Bond et al., 2000; Movsesyan and Faden, 2006; Beraudi et al., 2007; Yoshioka et al., 2009). Interestingly, according to those studies, group II mGluR agonists also have a neuroprotective effect in different models of neuronal injury, such as ischemia and TBI. However, because the agonists reduce synaptic release of glutamate (including the injury-mediated release) (Beraudi et al., 2007; Yoshioka et al., 2009), it has been proposed that the neuroprotection is via activation of presynaptic group II mGluRs that decreases glutamate release and ultimately reduces the amount of neuronal death. In contrast, according to our data, the regulation of Cx36 is via postsynaptic receptors (see above). Furthermore, activation of group II mGluRs does not cause significant increases in neuronal gap junction coupling and Cx36 expression above the levels that already are elevated as a consequence of injury (Figs. 2A–C, 4C, 10B, 11B, 12B). This suggests the possibility that administration of group II mGluR agonists following injury would not induce additional gap junction-dependent neuronal death. Thus, both inactivation and activation of group II mGluRs may be neuroprotective, but via different mechanisms [i.e., via decrease in Cx36 expression caused by inactivation of the receptors (likely postsynaptic) (present study) and via decrease in glutamate release caused by activation of the receptors (likely presynaptic) (Beraudi et al., 2007; Yoshioka et al., 2009)].
It has been suggested previously (Choi, 1988; Arundine and Tymianski, 2004; Hazell, 2007) that glutamate-dependent excitotoxicity plays critical role in the secondary (delayed) neuronal death during different types of neuronal injury. The excitotoxic mechanisms of glutamate are well characterized and include hyperactivation of glutamate receptors (primarily NMDARs), massive influx of Ca2+ ions, and overactivation of Ca2+-dependent signaling pathways that eventually causes death of neurons (Fig. 13A) (Choi, 1988; Arundine and Tymianski, 2004; Hazell, 2007). However, as discussed above, according to our studies, NMDAR-mediated neuronal death is reduced dramatically if neuronal gap junction coupling also is reduced (de Rivero Vaccari et al., 2007; Wang et al., 2010; Park et al., 2011). Based on our previous data and the results presented here, we propose a novel model for the mechanisms of glutamate-dependent excitotoxicity (Fig. 13B). We postulate that, during neuronal injury, the main determinant of massive glutamate-dependent neuronal death is not overactivation of NMDARs per se, but rather the presence of neuronal gap junctions. While overactivation of NMDARs triggers neurodegenerative processes, in the absence of neuronal gap junctions the neurodegeneration is limited to a small group of neurons [which, for various reasons, may be especially sensitive to excitotoxicity; see discussion in the study by de Rivero Vaccari et al. (2007)]. However, in the presence of neuronal gap junctions, the amount of NMDAR-mediated neuronal death is greatly multiplied and death occurs in nearly all (or all) coupled neurons. In this respect, the background level of neuronal gap junction coupling, normally found in many mature brain regions (Bennett and Zukin, 2004), is critical for NMDAR-mediated excitotoxicity to occur and expand. In addition, the activation of group II mGluRs (caused by injury-mediated glutamate release) induces synthesis of new neuronal gap junctions and these new gap junctions enhance the extent of neuronal death.
Figure 13.

Glutamate-dependent excitotoxicity during neuronal injuries. A, Traditional model of the mechanisms for glutamate-dependent excitotoxicity. B, Novel model of the mechanisms of glutamate-dependent excitotoxicity. b1, Existing neuronal gap junctions (GJ) contribute substantially to neuronal death caused by overactivation of NMDARs. b2, New neuronal gap junctions are induced by activation of group II mGluRs (IImGluRs) and also contribute to glutamate-dependent neuronal death. ⊕, This sign indicates the increase. See text for details.
We postulate that this is a universal mechanism for glutamate-dependent neuronal death regardless of the type of neuronal injury: this mechanism operates when glutamate is released by injured cells. The contribution of gap junctions to cell death likely is through propagation between the coupled neurons of gap junction-permeable neurodegenerative signals, for example NMDAR-, inflammation-, and apoptosis-dependent signals such as Ca2+, Na+, and IP3 and presumably others (Cusato et al., 2006; Decrock et al., 2009; Peixoto et al., 2009). Obviously, non-Cx36-containing and pannexin-containing gap junctions and/or hemichannels also may play a role in neuronal death during injury (Oguro et al., 2001; Frantseva et al., 2002; Thompson et al., 2008). In fact, the ischemia-mediated neuronal death that is seen in Cx36 knock-out cultures/animals in Figure 9, B and H, may potentially be due to necrotic and apoptotic mechanisms, with contribution of non-Cx36-containing gap junctions and hemichannels. However, given the amount of neuroprotection provided by elimination of Cx36 and/or inactivation of the mechanisms for increase in Cx36 expression, the role for neuronal gap junctions in injury-mediated neuronal death seems to be substantial.
In conclusion, this study provides new mechanisms for excitotoxicity beyond ionotropic glutamate receptors that include neuronal gap junctions as a critical part of these mechanisms. Because clinical trials for NMDAR antagonists as neuroprotective agents largely failed (Ikonomidou and Turski, 2002), our study suggests that another important therapeutic target for the development of new neuroprotective agents can be neuronal gap junction coupling.
Footnotes
This work was supported by NIH Grant R01 NS064256 and the University of Kansas Medical Center funds (A.B.B.). Core support was provided by NIH Grant HD002528. We are grateful to Drs. D. Paul and M. Feller for providing us with the Cx36 knock-out mice and Dr. R. Nudo for help with establishing ischemic models.
References
- Arumugam H, Liu X, Colombo PJ, Corriveau RA, Belousov AB. NMDA receptors regulate developmental gap junction uncoupling via CREB signaling. Nat Neurosci. 2005;8:1720–1726. doi: 10.1038/nn1588. [DOI] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61:657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bani-Yaghoub M, Underhill TM, Naus CC. Gap junction blockage interferes with neuronal and astroglial differentiation of mouse P19 embryonal carcinoma cells. Dev Genet. 1999;24:69–81. doi: 10.1002/(SICI)1520-6408(1999)24:1/2<69::AID-DVG8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Belluardo N, Mudò G, Trovato-Salinaro A, Le Gurun S, Charollais A, Serre-Beinier V, Amato G, Haefliger JA, Meda P, Condorelli DF. Expression of connexin36 in the adult and developing rat brain. Brain Res. 2000;865:121–138. doi: 10.1016/s0006-8993(00)02300-3. [DOI] [PubMed] [Google Scholar]
- Bennett MV, Zukin RS. Electrical coupling and neuronal synchronization in the mammalian brain. Neuron. 2004;41:495–511. doi: 10.1016/s0896-6273(04)00043-1. [DOI] [PubMed] [Google Scholar]
- Beraudi A, Bruno V, Battaglia G, Biagioni F, Rampello L, Nicoletti F, Poli A. Pharmacological activation of mGlu2/3 metabotropic glutamate receptors protects retinal neurons against anoxic damage in the goldfish Carassius auratus. Exp Eye Res. 2007;84:544–552. doi: 10.1016/j.exer.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Bond A, Jones NM, Hicks CA, Whiffin GM, Ward MA, O'Neill MF, Kingston AE, Monn JA, Ornstein PL, Schoepp DD, Lodge D, O'Neill MJ. Neuroprotective effects of LY379268, a selective mGlu2/3 receptor agonist: investigations into possible mechanism of action in vivo. J Pharmacol Exp Ther. 2000;294:800–809. [PubMed] [Google Scholar]
- Chang Q, Pereda A, Pinter MJ, Balice-Gordon RJ. Nerve injury induces gap junctional coupling among axotomized adult motor neurons. J Neurosci. 2000;20:674–684. doi: 10.1523/JNEUROSCI.20-02-00674.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Battaglia G, Marino MJ, Nicoletti F. Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat Rev Neurosci. 2005;6:787–798. doi: 10.1038/nrn1763. [DOI] [PubMed] [Google Scholar]
- Connors BW, Benardo LS, Prince DA. Coupling between neurons of the developing rat neocortex. J Neurosci. 1983;3:773–782. doi: 10.1523/JNEUROSCI.03-04-00773.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusato K, Bosco A, Rozental R, Guimarães CA, Reese BE, Linden R, Spray DC. Gap junctions mediate bystander cell death in developing retina. J Neurosci. 2003;23:6413–6422. doi: 10.1523/JNEUROSCI.23-16-06413.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusato K, Ripps H, Zakevicius J, Spray DC. Gap junctions remain open during cytochrome c-induced cell death: relationship of conductance to “bystander” cell killing. Cell Death Differ. 2006;13:1707–1714. doi: 10.1038/sj.cdd.4401876. [DOI] [PubMed] [Google Scholar]
- Danesh-Meyer HV, Huang R, Nicholson LF, Green CR. Connexin43 antisense oligodeoxynucleotide treatment down-regulates the inflammatory response in an in vitro interphase organotypic culture model of optic nerve ischaemia. J Clin Neurosci. 2008;15:1253–1263. doi: 10.1016/j.jocn.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Deans MR, Gibson JR, Sellitto C, Connors BW, Paul DL. Synchronous activity of inhibitory networks in neocortex requires electrical synapses containing connexin36. Neuron. 2001;31:477–485. doi: 10.1016/s0896-6273(01)00373-7. [DOI] [PubMed] [Google Scholar]
- De Cristóbal J, Moro MA, Dávalos A, Castillo J, Leza JC, Camarero J, Colado MI, Lorenzo P, Lizasoain I. Neuroprotective effect of aspirin by inhibition of glutamate release after permanent focal cerebral ischaemia in rats. J Neurochem. 2001;79:456–459. doi: 10.1046/j.1471-4159.2001.00600.x. [DOI] [PubMed] [Google Scholar]
- Decrock E, Vinken M, De Vuyst E, Krysko DV, D'Herde K, Vanhaecke T, Vandenabeele P, Rogiers V, Leybaert L. Connexin-related signaling in cell death: to live or let die? Cell Death Differ. 2009;16:524–536. doi: 10.1038/cdd.2008.196. [DOI] [PubMed] [Google Scholar]
- de Pina-Benabou MH, Szostak V, Kyrozis A, Rempe D, Uziel D, Urban-Maldonado M, Benabou S, Spray DC, Federoff HJ, Stanton PK, Rozental R. Blockade of gap junctions in vivo provides neuroprotection after perinatal global ischemia. Stroke. 2005;36:2232–2237. doi: 10.1161/01.STR.0000182239.75969.d8. [DOI] [PubMed] [Google Scholar]
- de Rivero Vaccari JC, Corriveau RA, Belousov AB. Gap junctions are required for NMDA receptor-dependent cell death in developing neurons. J Neurophysiol. 2007;98:2878–2886. doi: 10.1152/jn.00362.2007. [DOI] [PubMed] [Google Scholar]
- Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci. 2002;22:644–653. doi: 10.1523/JNEUROSCI.22-03-00644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajda Z, Gyengési E, Hermesz E, Ali KS, Szente M. Involvement of gap junctions in the manifestation and control of the duration of seizures in rats in vivo. Epilepsia. 2003;44:1596–1600. doi: 10.1111/j.0013-9580.2003.25803.x. [DOI] [PubMed] [Google Scholar]
- Garrett FG, Durham PL. Differential expression of connexins in trigeminal ganglion neurons and satellite glial cells in response to chronic or acute joint inflammation. Neuron Glia Biol. 2008;4:295–306. doi: 10.1017/S1740925X09990093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton GI. Synaptic modulation of neuronal coupling. Cell Biol Int. 1998;22:765–780. doi: 10.1006/cbir.1998.0386. [DOI] [PubMed] [Google Scholar]
- Hazell AS. Excitotoxic mechanisms in stroke: an update of concepts and treatment strategies. Neurochem Int. 2007;50:941–953. doi: 10.1016/j.neuint.2007.04.026. [DOI] [PubMed] [Google Scholar]
- Hormuzdi SG, Pais I, LeBeau FE, Towers SK, Rozov A, Buhl EH, Whittington MA, Monyer H. Impaired electrical signaling disrupts gamma frequency oscillations in connexin 36-deficient mice. Neuron. 2001;31:487–495. doi: 10.1016/s0896-6273(01)00387-7. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Kandler K, Katz LC. Neuronal coupling and uncoupling in the developing nervous system. Curr Opin Neurobiol. 1995;5:98–105. doi: 10.1016/0959-4388(95)80093-x. [DOI] [PubMed] [Google Scholar]
- Lobner D, Choi DW. Dipyridamole increases oxygen-glucose deprivation-induced injury in cortical cell culture. Stroke. 1994;25:2085–2089. doi: 10.1161/01.str.25.10.2085. discussion 2089–2090. [DOI] [PubMed] [Google Scholar]
- Lo Turco JJ, Kriegstein AR. Clusters of coupled neuroblasts in embryonic neocortex. Science. 1991;252:563–566. doi: 10.1126/science.1850552. [DOI] [PubMed] [Google Scholar]
- Morrison B, 3rd, Saatman KE, Meaney DF, McIntosh TK. In vitro central nervous system models of mechanically induced trauma: a review. J Neurotrauma. 1998;15:911–928. doi: 10.1089/neu.1998.15.911. [DOI] [PubMed] [Google Scholar]
- Movsesyan VA, Faden AI. Neuroprotective effects of selective group II mGluR activation in brain trauma and traumatic neuronal injury. J Neurotrauma. 2006;23:117–127. doi: 10.1089/neu.2006.23.117. [DOI] [PubMed] [Google Scholar]
- Oguro K, Jover T, Tanaka H, Lin Y, Kojima T, Oguro N, Grooms SY, Bennett MV, Zukin RS. Global ischemia-induced increases in the gap junctional proteins connexin 32 (Cx32) and Cx36 in hippocampus and enhanced vulnerability of Cx32 knock-out mice. J Neurosci. 2001;21:7534–7542. doi: 10.1523/JNEUROSCI.21-19-07534.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park WM, Wang Y, Park S, Denisova JV, Fontes JD, Belousov AB. Interplay of chemical neurotransmitters regulates developmental increase in electrical synapses. J Neurosci. 2011;31:5909–5920. doi: 10.1523/JNEUROSCI.6787-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado A, Yuste R, Katz LC. Extensive dye coupling between rat neocortical neurons during the period of circuit formation. Neuron. 1993;10:103–114. doi: 10.1016/0896-6273(93)90246-n. [DOI] [PubMed] [Google Scholar]
- Peixoto PM, Ryu SY, Pruzansky DP, Kuriakose M, Gilmore A, Kinnally KW. Mitochondrial apoptosis is amplified through gap junctions. Biochem Biophys Res Commun. 2009;390:38–43. doi: 10.1016/j.bbrc.2009.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Velazquez JL, Valiante TA, Carlen PL. Modulation of gap junctional mechanisms during calcium-free induced field burst activity: a possible role for electrotonic coupling in epileptogenesis. J Neurosci. 1994;14:4308–4317. doi: 10.1523/JNEUROSCI.14-07-04308.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Velazquez JL, Frantseva MV, Naus CC. Gap junctions and neuronal injury: protectants or executioners? Neuroscientist. 2003;9:5–9. doi: 10.1177/1073858402239586. [DOI] [PubMed] [Google Scholar]
- Personius K, Chang Q, Bittman K, Panzer J, Balice-Gordon R. Gap junctional communication among motor and other neurons shapes patterns of neural activity and synaptic connectivity during development. Cell Commun Adhes. 2001;8:329–333. doi: 10.3109/15419060109080748. [DOI] [PubMed] [Google Scholar]
- Rash JE, Staines WA, Yasumura T, Patel D, Furman CS, Stelmack GL, Nagy JI. Immunogold evidence that neuronal gap junctions in adult rat brain and spinal cord contain connexin-36 but not connexin-32 or connexin-43. Proc Natl Acad Sci U S A. 2000;97:7573–7578. doi: 10.1073/pnas.97.13.7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samoilova M, Li J, Pelletier MR, Wentlandt K, Adamchik Y, Naus CC, Carlen PL. Epileptiform activity in hippocampal slice cultures exposed chronically to bicuculline: increased gap junctional function and expression. J Neurochem. 2003;86:687–699. doi: 10.1046/j.1471-4159.2003.01893.x. [DOI] [PubMed] [Google Scholar]
- Söhl G, Maxeiner S, Willecke K. Expression and functions of neuronal gap junctions. Nat Rev Neurosci. 2005;6:191–200. doi: 10.1038/nrn1627. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Jackson MF, Olah ME, Rungta RL, Hines DJ, Beazely MA, MacDonald JF, MacVicar BA. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science. 2008;322:1555–1559. doi: 10.1126/science.1165209. [DOI] [PubMed] [Google Scholar]
- Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129:1021–1029. doi: 10.1016/j.neuroscience.2004.06.046. [DOI] [PubMed] [Google Scholar]
- Wang Y, Denisova JV, Kang KS, Fontes JD, Zhu BT, Belousov AB. Neuronal gap junctions are required for NMDA receptor-mediated excitotoxicity: implications in ischemic stroke. J Neurophysiol. 2010;104:3551–3556. doi: 10.1152/jn.00656.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Yamada KA. Developmental characteristics of epileptiform activity in immature rat neocortex: a comparison of four in vitro seizure models. Brain Res Dev Brain Res. 2001;128:113–120. doi: 10.1016/s0165-3806(01)00149-3. [DOI] [PubMed] [Google Scholar]
- Yoshioka H, Sugita M, Kinouchi H. Neuroprotective effects of group II metabotropic glutamate receptor agonist DCG-IV on hippocampal neurons in transient forebrain ischemia. Neurosci Lett. 2009;461:266–270. doi: 10.1016/j.neulet.2009.06.056. [DOI] [PubMed] [Google Scholar]