

Abstract
Aims
Detection of atherosclerosis has generally been limited to the late stages of development, after cardiovascular symptoms present or a clinical event occurs. One possibility for early detection is the use of functionalized nanoparticles. The aim of this study was the early imaging of atherosclerosis using nanoparticles with a natural affinity for inflammatory cells in the lesion.
Materials & methods
We investigated uptake of cowpea mosaic virus by macrophages and foam cells in vitro and correlated this with vimentin expression. We also examined the ability of cowpea mosaic virus to interact with atherosclerotic lesions in a murine model of atherosclerosis.
Results & conclusions
We found that uptake of cowpea mosaic virus is increased in areas of atherosclerotic lesion. This correlated with increased surface vimentin in the lesion compared with nonlesion vasculature. In conclusion, cowpea mosaic virus and its vimentin-binding region holds potential for use as a targeting ligand for early atherosclerotic lesions, and as a probe for detecting upregulation of surface vimentin during inflammation.
Keywords: aorta, atherosclerosis, cowpea mosaic virus, foam cell, imaging, lesion, macrophage, surface vimentin, targeting, viral nanoparticle
Cardiovascular disease, including atherosclerosis, is the leading cause of death in developed countries [1]. In the initial stages of atherosclerosis, high levels of low-density lipoprotein (LDL) in the blood enter the extracellular matrix of the vascular wall in areas of disrupted blood flow [2]. This build-up of lipid can lead to inflammation in the area and the eventual accumulation of apoptotic cells, which results in the formation of a necrotic core [3,4]. As the lesion develops, a thick cap rich in collagen forms over the lesion [5-7]. A destabilized lesion can detach and enter the bloodstream, causing myocardial infarction or ischemic stroke [8]. The risk of such clinical events has been shown to depend more on the composition of the lesion as opposed to its size, and therefore destabilization of the lesion can occur early during lesion development [9-11]. Disruption of atherosclerotic lesion is associated with 75% of acute coronary events, such as myocardial infarction or stroke [12].
Evaluation of atherosclerosis with techniques such as elastography [13], thermography [14], ultrasound [15] and angioscopy [16] is typically initiated at later stages of atherosclerosis, when patients are symptomatic. There has been relatively little success in the detection of early atherosclerosis by conventional imaging modal ities. Even advanced high-resolution techniques such as ultrasound, computed tomography and MRI are limited by the sensitivity of detection, and generally only provide information about vascular anatomy. In complex cardiovascular disease, imaging methods that illuminate the molecular aspects of disease development and progression, such as inflammation in the vascular wall, inflammatory cell infiltration [17,18] and the production of extracellular molecular markers [19-22], are important for staging disease and monitoring therapy and lifestyle changes [23,24]. Thus, the development of tools that can detect and image these earlier stage events is essential for effective diagnosis and therapy.
Nanoparticles have the properties of appropriate size, multivalency and functionality needed for rapid and sensitive theranostics of diseased tissues. Various natural and synthetic nanoparticle platforms are being developed for such applications that include functionalities such as imaging agents, tissue-specific targeting ligands and drug delivery [25,26]. Nanoscale contrast agents being developed for the early detection and monitoring of atherosclerosis include fluorescent, radioactive, paramagnetic, electron-dense and lightscattering agents [27]. Virus-based nanoparticles are also being studied as contrast agents [28,29]. Cowpea mosaic virus (CPMV) is a plant virus in the Comoviridae family that has recently been investigated as a nanoparticle platform for attachment of imaging and targeting agents. The CPMV capsid is formed by repeated copies of two proteins that combine to form a 31-nm capsid with pseudo T = 3 symmetry.
Previous work has demonstrated that fluorescent-labeled CPMV can be used in intravital imaging of healthy and tumor vasculature [30,31]. Interestingly, endothelial cells readily internalize CPMV in vivo [31]. The uptake of CPMV into vascular endothelial cells is mediated by specific interaction with a surface-displayed form of the cytoskeletal protein vimentin [32,33]. Surface vimentin expression has been detected in endothelial cells in vivo, as well as in activated macrophages [34]. Uptake of CPMV has also been demonstrated in cells of the immune system including dendritic cells and macrophages in vivo, and the ability of cells to internalize CPMV is correlated with the presence of surface vimentin on these cell types [35]. In atherosclerosis, macrophages are the major immune cell within the lesion area and are required for disease progression [23,36]. In early atherosclerosis, monocytes are recruited from the blood vessel into the vascular wall where they differentiate into macrophages [37,38]. Macrophages internalize the lipid that accumulates in the wall of the vessel, and further differentiate into foam cells filled with lipid [39,40].
Since CPMV naturally interacts with macrophages via surface vimentin, we investigated whether CPMV could be used as a probe for studying atherosclerotic lesions as well as upregulation of surface vimentin within lesion areas. To do so, we studied the uptake of CPMV by macrophages and foam cells in vitro and correlated this with vimentin expression. In addition, we examined the ability of CPMV to interact with atherosclerotic lesions in the murine LDL-receptor knockout (LDLR−/−) model of atherosclerosis. The LDLR is expressed on many cell types and facilitates the endocytosis of LDL, particularly by liver cells, which removes the LDL from circulation [41]. In LDLR−/− mice fed a high-fat diet, accumulation of lipid and development of atherosclerotic lesions is observed [42,43]. The expression of surface vimentin in lesions, and the association of intravenously administered CPMV with atherosclerosis lesions at various stages of development was assessed by fluorescence confocal microscopy.
Materials & methods
Animals
Male LDLR−/− mice (B6.129S7-Ldlr tm1Her/J) and control mice (C57Bl6/J) were purchased from Jackson laboratory (ME, USA). Mice were fed ad libitum either normal chow or a high-fat diet (western diet from Newco, Purina [CA, USA] test diet #21551) starting at 8 weeks of age, and continuing for 12–20 weeks. All animal studies were reviewed and approved by the University of California, San Diego Institutional Animal Care and Use Committee (CA, USA).
Cell culture
RAW 264.7 macrophages were grown in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 2 mM l-glutamine, 100 μ/ml penicillin and 100 μg/ml strepto mycin. Thioglycolate-stimulated peritoneal macrophages (TSPM) were obtained using methods previously described [44].
Propagation & purification of CPMV
CPMV-infected leaves and potassium phosphate buffer (pH 7.0) were mixed and ground with a mortar and pestle. The resulting slurry was used as an inoculum for passage to new cowpea plants to produce stocks of virus. CPMV was purified from infected secondary leaves by standard methods described previously, and resuspended in sterile phosphate-buffered saline (PBS) [45]. Virus concentration was measured using absorbance spectroscopy. Virus concentration was calculated using the absorbance at a wavelength of 260 nm and the extinction coefficient of CPMV at that wavelength (8.1 ml/mg/cm). To confirm that CPMV particles were pure and intact, particles were analyzed on an ÄKTA™ Superose-6 size-exclusion column (Amersham Pharmacia, Amersham, UK). Particles were eluted on the column at 0.4 ml/min in 0.1 M potassium phosphate buffer at pH 7.0.
Conjugation of Alexa Fluor® 555 & 647 dyes to CPMV
Alexa Fluor® carboxylic acid, succinimidyl ester dyes (Molecular Probes, OR, USA) were dissolved in dimethyl sulfoxide and combined with buffer-containing virus (concentration of virus 2–3 mg/ml, final dye-to-particle ratio of 2000:1) so that the final mixture was 90% buffer and 10% dimethyl sulfoxide. The solution was protected from light and rotated at room temperature for 24 h. The solution was then filtered through Amicon® Ultra-0.5 ml (Millipore, MA, USA; #UFC510024) with buffer until flow through was clear of fluorescence. Relative concentrations of virus and dye were determined by absorbance spectroscopy. We determined virus concentration by measuring absorbance at 260 nm. Fluorochrome concentration was established by measuring absorbance at emission maximum and using the extinction coefficient for each dye (Alexa Fluor 555 ε = 150,000, Alexa Fluor 647 ε = 239,000). Average labeling of 60–90 dyes per particle was obtained.
CPMV uptake into macrophages
Macrophages were removed from the flask using cell dissociation buffer and suspended in media in 96-well plates at 5 × 105 cells/well. Cells were incubated for 3 h with 100,000 wild-type CPMV particles per cell. For confocal microscopy, cells were plated for the last hour on confocal dishes. Cells were washed and fixed in 4% electrom microscopy-grade formaldehyde. CPMV was detected by incubation for 1 h with a polyclonal CPMV antibody, washed in PBS, followed by incubation for 1 h with goat antirabbit Alexa Fluor 488 fluorescent secondary antibody.
Differentiation of macrophages into foam cells
TSPM, or the macrophage cell line RAW 264.7, were incubated for 24–72 h in media with 20 μg/ml modified human oxidized LDL (oxLDL; Intracel Resources, MD, USA; RP-049). Cells were then incubated with 1 × 106 unlabeled CPMV particles/cell for 3 h. Cells were fixed and CPMV was stained with a polyclonal antibody followed by goat antirabbit Alexa Fluor 488 fluorescent secondary antibody (Invitrogen, CA, USA).
Oil Red O staining of lipid
OxLDL uptake was confirmed by staining cells for lipid using Oil Red O (ORO). Fixed cells were incubated in 60% isopropanol for 5 min. Cells were then incubated for 5 min in Whatman-filtered 0.3% ORO in 60% iso propanol. Cells were then washed again in 60% isopropanol for 5 min and rehydrated in PBS for 10 min.
In vivo interaction of CPMV with atherosclerotic lesion
LDLR−/− mice were fed a high-fat diet for 12–20 weeks to initiate development of atherosclerosis. As controls, C57Bl6/J mice were fed a high-fat diet and LDLR−/− mice or C57Bl6/J mice were fed a normal chow diet. None of these controls developed atherosclerosis. CPMV-AF555 conjugate or 0.5 mg of CPMV-AF647 was injected by intraorbital injection and allowed to circulate for 1 h. Mice were sacrificed and perfused with cold sterile PBS. The liver and spleen were harvested to confirm injection of particles. The aorta was removed and divided into approximately 10-mm sections that were incubated in antivimentin antibody (Sigma, MO, USA; goat V4630) to stain vimentin on the lumenal surface of endothelial cells. Sections were then washed twice in PBS to remove excess antibody, imbedded in Tissue-Tek® OCT Compound (Sakura Finetek, CA, USA) and frozen in dry ice before storage at −80°C. Tissue was cut into 10-μm sections perpendicular to aorta on a Leica (IL, USA) cryomicrotome and mounted on slides. Primary antibodies to macrophages (MOMA2) and endothelium (CD31) and corresponding secondary fluorophore-conjugated antibodies were used for staining aortic sections.
Confocal microscopy
Images were acquired on an Olympus (PA, USA) FV1000 confocal microscope at 40×. Images were processed using ImageJ software. Fluorescence analysis of images was performed using the Threshold Color Plugin for ImageJ software [101].
Sirius red collagen staining of aorta
The 10-μm sections of aorta were fixed on slides in 4% electrom microscopy-grade formaldehyde for 1 h (Polysciences, Inc., PA, USA). Sections were stained with picro-sirius red (0.1% direct red 80 in saturated aqueous solution of picric acid, both from Sigma) for 1 h. Sections were washed twice in acidified water and dehydrated in three changes of 10% ethanol. Sections were mounted in Vectamount™ (Vector Laboratories, CA, USA).
Whole-mount aorta isolation & staining
LDLR−/− mice on a high-fat diet for approximately 9 months were injected with 100 μg of Alexa Fluor 555-labeled CPMV, and after 90 min were perfused with PBS, and the aorta was removed as described above. A section of aorta without atherosclerotic lesion was cut along the side and opened to reveal the lumen. Endothelial cells were stained using PECAM (anti-CD31) antibody and corresponding Alexa488 secondary antibody. Entire sections of aorta were mounted for analysis on a Bio-Rad (Zeiss; MA, USA) Radiance 2100 laser scanning confocal microscope. From Z-sections, 3D images were compiled using [102].
Antibody staining of surface vimentin on RAW macrophages
RAW macrophages were incubated in a flask of media with 20 μg/ml oxLDL for 0 h (control), 24 h and 72 h. Cells were removed from the flask using cell dissociation buffer, plated in a 96-well plate at 5 × 105 cells/well, and fixed using 4% EM-grade formaldehyde. Cells were stained with antivimentin antibody V4630 followed by staining with Alexa Fluor 647 conjugated to donkey antigoat antibody. Cells were analyzed on a digital LSR II flow cytometer (BD Biosciences, CA, USA) using FlowJo software (Treestar, CA, USA).
Results
CPMV production & labeling
CPMV was produced, purified and used either unlabeled or labeled with fluorescent dyes for ease of detection in vivo by fluorescence microscopy and FACS. Figure 1A shows a space-filling model of the CPMV capsid structure (PDB 1NY7) with the 300 naturally occurring addressable lysines that are on the surface of the capsid highlighted in red. For the experiments, the lysine residues on the capsid were conjugated with either NHS-AF488 or NHS-AF647, obtaining an average of 60–90 dyes per particle. UV spectroscopy and size-exclusion fast protein liquid chromatography (Figure 1B) were used to confirm the particles were intact, and the dye directly conjugated.
Figure 1. Characterization of labeled cowpea mosaic virus.
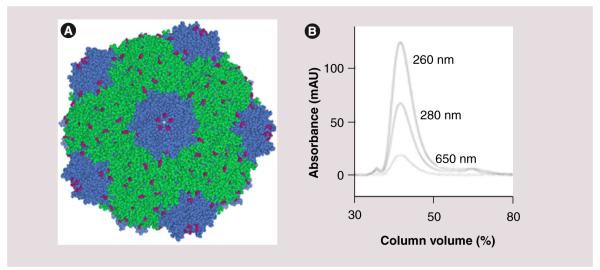
(A) Space-filling model of the cowpea mosaic virus capsid structure assembled in Chimera, using coordinates obtained from [103]. The large capsid subunit is in green, small capsid subunit is in blue and surface-accessible lysines are highlighted in pink. (B) Fast protein liquid chromatography chromatogram of Alexa Fluor® 647-labeled cowpea mosaic virus particles. The three traces reflect the absorbance in mAU at three wavelengths (from top to bottom: 260, 280 and 650 nm, respectively).
Association of CPMV with vessels is limited to the endothelium in nonlesion aorta
In this study, the LDLR−/− mouse model was used to study CPMV-vasculature interactions in the context of atherosclerosis. Atherosclerosis development was monitored by staining the endothelial cell layer and collagen in the lesion. LDLR−/− lesion tissue was stained with CD31 to check for intactness of the endothelial layers. As seen in Figure 2e, even after 20 weeks on a high-fat diet, the endothelial layer (stained in red) was present over lesion areas (white in inset). Staining for collagen in nonlesion (Figure 2F) and lesion tissue revealed the presence of collagen beginning at 12 weeks (Figure 2g) and becoming thicker by 20 weeks (Figure 2H).
Figure 2. Development and progression of atherosclerotic lesions.

(A) Design schematic for development and study of atherosclerotic lesion in vivo. (B-D) Shows three orientations of 3D confocal stacks. CPMV-A555 particles (red) interact with endothelial cells (CD31, green), but do not progress into deeper smooth muscles layers, best seen in (D), where the lumen side is marked with a white asterisk (4′,6-diamidino-2-phenylindole, blue). (E) Confocal image (40×) of endothelial staining (CD31, red) of aorta atherosclerotic lesion (white in small panel) from a low-density lipoprotein-receptor knockout mouse on western diet for 20 weeks. (F-H) Brightfield image (40×) of picrosirius red staining of collagen in control aorta from C57Bl/6J mice (F) and in aortic atherosclerotic lesions from low-density lipoprotein-receptor knockout mice on diet for (G) 12 and (H) 20 weeks.
CPMV: Cowpea mosaic virus.
To study the interaction of CPMV with vascular endothelium, LDLR−/− mice on a high-fat diet for 9 months were injected intravenously with 100 μg of CPMV-A555. Particles were allowed to circulate for 90 min and sections of aorta without lesion were removed and cut lengthwise to reveal the lumen. Sections were stained for endothelial cells using anti-CD31, and mounted for Z-series imaging on a confocal microscope (Figure 2B-D). CPMV fluorescence (red) was localized to the endothelial layer of cells, which are positive for CD31 (green). Z-series images revealed nuclei (blue) in the smooth muscle layer of the aorta. CPMV particles remained co localized with CD31 in the endothelium and did not penetrate through the endothelial layer to deeper layers in nonlesion tissue (Figure 2D).
CPMV colocalizes with endothelial cells & macrophages in atherosclerotic lesion in vivo
The schematic in Figure 2A shows the experimental setup for testing the interaction of CPMV with the vasculature in atherosclerotic mice. LDLR−/− or control C57Bl/6J mice were fed either normal chow or a high-fat (western) diet for 12-20 weeks. Mice received intraorbital injection of 0.5-mg CPMV-AF488 or CPMV-AF647 and tissues were harvested after 1 h of particle circulation.
Development of atherosclerotic lesions in mice was determined by ORO staining (Figure 3C; red). As expected, accumulation of lipid was minimal after 12 weeks on the diet, but was much more extensive by 20 weeks. The accumulation of macrophages in lesion areas was determined by staining the tissues with the macrophage marker MOMA-2. Macrophages were apparent in the lesion area (Figure 3B & D; inset, white) by 12 weeks, and were extensive by 20 weeks (Figure 3D). In addition, at 20 weeks, the colocalization of lipid staining with macrophage staining was more extensive (Supplementary Figure 1; see online at www.futuremedicine.com/doi/suppl/10.2217/NMM.11.185), suggesting that more of the macrophages present were internalizing lipids and differentiated into foam cells.
Figure 3. Vimentin, macrophages and cowpea mosaic virus uptake in atherosclerotic lesion.

(A) Vimentin (red) on the surface of endothelial cells in nonlesion tissue from low-density lipoprotein receptor knockout (LDLR−/−) mice (40×). (B) Vimentin (red) on the surface of endothelial cells in atherosclerotic lesion tissue from LDLR−/− mice (40×, white in small panel). (C) Lipid staining (Oil Red O, red) and CPMV (green) in atherosclerotic lesions from LDLR−/− mouse on western diet for 20 weeks (40×). (D) Macrophage staining (MOMA-2, red) and CPMV (green) in atherosclerotic lesions from LDLR−/− mouse on western diet for 20 weeks (40×).
CPMV: Cowpea mosaic virus; DAPI: 4′,6-diamidino-2-phenylindole.
Surface vimentin was identified by staining the lumen surface of a section of aorta with antivimentin antibody, followed by embedding and sectioning. Following sectioning, the bound antibody was detected by incubation with secondary antigoat fluorescent antibody. In normal tissue, vimentin was present on the surface of the endothelial layer in all controls (Supplementary Figure 2; red). Figure 3A shows a representative image of nonlesion tissue from LDLR−/− on a high-fat diet. Interestingly, an increase in surface vimentin was observed in lesion areas (Figure 3B; lesion area white in inset) by 12 weeks. The amount of vimentin in the lesion areas was quantified using ImageJ analysis software. Surface vimentin mean fluorescence intensity (MFI) was increased in LDLR−/− lesion tissue (MFI of 1104) compared with nonlesion tissue from LDLR−/− after 12 weeks on a high-fat diet (MFI of 557) (Figure 4A). These results suggested that upregulation of surface vimentin occurs within the inflammatory environment of the lesion, and that the natural CPMV-vimentin interaction may be exploited to detect atherosclerotic lesions in vivo.
Figure 4. Quantification of vimentin and cowpea mosaic virus in lesion versus nonlesion tissue.

Analysis of vimentin and CPMV in tissue sections using ImageJ. (A) Mean fluorescence of vimentin staining in the endothelium for C57Bl/6J control mice on normal chow, and lesion and nonlesion areas of LDLR−/− mice on a high-fat diet for 12 weeks. Mean fluorescence of vimentin was statistically significant between control C57Bl6/J tissue and LDLR−/− lesion (p < 0.01) and not statistically significant between LDLR−/− nonlesion and lesion tissue. (B) Mean fluorescence of CPMV in endothelium for control C57BL6/J mice on normal chow and lesion and nonlesion areas of LDLR−/− mice on high-fat diet for 20 weeks (p < 0.0001, between LDLR−/− lesion and both LDLR−/− nonlesion and control). (C) Percent of endothelial area positive for CPMV particle fluorescence in control and atherosclerotic mouse aorta after 12 and 20 weeks on a high-fat diet using the color threshold plugin for ImageJ. LDLR−/− lesion tissue compared with both nonlesion LDLR−/− tissue and control tissue was statistically significant (p < 0.001). (D) Percent total lesion area positive for CPMV particle fluorescence using color threshold plugin for ImageJ.
CPMV: Cowpea mosaic virus; LDLR−/−: Low-density lipoprotein receptor knockout; MFI: Median fluorescence intensity.
In order to characterize the ability of CPMV to interact with atherosclerotic lesions in vivo, CPMV was administered to LDLR−/− mice fed a high-fat diet. Following 1 h of CPMV circulation in LDLR−/− animals on a high-fat diet for 20 weeks, mean CPMV fluorescence was more than doubled in LDLR−/− lesion tissue (MFI 435) compared with nonlesion tissue, although CPMV was present in endothelial cells of all controls (Supplementary Figure 2; green). Fluorescence in nonlesion LDLR−/− tissue was not significantly different from control tissue (C57Bl6/J on normal chow, MFI 190) (Figure 4B). This correlated with the observed upregulation of surface vimentin (Figure 3B). To determine the percent of endothelial area that was positive for CPMV fluorescence, the Color Threshold ImageJ plug in was utilized. The percent of endothelial area that was determined to be positive for CPMV in LDLR−/− lesions compared with control vascular tissue (both nonlesion LDL−/− on a high-fat diet and C57Bl6/J on normal chow) was found to be 27-times higher at 12 weeks and 32-times higher at 20 weeks (Figure 4C) (p < 0.0001 vs controls at both 12 and 20 weeks). Together these results indicate that upregulation of surface vimentin occurs in the vicinity of atherosclerotic lesion in vivo. This upregulation in turn correlates with specific internalization of CPMV particles in lesion areas.
It is interesting that the amount of CPMV in the endothelial area does not increase appreciably between 12 and 20 weeks compared with controls (Figure 4C). In addition, when the entire lesion area was analyzed instead of just the endothelium, there was a trend toward reduced CPMV in the lesion at 20 weeks compared with 12 weeks (Figure 4D, not significant). As the lesion develops and has more endothelial surface area and volume, relatively less CPMV associates with the lesion area.
CPMV uptake in immortalized & primary macrophages & differentiated foam cells
To further investigate the ability of macrophages and differentiated foam cells to capture and internalize CPMV particles in vitro, we used the immortalized murine cell line RAW 264.7, and primary TSPM. Cells were differentiated into foam cells by treating with a modified lipid, oxLDL (Figure 5; right column of images) and compared with untreated macrophages (left column). RAW macrophages (Figure 5A-D) and TSPM (Figure 5e-H) were stained with ORO (red) to detect the uptake of lipid (Figure 5A, B, e & F), indicating a foam-cell phenotype. To determine whether foam cells could take up CPMV, both untreated and treated RAW and TSPM cells were suspended in cell dissociation buffer and incubated for 3 h with 1 × 105 wild-type CPMV particles per cell. Untreated RAW (Figure 5C) and TSPM (Figure 5g) could internalize CPMV (green), consistent with our previous studies in mice (Figure 5A) [35]. Following oxLDL treatment, confocal microscopy demonstrated that treated RAW (Figure 5D) and TSPM (Figure 5H) cells showed reduced uptake of CPMV compared with untreated cells. Flow cytometry analysis was also used to quantify the ability of RAW macrophages to internalize CPMV. RAW macrophages exposed to oxLDL followed by CPMV showed a reduction in CPMV uptake from an average of 78 to 25% (Figure 5i). Together these results suggest that macrophage differentiation state affects the ability to internalize CPMV.
Figure 5. Reduced cowpea mosaic virus interaction with differentiated macrophages in vitro.

(A) Confocal image of lipid staining of untreated RAW macrophages with ORO (red). (B) Confocal image of lipid staining of oxLDL-treated RAW macrophages with ORO (red). (C) Confocal image of CPMV uptake by untreated RAW macrophages (green). (D) Confocal image of CPMV uptake by oxLDL-treated RAW macrophages (green). (E) Confocal image of lipid staining of untreated TSPM with ORO (red). (F) Confocal image of lipid staining of oxLDL treated TSPM with ORO (red). (G) Confocal image of CPMV uptake by untreated TSPM (green). (H) Confocal image of CPMV uptake by oxLDL-treated TSPM (green). All confocal images were obtained at 60× and the nucleus has been stained with 4′,6-diamidino-2-phenylindole (blue). (I) FACS analysis of CPMV uptake in RAW macrophages with and without oxLDL treatment. (J) FACS analysis of number of RAW macrophages stained positive for surface vimentin with oxLDL treatment for 24 h (p = 0.22, not significant) and 72 h (p = 0.09, not significant).
CPMV: Cowpea mosaic virus; ORO: Oil Red O; oxLDL: Oxidized low-density lipoprotein;
TSPM: Thioglycolate-stimulated mouse peritoneal macrophages.
Amount of surface vimentin changes following treatment with oxLDL
Flow cytometry analysis was used to quantify the amount of vimentin on the surface of oxLDL-treated RAW 264.7 macrophages. Analysis showed a trend of decreased surface vimentin with the accumulation of oxLDL over 24 and 72 h, although the results were not statistically significant (Figure 5J). A reduction in surface vimentin as atherosclerotic lesion develops would also explain the relative plateau in the amount of CPMV associating with the growing lesion during later stages in lesion development.
Combined with the in vivo findings, these results support the potential use of CPMV for targeting early atherosclerosis. These results suggest that during the early stages of lesion development, surface vimentin expression and CPMV uptake is higher, and as macrophages undergo the differentiation process into foam cells, their ability to display surface vimentin and take up CPMV decreases.
Discussion
This study reveals that surface vimentin is upregulated in atherosclerotic lesions, and this provides a potential target for the delivery of imaging or therapeutic agents to the lesion. The natural interaction of CPMV with vimentin can be utilized by using CPMV as an atherosclerosis-targeting agent or the vimentin-binding region as a targeting ligand attached to other platforms. Furthermore, CPMV may be particularly sensitive at detecting early atherosclerotic lesions, a time point that is difficult to detect with conventional imaging technologies.
Previous studies have shown that CPMV interacts with various cell types, including immune cells such as macrophages [32,35]. In this study, we report on the interaction of CPMV with macrophages during atherosclerotic lesion formation, a pathology where macrophages play a direct role in disease development.
In nonlesion tissues, CPMV enters cells of the vascular endothelium, but does not penetrate into deeper layers of the vascular wall (Figure 2D). Due to the many pathological changes that take place during early stages of atherosclerosis, such as endothelial dysfunction [46] and the narrowing of the artery lumen due to accumulation of lipid and inflammatory cells [47], the lesion environment is very different from non-lesion vascular tissue. These alterations could make areas with lesions more susceptible to deeper penetration by the circulating particles (Figure 3D), similar to what has been observed as the enhanced permeability and retention effect in tumor vasculature [48]. The development of atherosclerotic lesions allows for the penetration of CPMV past the endothelial layer where it associates with macrophages and foam cells. In an alternative hypothesis, the CPMV could interact with macrophages in the blood or in organs of the immune system before they enter the lesion area. However, because a substantial number of particles are already associated with the lesion after 1 h of circulation, this is unlikely to be the major method of association.
These results confirm that, similar to other developing therapies that target macrophages for imaging and therapy [49-51], CPMV can associate with macrophages during early lesion development, when macrophages begin to accumulate within the lesion. Gadolinium-containing lipid-based nanoparticles that are targeted to macrophage scavenger receptor-B have shown potential for identifying lesions before an atherothrombotic event [52]. In addition, similarly targeted and functionalized polysaccharide-containing nanoparticles have found success in PET imaging of macrophages in atherosclerotic lesions [49]. This suggests that nanoparticle association with early pathological alterations in lesions (i.e., macrophage accumulation) may facilitate their detection.
We discovered that lesion tissue is inundated with CPMV, compared with control vascular areas (Figure 4C). This increased association with lesions correlates well with the elevated levels of surface vimentin in the lesion area. Many examples of surface-expressed and even secreted forms of vimentin exist, but little is known about the role of vimentin outside the cell or its regulation in disease states [34,53-61]. Based on the expression pattern of vimentin, it has been suggested that the protein may moderate inflammatory responses through its interaction with various plasma proteins [58,59,62-64]. It is possible that vimentin is playing a role in inflammation in the lesion, and is thus upregulated. Because CPMV binds to vimentin on the cell surface [32,33], it could be used as a tool to more specifically assess the expression of surface vimentin in vivo.
The main finding of the present study is that CPMV interacts with macrophages in the lesion. In our in vivo experiments, this interaction occurs early during lesion development, even before appreciable amounts of lipid are present in the lesion. Later during lesion development, lesion areas continue to show CPMV uptake, but not to the degree expected based on the increase in lesion surface area. The lack of scaling of CPMV uptake to lesion area is supported by in vitro studies involving RAW and peritoneal macrophages. These studies suggest that once lipid is accumulated in macrophages, the uptake of CPMV particles is reduced, which correlates with a reduction in surface vimentin. Late during lesion development, a population of macrophages, particularly macrophages that have recently been recruited into the lesion, may retain the ability to interact with CPMV. However, a portion of the macrophages may have taken up large amounts of lipid, which could account for the plateau in CPMV uptake compared with lesion surface area and volume. Furthermore, alterations in regional blood flow in areas where the lesion obscures portions of the lumen, resulting in narrowed vessels [2] must be considered as a potential cause for increased CPMV in these areas.
Other protein-based nanoparticles are being developed for targeting of atherosclerosis [65-68]. A protein cage nanoparticle bearing Lyp-1 peptide for macrophage targeting was shown to traffic to macrophage-rich carotid lesions in mice [69]. Although targeting of macrophages holds promise for early characterization of atherosclerotic lesions, carotid ligation used in this study constricts blood flow significantly in this area, which could enhance particle uptake. The use of whole CPMV nanoparticles for imaging atherosclerotic lesion poses a few problems. Optical imaging at the depth of the aorta in humans is not possible. Other methods of imaging, including MRI using gadolinium-loaded CPMV particles as contrast agents, are being considered. However, there are many difficulties, including small size and movement of the vessels, inherent to imaging vasculature with this method.
The finding that CPMV efficiently targets atherosclerotic lesions in relatively early stages prior to gross morphological changes suggests that using CPMV or similar multivalent strategies for targeting surface vimentin may be an exciting strategy for imaging and monitoring the development and therapy of atherosclerosis.
Conclusion
We determined that CPMV holds significant potential for early detection of atherosclerosis, which is important for preventing the development of symptomatic disease. In addition, the particles can be utilized as a probe for increased surface vimentin in the lesion.
Supplementary Material
Executive summary.
-
■
Cowpea mosaic virus (CPMV) holds significant potential for early detection of atherosclerosis.
-
■
CPMV can be utilized as a probe for increased surface vimentin in the lesion.
-
■
Treatment of RAW macrophages with lipopolysaccharide or oxidized low-density lipoprotein alters the amount of surface vimentin.
-
■
Association of CPMV with vessels is limited to the endothelium in nonlesion aorta.
-
■
CPMV colocalizes with endothelial cells and macrophages in atherosclerotic lesion in vivo.
-
■
CPMV has heightened specificity for lesion area, particularly early in lesion development.
Acknowledgements
The authors would like to thank the Bruce Beutler laboratory for providing murine peritoneal macrophages.
Financial & competing interests disclosure
The work was supported by NIH R01CA112075 to M Manchester and the American Heart Association Western States Affiliate Predoctoral Fellowship. The authors acknowledge use of the UCSD Neuroscience Microscopy core facility supported by NIH P30NS047101. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Footnotes
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
References
- 1.Slevin M, Badimon L, Grau-Olivares M, et al. Combining nanotechnology with current biomedical knowledge for the vascular imaging and treatment of atherosclerosis. Mol. Biosyst. 2010;6(3):444–450. doi: 10.1039/b916175a. [DOI] [PubMed] [Google Scholar]
- 2.Cybulsky MI, Jongstra-Bilen J. Resident intimal dendritic cells and the initiation of atherosclerosis. Curr. Opin. Lipidol. 2010;21(5):397–403. doi: 10.1097/MOL.0b013e32833ded96. [DOI] [PubMed] [Google Scholar]
- 3.Tavora F, Cresswell N, Li L, Ripple M, Burke A. Immunolocalisation of fibrin in coronary atherosclerosis: implications for necrotic core development. Pathology. 2010;42(1):15–22. doi: 10.3109/00313020903434348. [DOI] [PubMed] [Google Scholar]
- 4.Ota H, Yu W, Underhill HR, et al. Hemorrhage and large lipid-rich necrotic cores are independently associated with thin or ruptured fibrous caps: an in vivo 3T MRI study. Arterioscler. Thromb. Vasc. Biol. 2009;29(10):1696–1701. doi: 10.1161/ATVBAHA.109.192179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ovchinnikova O, Gylfe A, Bailey L, et al. Osteoprotegerin promotes fibrous cap formation in atherosclerotic lesions of ApoE-deficient mice--brief report. Arterioscler. Thromb. Vasc. Biol. 2009;29(10):1478–1480. doi: 10.1161/ATVBAHA.109.188185. [DOI] [PubMed] [Google Scholar]
- 6.Van Den Diepstraten C, Papay K, Bolender Z, Brown A, Pickering JG. Cloning of a novel prolyl 4-hydroxylase subunit expressed in the fibrous cap of human atherosclerotic plaque. Circulation. 2003;108(5):508–511. doi: 10.1161/01.CIR.0000080883.53863.5C. [DOI] [PubMed] [Google Scholar]
- 7.Heeneman S, Cleutjens JP, Faber BC, et al. The dynamic extracellular matrix: intervention strategies during heart failure and atherosclerosis. J. Pathol. 2003;200(4):516–525. doi: 10.1002/path.1395. [DOI] [PubMed] [Google Scholar]
- 8.McNeill E, Channon KM, Greaves DR. Inflammatory cell recruitment in cardiovascular disease: murine models and potential clinical applications. Clin. Sci. (Lond.) 2010;118(11):641–655. doi: 10.1042/CS20090488. [DOI] [PubMed] [Google Scholar]
- 9.Yla-Herttuala S, Bentzon JF, Daemen M, et al. Stabilisation of atherosclerotic plaques. Position paper of the European Society of Cardiology (ESC) Working Group on atherosclerosis and vascular biology. Thromb. Haemost. 2011;106(1):1–19. doi: 10.1160/TH10-12-0784. [DOI] [PubMed] [Google Scholar]
- 10.Ambrose JA. In search of the ‘vulnerable plaque’: can it be localized and will focal regional therapy ever be an option for cardiac prevention? J. Am. Coll. Cardiol. 2008;51(16):1539–1542. doi: 10.1016/j.jacc.2007.12.041. [DOI] [PubMed] [Google Scholar]
- 11.Kubo T, Imanishi T, Takarada S, et al. Assessment of culprit lesion morphology in acute myocardial infarction: ability of optical coherence tomography compared with intravascular ultrasound and coronary angioscopy. J. Am. Coll. Cardiol. 2007;50(10):933–939. doi: 10.1016/j.jacc.2007.04.082. [DOI] [PubMed] [Google Scholar]
- 12.Kolodgie FD, Narula J, Yuan C, Burke AP, Finn AV, Virmani R. Elimination of neoangiogenesis for plaque stabilization: is there a role for local drug therapy? J. Am. Coll. Cardiol. 2007;49(21):2093–2101. doi: 10.1016/j.jacc.2006.10.083. [DOI] [PubMed] [Google Scholar]
- 13.Carlier SG, De Korte CL, Brusseau E, Schaar JA, Serruys PW, Van Der Steen AF. Imaging of atherosclerosis. Elastography. J. Cardiovasc. Risk. 2002;9(5):237–245. [PubMed] [Google Scholar]
- 14.Madjid M, Naghavi M, Malik BA, Litovsky S, Willerson JT, Casscells W. Thermal detection of vulnerable plaque. Am. J. Cardiol. 2002;90(10C):36L–39L. doi: 10.1016/s0002-9149(02)02962-4. [DOI] [PubMed] [Google Scholar]
- 15.Demos SM, Alkan-Onyuksel H, Kane BJ, et al. In vivo targeting of acoustically reflective liposomes for intravascular and transvascular ultrasonic enhancement. J. Am. Coll. Cardiol. 1999;33(3):867–875. doi: 10.1016/s0735-1097(98)00607-x. [DOI] [PubMed] [Google Scholar]
- 16.Unno N, Mitsuoka H, Takei Y, et al. Virtual angioscopy using 3-dimensional rotational digital subtraction angiography for endovascular assessment. J. Endovasc. Ther. 2002;9(4):529–534. doi: 10.1177/152660280200900423. [DOI] [PubMed] [Google Scholar]
- 17.Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 18.Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2006;26(5):987–994. doi: 10.1161/01.ATV.0000214999.12921.4f. [DOI] [PubMed] [Google Scholar]
- 19.Nahrendorf M, Jaffer FA, Kelly KA, et al. Noninvasive vascular cell adhesion molecule-1 imaging identifies inflammatory activation of cells in atherosclerosis. Circulation. 2006;114(14):1504–1511. doi: 10.1161/CIRCULATIONAHA.106.646380. [DOI] [PubMed] [Google Scholar]
- 20.Laitinen I, Saraste A, Weidl E, et al. Evaluation of αvβ3 integrin-targeted positron emission tomography tracer 18F-galacto-RGD for imaging of vascular inflammation in atherosclerotic mice. Circ. Cardiovasc. Imaging. 2009;2(4):331–338. doi: 10.1161/CIRCIMAGING.108.846865. [DOI] [PubMed] [Google Scholar]
- 21.Tawakol A, Castano AP, Gad F, et al. Intravascular detection of inflamed atherosclerotic plaques using a fluorescent photosensitizer targeted to the scavenger receptor. Photochem. Photobiol. Sci. 2008;7(1):33–39. doi: 10.1039/b710746c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cole JE, Mitra AT, Monaco C. Treating atherosclerosis: the potential of Toll-like receptors as therapeutic targets. Expert Rev. Cardiovasc. Ther. 2010;8(11):1619–1635. doi: 10.1586/erc.10.149. [DOI] [PubMed] [Google Scholar]
- 23.Quillard T, Croce K, Jaffer FA, Weissleder R, Libby P. Molecular imaging of macrophage protease activity in cardiovascular inflammation in vivo. Thromb. Haemost. 2010;105(5):828–836. doi: 10.1160/TH10-09-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winter PM, Morawski AM, Caruthers SD, et al. Molecular imaging of angiogenesis in early-stage atherosclerosis with α(v) β3-integrin-targeted nanoparticles. Circulation. 2003;108(18):2270–2274. doi: 10.1161/01.CIR.0000093185.16083.95. [DOI] [PubMed] [Google Scholar]
- 25.Kuroda S, Mukohyama H, Kondo H, et al. Bone mineral density of the mandible in ovariectomized rats: analyses using dual energy X-ray absorptiometry and peripheral quantitative computed tomography. Oral Dis. 2003;9(1):24–28. doi: 10.1034/j.1601-0825.2003.01810.x. [DOI] [PubMed] [Google Scholar]
- 26.Sajja HK, East MP, Mao H, Wang YA, Nie S, Yang L. Development of multifunctional nanoparticles for targeted drug delivery and noninvasive imaging of therapeutic effect. Curr. Drug Discov. Technol. 2009;6(1):43–51. doi: 10.2174/157016309787581066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cormode DP, Skajaa T, Fayad ZA, Mulder WJ. Nanotechnology in medical imaging: probe design and applications. Arterioscler. Thromb. Vasc. Biol. 2009;29(7):992–1000. doi: 10.1161/ATVBAHA.108.165506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen M, Bulte JW, Liepold L, et al. Paramagnetic viral nanoparticles as potential high-relaxivity magnetic resonance contrast agents. Magn. Reson. Med. 2005;54(4):807–812. doi: 10.1002/mrm.20614. [DOI] [PubMed] [Google Scholar]
- 29.Cormode DP, Jarzyna PA, Mulder WJ, Fayad ZA. Modified natural nanoparticles as contrast agents for medical imaging. Adv. Drug Deliv. Rev. 2010;62(3):329–338. doi: 10.1016/j.addr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis JD, Destito G, Zijlstra A, et al. Viral nanoparticles as tools for intravital vascular imaging. Nat. Med. 2006;12(3):354–360. doi: 10.1038/nm1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leong HS, Steinmetz NF, Ablack A, et al. Intravital imaging of embryonic and tumor neovasculature using viral nanoparticles. Nat. Protoc. 2010;5(8):1406–1417. doi: 10.1038/nprot.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koudelka KJ, Destito G, Plummer EM, Trauger SA, Siuzdak G, Manchester M. Endothelial targeting of cowpea mosaic virus (CPMV) via surface vimentin. PLoS Pathog. 2009;5(5):e1000417. doi: 10.1371/journal.ppat.1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koudelka KJ, Rae CS, Gonzalez MJ, Manchester M. Interaction between a 54-kilodalton mammalian cell surface protein and cowpea mosaic virus. J. Virol. 2007;81(4):1632–1640. doi: 10.1128/JVI.00960-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. Vimentin is secreted by activated macrophages. Nat. Cell Biol. 2003;5(1):59–63. doi: 10.1038/ncb898. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez MJ, Plummer EM, Rae CS, Manchester M. Interaction of cowpea mosaic virus (CPMV) nanoparticles with antigen presenting cells in vitro and in vivo. PLoS ONE. 2009;4(11):e7981. doi: 10.1371/journal.pone.0007981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat. Rev. Cardiol. 2010;7(2):77–86. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pamukcu B, Lip GY, Devitt A, Griffiths H, Shantsila E. The role of monocytes in atherosclerotic coronary artery disease. Ann. Med. 2010;42(6):394–403. doi: 10.3109/07853890.2010.497767. [DOI] [PubMed] [Google Scholar]
- 38.Lundberg AM, Hansson GK. Innate immune signals in atherosclerosis. Clin. Immunol. 2010;134(1):5–24. doi: 10.1016/j.clim.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 39.Webb NR, Moore KJ. Macrophage-derived foam cells in atherosclerosis: lessons from murine models and implications for therapy. Curr. Drug Targets. 2007;8(12):1249–1263. doi: 10.2174/138945007783220597. [DOI] [PubMed] [Google Scholar]
- 40.Siegel-Axel D, Daub K, Seizer P, Lindemann S, Gawaz M. Platelet lipoprotein interplay: trigger of foam cell formation and driver of atherosclerosis. Cardiovasc. Res. 2008;78(1):8–17. doi: 10.1093/cvr/cvn015. [DOI] [PubMed] [Google Scholar]
- 41.Zaragoza C, Gomez-Guerrero C, Martin-Ventura JL, et al. Animal models of cardiovascular diseases. J. Biomed. Biotechnol. 2011;2011:497841. doi: 10.1155/2011/497841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merkel M, Velez-Carrasco W, Hudgins LC, Breslow JL. Compared with saturated fatty acids, dietary monounsaturated fatty acids and carbohydrates increase atherosclerosis and VLDL cholesterol levels in LDL receptor-deficient, but not apolipoprotein E-deficient, mice. Proc. Natl Acad. Sci. USA. 2001;98(23):13294–13299. doi: 10.1073/pnas.231490498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Babaev VR, Patel MB, Semenkovich CF, Fazio S, Linton MF. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in low density lipoprotein receptor-deficient mice. J. Biol. Chem. 2000;275(34):26293–26299. doi: 10.1074/jbc.M002423200. [DOI] [PubMed] [Google Scholar]
- 44.Jiang Z, Georgel P, Du X, et al. CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 2005;6(6):565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 45.Rae CS, Khor IW, Wang Q, et al. Systemic trafficking of plant virus nanoparticles in mice via the oral route. Virology. 2005;343(2):224–235. doi: 10.1016/j.virol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 46.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109(23 Suppl. 1):III27–III32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 47.Eefting FD, Pasterkamp G, Clarijs RJ, Van Leeuwen TG, Borst C. Remodeling of the atherosclerotic arterial wall: a determinant of luminal narrowing in human coronary arteries. Coron. Artery Dis. 1997;8(7):415–421. doi: 10.1097/00019501-199707000-00003. [DOI] [PubMed] [Google Scholar]
- 48.Steinmetz NF, Cho CF, Ablack A, Lewis JD, Manchester M. Cowpea mosaic virus nanoparticles target surface vimentin on cancer cells. Nanomedicine (Lond) 2011;6(2):351–364. doi: 10.2217/nnm.10.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nahrendorf M, Zhang H, Hembrador S, et al. Nanoparticle PET-CT imaging of macrophages in inflammatory atherosclerosis. Circulation. 2008;117(3):379–387. doi: 10.1161/CIRCULATIONAHA.107.741181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tearney GJ, Yabushita H, Houser SL, et al. Quantification of macrophage content in atherosclerotic plaques by optical coherence tomography. Circulation. 2003;107(1):113–119. doi: 10.1161/01.cir.0000044384.41037.43. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Z, Machac J, Helft G, et al. Non-invasive imaging of atherosclerotic plaque macrophage in a rabbit model with F-18 FDG PET: a histopathological correlation. BMC Nucl. Med. 2006;6:3. doi: 10.1186/1471-2385-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lipinski MJ, Frias JC, Amirbekian V, et al. Macrophage-specific lipid-based nanoparticles improve cardiac magnetic resonance detection and characterization of human atherosclerosis. JACC Cardiovasc. Imaging. 2009;2(5):637–647. doi: 10.1016/j.jcmg.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bryant AE, Bayer CR, Huntington JD, Stevens DL. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J. Infect. Dis. 2006;193(12):1685–1692. doi: 10.1086/504261. [DOI] [PubMed] [Google Scholar]
- 54.Garg A, Barnes PF, Porgador A, et al. Vimentin expressed on Mycobacterium tuberculosis-infected human monocytes is involved in binding to the NKp46 receptor. J. Immunol. 2006;177(9):6192–6198. doi: 10.4049/jimmunol.177.9.6192. [DOI] [PubMed] [Google Scholar]
- 55.Huet D, Bagot M, Loyaux D, et al. SC5 mAb represents a unique tool for the detection of extracellular vimentin as a specific marker of Sezary cells. J. Immunol. 2006;176(1):652–659. doi: 10.4049/jimmunol.176.1.652. [DOI] [PubMed] [Google Scholar]
- 56.Leong HS, Mahesh BM, Day JR, et al. Vimentin autoantibodies induce platelet activation and formation of platelet-leukocyte conjugates via platelet-activating factor. J. Leukoc. Biol. 2008;83(2):263–271. doi: 10.1189/jlb.0607339. [DOI] [PubMed] [Google Scholar]
- 57.Moisan E, Girard D. Cell surface expression of intermediate filament proteins vimentin and lamin B1 in human neutrophil spontaneous apoptosis. J. Leukoc. Biol. 2006;79(3):489–498. doi: 10.1189/jlb.0405190. [DOI] [PubMed] [Google Scholar]
- 58.Nieminen M, Henttinen T, Merinen M, Marttila-Ichihara F, Eriksson Je, Jalkanen S. Vimentin function in lymphocyte adhesion and transcellular migration. Nat. Cell. Biol. 2006;8(2):156–162. doi: 10.1038/ncb1355. [DOI] [PubMed] [Google Scholar]
- 59.Podor TJ, Singh D, Chindemi P, et al. Vimentin exposed on activated platelets and platelet microparticles localizes vitronectin and plasminogen activator inhibitor complexes on their surface. J. Biol. Chem. 2002;277(9):7529–7539. doi: 10.1074/jbc.M109675200. [DOI] [PubMed] [Google Scholar]
- 60.Van Beijnum JR, Dings RP, Van Der Linden E, et al. Gene expression of tumor angiogenesis dissected: specific targeting of colon cancer angiogenic vasculature. Blood. 2006;108(7):2339–2348. doi: 10.1182/blood-2006-02-004291. [DOI] [PubMed] [Google Scholar]
- 61.Xu B, Dewaal RM, Mor-Vaknin N, Hibbard C, Markovitz DM, Kahn ML. The endothelial cell-specific antibody PAL-E identifies a secreted form of vimentin in the blood vasculature. Mol. Cell. Biol. 2004;24(20):9198–9206. doi: 10.1128/MCB.24.20.9198-9206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levin N, Bischoff ED, Daige CL, et al. Macrophage liver X receptor is required for antiatherogenic activity of LXR agonists. Arterioscler. Thromb. Vasc. Biol. 2005;25(1):135–142. doi: 10.1161/01.ATV.0000150044.84012.68. [DOI] [PubMed] [Google Scholar]
- 63.Liu C, Bhattacharjee G, Boisvert W, Dilley R, Edgington T. In vivo interrogation of the molecular display of atherosclerotic lesion surfaces. Am. J. Pathol. 2003;163(5):1859–1871. doi: 10.1016/S0002-9440(10)63545-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lomonossoff GP, Shanks M. The nucleotide sequence of cowpea mosaic virus B RNA. EMBO J. 1983;2(12):2253–2258. doi: 10.1002/j.1460-2075.1983.tb01731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chacko AM, Hood ED, Zern BJ, Muzykantov VR. Targeted nanocarriers for imaging and therapy of vascular inflammation. Curr. Opin. Colloid Interface Sci. 2011;16(3):215–227. doi: 10.1016/j.cocis.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lewis DR, Kamisoglu K, York AW, Moghe PV. Polymer-based therapeutics: nanoassemblies and nanoparticles for management of atherosclerosis. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011;3(4):400–420. doi: 10.1002/wnan.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Douma K, Megens RT, Van Zandvoort MA. Optical molecular imaging of atherosclerosis using nanoparticles: shedding new light on the darkness. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011;3(4):376–388. doi: 10.1002/wnan.139. [DOI] [PubMed] [Google Scholar]
- 68.Terashima M, Uchida M, Kosuge H, et al. Human ferritin cages for imaging vascular macrophages. Biomaterials. 2011;32(5):1430–1437. doi: 10.1016/j.biomaterials.2010.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uchida M, Kosuge H, Terashima M, et al. Protein cage nanoparticles bearing the LyP-1 peptide for enhanced imaging of macrophage-rich vascular lesions. ACS Nano. 2011;5(4):2493–2502. doi: 10.1021/nn102863y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Websites
- 101.ImageJ: Image processing and analysis in Java. http://rsbweb.nih.gov/ij/
- 102.Imaris (bitplane) software. www.bitplane.com/go/products/imaris.
- 103.Virus particle explorer. http://viperDB.scripps.edu.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.