

Background: Isocitrate dehydrogenase (IDH) 1 and IDH2 mutations can lead to 2-hydroxyglutarate (2HG) accumulation in cancer.
Results: 2HG production from IDH mutants varies with subcellular localization and dependence on substrate production from the persistent wild-type IDH allele.
Conclusion: IDH1 but not IDH2 mutants require a wild-type IDH partner for 2HG production.
Significance: Differential 2HG production may explain the prognosis of IDH1/2 mutant cancers.
Keywords: Cancer Biology, Cell Metabolism, Leukemia, Mitochondria, Tumor Metabolism
Abstract
Monoallelic point mutations in cytosolic isocitrate dehydrogenase 1 (IDH1) and its mitochondrial homolog IDH2 can lead to elevated levels of 2-hydroxyglutarate (2HG) in multiple cancers. Here we report that cellular 2HG production from cytosolic IDH1 mutation is dependent on the activity of a retained wild-type IDH1 allele. In contrast, expression of mitochondrial IDH2 mutations led to robust 2HG production in a manner independent of wild-type mitochondrial IDH function. Among the recurrent IDH2 mutations at Arg-172 and Arg-140, IDH2 Arg-172 mutations consistently led to greater 2HG accumulation than IDH2 Arg-140 mutations, and the degree of 2HG accumulation correlated with the ability of these mutations to block cellular differentiation. Cytosolic IDH1 Arg-132 mutations, although structurally analogous to mutations at mitochondrial IDH2 Arg-172, were only able to elevate intracellular 2HG to comparable levels when an equivalent level of wild-type IDH1 was co-expressed. Consistent with 2HG production from cytosolic IDH1 being limited by substrate production from wild-type IDH1, we observed 2HG levels to increase in cancer cells harboring an endogenous monoallelic IDH1 mutation when mitochondrial IDH flux was diverted to the cytosol. Finally, expression of an IDH1 construct engineered to localize to the mitochondria rather than the cytosol resulted in greater 2HG accumulation. These data demonstrate that allelic and subcellular compartment differences can regulate the potential for IDH mutations to produce 2HG in cells. The consequences of 2HG elevation are dose-dependent, and the non-equivalent 2HG accumulation resulting from IDH1 and IDH2 mutations may underlie their differential prognosis and prevalence in various cancers.
Introduction
Monoallelic point mutations in cytosolic NADP+-dependent isocitrate dehydrogenase 1 (IDH1)3 were first discovered in exon sequencing studies of glioma and acute myeloid leukemia (AML) (1–3). All of the reported mutations were missense and at a single residue in the active site, Arg-132. These mutations were initially hypothesized to promote tumorigenesis through loss-of-function for the conversion of isocitrate to α-ketoglutarate, coupled with dominant-negative activity against the retained wild-type allele (4). However, subsequent work found a novel gain-of-function for Arg-132 mutations in IDH1, as well as for structurally analogous mutations at Arg-172 of IDH2, the mitochondrial homolog of IDH1. This shared neomorphic enzymatic activity is the NADPH-dependent reduction of α-ketoglutarate to R(−)-2-hydroxyglutarate (2HG) (5–7). Metabolite screening of AML samples revealed that 2HG-producing mutations can also occur at the non-orthologous Arg-140 residue of mitochondrial IDH2 (7). Collectively, 2HG-producing IDH1 Arg-132, IDH2 Arg-172, and IDH2 Arg-140 mutations are now known to be present in a large fraction of glioma, AML, chondrosarcoma, cholangiocarinoma, and T-cell angioimmunoblastic lymphoma samples, as well as isolated cases of colorectal and prostate cancer (3, 8–13). Additional 2HG-producing IDH mutations beyond those at IDH1 Arg-132 and IDH2 Arg-172/Arg-140 exist, but appear to be very rare and do not include rare IDH mutations reported in thyroid cancer and melanoma nor common IDH single nucleotide polymorphisms (14).
Through production of the oncometabolite 2HG, IDH mutations have been proposed to have numerous pro-tumorigenic effects, most notably the competitive inhibition of α-ketoglutarate-dependent dioxygenase enzymes that modify chromatin (15–19). These effects have been associated with a block in cell differentiation. Although Arg-132 mutations in cytosolic IDH1, the analogous Arg-172 mutations in mitochondrial IDH2, and the non-analogous IDH2 Arg-140 mutations all demonstrate the ability to elevate 2HG oncometabolite levels, it is becoming increasingly clear that these mutations are not functionally equivalent. The best evidence to date for this concept may be found in AML, where a better prognosis for patients with IDH2 Arg-140 mutations versus those with IDH2 Arg-172 mutations or IDH1 Arg-132 mutations has been reported by multiple groups (20–22). IDH2 Arg-140 mutations have yet to be described in glioma, chondrosarcoma, or cholangiocarinoma, despite the established prevalence of both IDH1 Arg-132 and IDH2 Arg-172 mutations in these cancers. In contrast, IDH2 Arg-140 mutations are the only IDH mutations found in the inborn error of metabolism d-2HG aciduria (23). The importance of subcellular localization differences between IDH1 and IDH2 proteins has also remained unexplored.
In this study, we have determined that there are distinct differences between the various 2HG-producing IDH1 and IDH2 mutations; both upstream regarding the metabolic pathways required to support 2HG production, and downstream regarding the cellular consequences of 2HG accumulation. The extent of 2HG production from mitochondrial IDH2 mutations depends on the particular site that is mutated. IDH2 Arg-140 mutations result in less cellular 2HG accumulation than IDH2 Arg-172 mutations under a variety of experimental conditions, correlating with the weaker ability of Arg-140 mutations to impair cell differentiation relative to Arg-172 mutations. Surprisingly, mutations in cytosolic IDH1 Arg-132, structurally analogous to mutations in mitochondrial IDH2 Arg-172, do not produce as much 2HG when overexpressed in cells at comparable levels. To a much greater extent than mitochondrial IDH2 mutations, cytosolic IDH1 mutations are substrate-limited for 2HG production in cells. Cellular 2HG accumulation from mutant IDH1 can be enhanced by co-expression of wild-type IDH1, diversion of wild-type IDH flux from mitochondria to cytosol, or forced re-localization of mutant IDH1 from cytosol to mitochondria. These results identify dose-dependent consequences of cellular 2HG accumulation and demonstrate that both allelic differences and the subcellular compartmentalization of metabolic flux can affect the ability of IDH mutations to result in cellular 2HG accumulation.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
293T cells, 3T3-L1 cells, JJ012 chondrosarcoma cells (24), and CS-1 chondrosarcoma cells (25) were cultured in Dulbecco's modified Eagle's medium (Invitrogen) with 10% fetal bovine serum (CellGro). JJ012 cells have a monoallelic endogenous IDH1 R132G mutation that has previously been reported (26), which we confirmed by Sequenom assay. CS-1 cells have a monoallelic endogenous IDH2 R172S mutation, which we determined by Sequenom assay. IDH mutation analysis in this cell line has not been previously reported. 3T3-L1 cells with stable expression of wild-type or mutant IDH2 were generated as described previously (17).
Cell Differentiation, Oil Red O Staining, Quantitative Real-time PCR
3T3-L1 cell differentiation, Oil Red O staining, and quantitative real-time PCR were performed as previously described (17). Experiments on primary murine bone marrow were performed according to previously published methods (16).
Protein Harvest and Quantitation and Western Blot
Cells were lysed 48 h following transfection with RIPA buffer or mammalian protein extraction reagent (Pierce) supplemented with protease inhibitor mixture (Roche Applied Science) and phosphatase inhibitor mixtures 2 and 3 (Sigma). Lysates were sonicated with 2 × 30-s pulses using the high setting on a Bioruptor300 (Diagenode) and then centrifuged at 14,000 × g for 20 min at 4 °C. Supernatants were subsequently collected and assessed for protein concentration with BCA Protein Assay (Pierce). α-Ketoglutarate-dependent NADPH consumption assays from cell lysates were performed as previously described (7). For cellular fractionation experiments, cells were lysed in isotonic buffer containing 200 mm mannitol, 68 mm sucrose, 10 mm HEPES-KOH, pH 7.4, 1 mm EGTA, and protease inhibitor mixture. Lysates were homogenized with 60 strikes in a Dounce homogenizer and then centrifuged at 600 × g for 10 min at 4 °C. The supernatant was collected and centrifuged again at 600 × g for 5 min at 4 °C. This supernatant was then centrifuged at 14,000 × g for 10 min at 4 °C. The pellet from this centrifugation was resuspended in RIPA buffer + protease inhibitor mixture and used as the heavy membrane fraction, whereas the supernatant layer was collected as the non-heavy membrane fraction. For Western blotting, lysates were separated by SDS-PAGE on Novex 4–12% BisTris gels (Invitrogen), transferred to nitrocellulose membranes, and blocked in 5% nonfat milk in PBS containing 0.2% Tween 20. Primary antibodies used were: anti-IDH1 (Santa Cruz sc-49996, 1:200 dilution), anti-IDH2 (Abcam ab55271, 1:500), anti-IDH3A (Abcam ab58641, 1:750), anti-ACO2 (Cell Signaling 6922, 1:1,000), anti-FLAG (Sigma F1804, 1:250), anti-Myc tag (Millipore 05–419, 1:1,000), anti-VDAC (Cell Signaling 4661S, 1:2,000), anti-α-tubulin (Sigma T6199, 1:10,000), and anti-S6 (Cell Signaling 2317 or 2217, 1:1,000). Detection was performed with horseradish peroxidase-conjugated anti-rabbit, anti-mouse, or anti-goat antibodies (GE Healthcare NA934V, GE Healthcare NA931V, and Santa Cruz sc-2020; all 1:10,000 dilution).
Metabolite Extraction
Following gentle removal of culture medium from proliferating cells, cells were rapidly quenched with 80% methanol pre-chilled to −80 °C and containing a M+5 internal standard of R(−)-2HG containing five deuterium atoms (d-2-hydroxyglutaric-2,3,3,4,4-d5 acid; details for synthesis can be found in supplemental “Experimental Procedures”). Following incubation at −80 °C for at least 30 min, cell extracts were collected, sonicated, and centrifuged at 14,000 × g for 20 min at 4 °C to remove precipitated protein. Supernatants were next dried under nitrogen gas, redissolved in 1:1 acetonitrile: N-methyl-N-tert-butyldimethylsilyltrifluoroacetamide (Regis, Morton Grove, IL), and heated at 70 °C for 80 min to derivatize metabolites. t-Butyl dimethyl silyl derivatives of organic acids were then analyzed by gas chromatography-mass spectrometry (GC-MS) as previously described (7, 27). 2HG levels were obtained by quantifying the peak area of the ion at m/z 433, formed through the loss of a t-butyl group (−57 atomic mass units) from the molecular ion tri-t-butyl dimethyl silyl-2HG, and normalizing to the peak area of the m/z 438 ion (representing the analogous derivative of the d5-2HG internal standard spiked at known concentration) and the total cellular protein as measured by BCA.
Plasmid Construction and Transfection
The cDNA clone of human IDH1 (BC012846.1) was obtained from American Type Culture Collection, and human IDH2 (BC009244) from Invitrogen. IDH1 R132H, IDH2 R172K, and IDH2 R140Q point mutations were generated as previously described (17). FLAG or Myc tags were added to the C termini of the open reading frames by standard PCR techniques. To introduce siRNA resistance, silent point mutations were generated as follows by site-directed mutagenesis: for IDH1, the cDNA sequence 5′-gt cac tac cgc atg tac ca-3′ was changed to 5′-gt cat tat agg atg tat ca-3′ (alterations underlined) to confer resistance to the human IDH1 siRNA with sense sequence 5′-gu cac uac cgc aug uac ca-3′ from Sigma-Proligo (SASI_Hs01_00340497). For IDH2, the cDNA sequence 5′-gt gat gag atg acc cgt at-3′ was changed to 5′-gc gac gaa aca agg at-3′ to confer resistance to the human IDH2 siRNA with sense sequence 5′-gu gau gag aug acc cgu au-3′ from Sigma-Proligo (SASI_Hs01_00151612). These changes are not predicted to alter the sequence of the translated IDH1/2 proteins. To generate the IDH1 construct that localizes to mitochondria, the cDNA sequence encoding the predicted N-terminal mitochondrial targeting sequence (MTS) of IDH2 plus 10 subsequent amino acids was amplified using primers 5′-tgt caa ggt tta ttg aag tca aaa tgg ccg gct acc tgc ggg t-3′ and 5′-tct cct tgc atc tct acc acg ggc ttc gcc acc ttg atc c-3′. The resulting PCR product was purified and subsequently used as a “super primer” in a standard QuikChange site-directed mutagenesis reaction (28) using the IDH1 cDNA as template (with the underlined segments of the above listed primers providing the necessary complementarity to the IDH1 cDNA for the super primer). The mutagenesis reaction was then transformed into TOP10 cells (Invitrogen) and clones were identified that had incorporated the desired N-terminal MTS and subsequent 10 residues of IDH2 in place of the first 9 residues of IDH1. A similar N-terminal IDH domain swapping strategy has previously been shown to be effective with yeast IDH enzymes (29). Integrity of constructs was confirmed by direct sequencing prior to transfection into 293T cells in pCMV-Sport6 or pCDNA3 expression vectors using Lipofectamine 2000 according to the manufacturer's instructions.
siRNA
For siRNA experiments in JJ012 and CS-1 cells, cells were reverse transfected with Lipofectamine RNAimax (Invitrogen) using 30 pmol of siRNA (final concentration of 10 nm) per 1.5 × 105 cells/well of 6-well plates. For plasmid and siRNA co-transfection experiments in 293T cells, forward transfection was performed using Lipofectamine 2000 using 50–60 pmol of siRNA (final concentration of 25–30 nm) per 7–8 × 105 cells/well of 6-well plates. In addition to the IDH1 and IDH2 siRNAs described above, additional siRNAs used were: universal negative control from Sigma-Proligo (SIC001), IDH1 5′-cugucuaaggguuggccuu-3′ from Sigma-Proligo (SASI_Hs01_00023490), IDH2 5′-caagucuucggguggcuuu-3′ from Sigma-Proligo (SASI_Hs02_00333382), IDH3A 5′-caggcaaggacauggcgaa-3′ from Sigma-Proligo (SASI_Hs02_00339757), ACO2 5′-cauccauuaugaccugcu-3′ and 5′-cauuaacauuguucgcaa-3′ from Ambion (Silencer Select 4390824, s921 and s922). All listed sequences are for the sense strand.
Statistical Analysis
Statistical analyses were performed with Student's t test. To analyze experiments comparing 2HG accumulation in pairs of transfectants over a range of IDH expression levels, a paired samples Student's t test was used.
RESULTS
Extent of 2HG Production from Mitochondrial IDH2 Mutations Depends on the Affected Residue but Not on Wild-type IDH Activity
Arg-172 mutations in mitochondrial IDH2 are found in glioma, chondrosarcoma, cholangiocarcinoma, and lymphoma, and are associated with a poor prognosis in AML. In contrast, Arg-140 mutations are not found in glioma, chondrosarcoma, and cholangiocarcinoma, and are associated with a better prognosis in AML. We hypothesized that these differences could be accounted for, in part, by quantitative differences in the extent of cellular 2HG accumulation resulting from Arg-140 versus Arg-172 mutations. To test this hypothesis, we compared the most common IDH2 mutations R140Q and R172K for their ability to produce 2HG at various levels of expression in cells lacking endogenous IDH1 or IDH2 mutations. Increasing expression of both mutants led to increasing amounts of 2HG (Fig. 1A). However, R172K mutant IDH2 expression consistently resulted in more 2HG accumulation than R140Q mutant expression (p < 0.005).
FIGURE 1.

Mitochondrial IDH2 Arg-140 mutations result in less cellular 2HG accumulation than IDH2 Arg-172 mutations, and 2HG accumulation from both mutations is insensitive to depletion of wild-type IDH. A, FLAG-tagged IDH2 R140Q and R172K cDNA constructs, or empty vector, were transfected into 293T cells at various doses. Cells were harvested 48 h post-transfection and assessed for 2HG accumulation by GC-MS (top) or protein expression by Western blot (bottom). Data are from a representative of 3 independent experiments. B, FLAG-tagged IDH2 R140Q and R172K constructs engineered to be resistant to siRNA knockdown, or empty vector, were transfected into cells along with 50 pmol of non-targeting siRNA (siCTRL) or 50 pmol of siRNA targeting only the endogenous wild-type IDH2 (siIDH2). C, siRNA-resistant IDH2 constructs were transfected into cells along with 60 pmol of siCTRL (2x siCTRL), 30 pmol of siRNA targeting the α subunit of IDH3 (siIDH3A) plus 30 pmol of siCTRL, or 30 pmol of siIDH3A plus 30 pmol of siIDH2. D, CS-1 chondrosarcoma cells with a naturally occurring, endogenous, monoallelic IDH2 R172S mutation were transfected with 30 pmol of siCTRL, siIDH2 (targeting both wild-type and mutant IDH2), one of two independent siRNAs targeting the mitochondrial aconitase ACO2 (siACO2#1 and #2), siIDH3A, or siIDH1. For B-D, data are representative of mean ± S.D. of 3 biological replicates from 2 independent experiments. *, p < 0.005; **, p < 0.001.
In most reported cases, both Arg-140 and Arg-172 mutations in IDH2 occur at only one allele, with retention of one wild-type IDH2 copy in tumors. Wild-type IDH2 can potentially produce both mitochondrial α-ketoglutarate and NADPH, the substrates for the neomorphic activity of mutant IDH2. To address whether differential dependence on wild-type IDH2 function could account for the differences in 2HG production we observed between Arg-140 and Arg-172 mutations, we expressed siRNA-resistant versions of the IDH2 R140Q and R172K mutants with or without simultaneous siRNA knockdown of the endogenous wild-type IDH2. The ability of both mutations to result in 2HG accumulation was insensitive to knockdown of the endogenous wild-type IDH2 (Fig. 1B). To address whether IDH3, which produces mitochondrial α-ketoglutarate and NADH, could compensate for the loss of wild-type IDH2, we expressed siRNA-resistant versions of IDH2 R140Q or R172K with simultaneous knockdown of either IDH3 alone or IDH3 together with endogenous wild-type IDH2 (Fig. 1C). Neither IDH3 knockdown alone nor the combined knockdown of endogenous wild-type IDH2 and IDH3 decreased cellular 2HG accumulation from IDH2 R140Q or R172K mutant expression. Directly upstream of the conversion of isocitrate to α-ketoglutarate by wild-type mitochondrial IDH2 and IDH3 is the mitochondrial aconitase ACO2, which isomerizes mitochondrial citrate to isocitrate. In a chondrosarcoma cell line with a naturally occurring, endogenous, monoallelic IDH2 R172S mutation, we examined the effect of impairing the pathway of mitochondrial citrate to α-ketoglutarate conversion. As expected, siRNA against IDH2 in this cell line, targeting both the mutant and wild-type IDH2, resulted in a sharp decrease in 2HG accumulation from mutant IDH2. However, knockdown of ACO2 did not impair 2HG accumulation, providing further evidence that the ability to convert citrate to isocitrate and then α-ketoglutarate in the mitochondria is not critical for the production of 2HG by mitochondrial IDH2 mutations (Fig. 1D). siRNA knockdown of either IDH3 or IDH1 was also unable to markedly impair 2HG accumulation in these cells. Collectively, these results demonstrate that IDH2 Arg-140 mutations consistently result in less 2HG accumulation in cells than IDH2 Arg-172 mutations, and that 2HG production by IDH2 mutations is unaffected by impaired production of α-ketoglutarate from mitochondrial citrate in multiple cell types.
Differential 2HG Production by IDH2 Arg-140 and Arg-172 Mutations Has Consequences for Gene Expression and Cell Differentiation
Recent work has highlighted the ability of 2HG to inhibit multiple α-ketoglutarate-dependent dioxygenase enzymes and impair growth factor/cytokine-induced gene expression and the ability of cells to differentiate. Previously we have reported that stable expression of an R172K IDH2 mutant in 3T3-L1 preadipocytes can impair their ability to differentiate into mature adipocytes following exposure to a well established differentiation mixture and this effect can be mimicked by adding cell penetrant forms of 2HG to the medium (17). To extend this observation, 3T3-L1 cells expressing either the R140Q or R172K mutant of IDH2 were compared. Although IDH2 R140Q expressing 3T3-L1 cells were impaired in their ability to differentiate into mature adipocytes, the extent of differentiation blockade was consistently less than that observed with IDH2 R172K expressing cells (Fig. 2, A-C). IDH2 R140Q expressing cells showed a more modest block in the ability to accumulate intracellular lipid as assessed by Oil Red O staining, and also demonstrated less silencing of the expression of the fat cell differentiation genes Glut4, FABP4, Adiponectin, and PPARγ than IDH2 R172K expressing cells. These effects correlated with the R140Q expressing cells accumulating 2HG to ∼⅓ of the level found in the R172K expressing cells (Fig. 2D). These findings demonstrate that IDH2 Arg-140 mutations can result in less 2HG accumulation than IDH2 Arg-172 mutations in non-transformed cells, and suggest that dosage differences in 2HG accumulation can account for the different cellular effects of IDH2 Arg-140 versus Arg-172 mutations.
FIGURE 2.

Extent of 2HG accumulation correlates with the degree of differentiation blockade in non-transformed cells expressing IDH2 Arg-140 or Arg-172 mutations. A, Western blot of 3T3-L1 pre-adipocytes stably expressing additional IDH2 WT, IDH2 R172K, IDH2 R140Q, or empty vector. IDH1 protein levels were used as a loading control. B, cells were treated with a mixture to induce differentiation into mature adipocytes for 7 days. Oil Red O staining was used to assess the accumulation of lipid droplets. C, at day 4 following differentiation induction, RNA was extracted and the expression of adipocyte-specific gene and transcription factors was measured by quantitative PCR with reverse transcription (RT-qPCR). B and C, data are representative of 3 independent experiments. D, cellular 2HG accumulation was measured by GC-MS. Data are representative of mean ± S.D. of 3 biological replicates from 2 independent experiments. *, p < 0.001.
Although we have utilized 3T3-L1 cells as a general model system for cell differentiation, reflecting the occurrence of IDH mutations in a wide variety of cancers and in metabolic disorders, we have also specifically examined the differential effects of IDH2 R140Q and IDH2 R172K in a hematopoietic cell model relevant to AML. We have previously reported that expression of IDH2 R140Q can increase the expression of the progenitor cell marker c-kit (stem cell factor receptor) in murine primary bone marrow cells (16). We report here an extension of this analysis to compare the relative abilities of IDH2 R140Q and IDH2 R172K to increase c-kit expression. Following 5 days of liquid culture in myeloid growth conditions, a higher percentage of c-kit expressing cells was observed in bone marrow expressing IDH2 R172K than in bone marrow expressing IDH2 R140Q (supplemental Fig. S1). These data provide further evidence that IDH2 Arg-172 and Arg-140 mutations can result in distinct degrees of cell differentiation impairment.
2HG Production from Cytosolic IDH1 Mutation in Cells Is Dependent on Retained Wild-type IDH1 Expression
With no exceptions reported to date, 2HG-producing mutations at Arg-132 of cytosolic IDH1 are monoallelic, with retention of the wild-type IDH1 allele. Arg-132 of cytosolic IDH1 is the analogous residue to Arg-172 of mitochondrial IDH2 (Fig. 3A). Both IDH1 Arg-132 and IDH2 Arg-172 play similar roles for substrate stability in the enzyme active site. To directly compare the cellular 2HG accumulation of cytosolic IDH1 Arg-132 mutations versus mitochondrial IDH2 mutations, we expressed a FLAG-tagged version of the most common IDH1 mutation, R132H, as well as both IDH2 R140Q and IDH2 R172K. Surprisingly, IDH1 R132H expression did not result in as much 2HG accumulation as expression of IDH2 R172K (Fig. 3B). The inability of IDH1 R132H expression to result in as much 2HG accumulation as IDH2 R172K expression was observed across a range in mutant IDH expression (Fig. 3C). However, when additional wild-type IDH1 was co-overexpressed, 2HG accumulation was reproducibly increased in cells expressing various levels of R132H mutant IDH1 (p < 0.05). With simultaneous transfection of exogenous wild-type and mutant IDH1, predicted to maximize the formation of wild-type:mutant IDH1 heterodimers, cells expressing R132H mutant IDH1 demonstrated comparable 2HG accumulation to cells expressing IDH2 R172K, and both IDH1 R132H and IDH2 R172K cells had greater 2HG levels than cells expressing IDH2 R140Q (Fig. 3C). The ability of co-overexpression of wild-type IDH1 to enhance cellular 2HG production from the R132H mutant IDH1 was also observed with untagged constructs, as well as different combinations of FLAG and Myc-tagged versions of IDH1 (supplemental Fig. S2, A and B). However, co-transfection of an IDH1 A134D mutant previously reported to be catalytically inactive (14) did not result in comparable augmentation of 2HG accumulation in IDH1 R132H-transfected cells (supplemental Fig. S2C).
FIGURE 3.
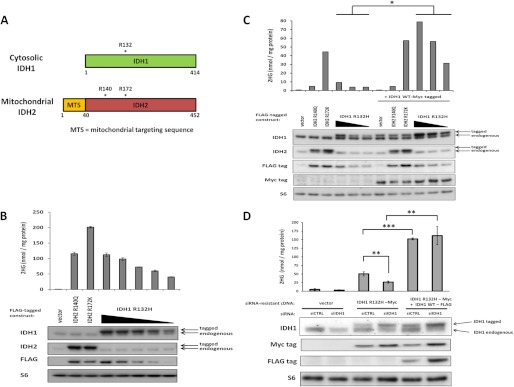
Cellular 2HG accumulation from cytosolic IDH1 mutation depends on maintained expression of wild-type IDH1. A, depiction of the open reading frames of the homologous proteins cytosolic IDH1 and mitochondrial IDH2. Recurrently mutated amino acid residues are marked with an asterisk (*). Arg-132 of cytosolic IDH1 is structurally analogous to Arg-172 of mitochondrial IDH2. B, FLAG-tagged IDH2 R140Q, IDH2 R172K, and IDH1 R132H (at various doses), or empty vector were transfected into 293T cells. Forty-eight hours post-transfection, cells were harvested and assessed for 2HG accumulation by GC-MS (top) or protein expression by Western blot (bottom). Data are representative of mean ± S.D. of 3 biological replicates from 2 independent experiments. C, a similar transfection as in B was performed with or without the co-transfection of Myc-tagged wild-type IDH1. D, Myc-tagged IDH1 R132H engineered to be siRNA-resistant, with or without FLAG-tagged IDH1 WT also engineered to be siRNA resistant, or empty vector, were transfected into 293T cells along with siCTRL or siRNA targeting only endogenous wild-type IDH1 (siIDH1). siIDH1 transfection to knockdown endogenous IDH1 levels was reproducibly associated with greater expression levels of Myc-tagged mutant IDH1, but this was still accompanied by a significant decrease in 2HG accumulation unless FLAG-tagged wild-type IDH1 was co-transfected. Data are representative of mean ± S.D. of 3 biological replicates from 4 independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To examine if depletion of endogenous wild-type IDH1 could impair 2HG production from mutant IDH1, we constructed a tagged version of IDH1 R132H engineered to be siRNA resistant, and co-transfected it along with siRNA against endogenous wild-type IDH1 (siIDH1). Although we reproducibly observed ∼2-fold higher expression of Myc-tagged IDH1 R132H when endogenous wild-type IDH1 levels were decreased, 2HG accumulation did not double but instead decreased by 50% (Fig. 3D). To confirm that this reduction in 2HG accumulation was due to the depletion of wild-type IDH1 and not an off-target siRNA effect, we co-transfected FLAG-tagged IDH1 WT that was also engineered to be siRNA resistant along with the siRNA-resistant Myc-tagged IDH1 R132H. With co-expression of IDH1 WT-FLAG along with IDH1 R132H-Myc, there was a 3-fold increase in 2HG production that was not diminished by endogenous wild-type IDH1 depletion with siIDH1 transfection. Cell proliferation was confirmed to be comparable under all of these transfection conditions (supplemental Fig. S2D). These results demonstrate that retention of wild-type IDH1 expression is critical for 2HG production from mutant IDH1 in the cytosol.
Impairing Mitochondrial IDH Flux Increases 2HG Production from Mutant IDH1 in the Cytosol
To examine whether increasing substrate flux through the existing wild-type IDH1 can enhance the ability of mutant IDH1 to produce 2HG in the cytosol, we examined the effect of impairing mitochondrial IDH activity in JJ012 chondrosarcoma cells harboring a naturally occurring, endogenous, monoallelic IDH1 R132G mutation. Depleting mitochondrial IDH2 or IDH3 can result in the diversion of citrate and/or isocitrate to the cytosol thus increasing substrate availability for wild-type IDH1 (Fig. 4A). As expected, transfection of siRNA targeting both wild-type and mutant IDH1 (siIDH1) impaired the ability of these cells to accumulate 2HG (Fig. 4B). However, depletion of either mitochondrial IDH2 or IDH3 increased 2HG accumulation in these cells harboring a monoallelic IDH1 mutation. Depletion of mitochondrial aconitase (ACO2) with two independent siRNAs also led to a reproducible increase in 2HG accumulation. These results provide further evidence that the ability of mutant IDH1 to produce 2HG is limited by the local supply of substrate, which can be increased by enhancing the isocitrate → α-ketoglutarate flux and cytosolic NADPH production through wild-type IDH1.
FIGURE 4.
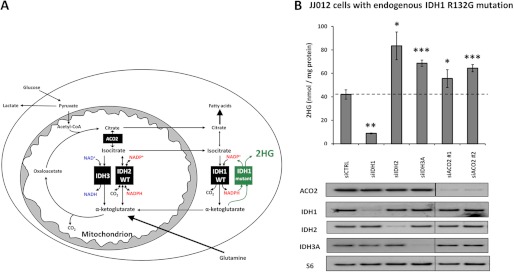
Impairing mitochondrial IDH flux results in increased 2HG accumulation in cells with endogenous cytosolic IDH1 mutation. A, model for metabolism in a cell harboring a monoallelic IDH1 mutation. B, JJ012 chondrosarcoma cells with a naturally occurring, endogenous, monoallelic IDH1 R132G mutation were transfected with 30 pmol of siCTRL, siIDH1 (targeting both wild-type and mutant IDH1), siIDH2, siIDH3A, or one of two independent siRNAs against ACO2. Forty-eight hours post-transfection cells were harvested and assessed for cellular 2HG accumulation by GC-MS (top) or protein expression by Western blot (bottom, images presented are panels from different areas of the same gel). Data are representative of mean ± S.D. of 3 biological replicates from 3 independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with siCTRL.
Mitochondrial Localization of Cytosolic IDH1 Mutation Results in Greater 2HG Accumulation
Collectively, the above results suggest that cytosolic IDH1 mutants are more limited in their supply of substrate for 2HG production than mitochondrial IDH2 mutants. To confirm that the differing substrate supply dependence between IDH1 and IDH2 mutants in cells is due to their subcellular localization differences, we generated an IDH1 construct that incorporated the N-terminal mitochondrial targeting sequence (MTS) of IDH2 (Fig. 5A). We confirmed that this “mito-IDH1” construct localized to the cellular heavy membrane fraction containing mitochondria, like mitochondrial IDH2 and unlike cytosolic IDH1 lacking the MTS (Fig. 5B). The tagged versions of IDH1 R132H and mito-IDH R132H were then transfected over a 40-fold range. We determined that cell lysates with equivalent expression levels of the mitochondrial and cytosolic localized IDH1 R132H proteins demonstrated equivalent α-ketoglutarate-dependent NADPH consumption rates when α-ketoglutarate and NAPH were provided in excess in vitro following cell lysis (data not shown). However, when assessing 2HG accumulation in cells with a range of expression levels of cytosolic IDH1 R132H or mito-IDH1 R132H, mito-IDH1 R132H-expressing cells consistently demonstrated greater 2HG accumulation than cells expressing comparable levels of cytosolic IDH1 R132H (p < 0.01, Fig. 5C). We performed another experiment to assess 2HG accumulation in cells expressing IDH1 R132H in the cytosol or mitochondria relative to cells expressing IDH2 R172K with its normal mitochondrial localization sequence intact. At comparable expression levels, both IDH2 R172K and mito-IDH1 R132H expressing cells had greater 2HG accumulation than cells expressing IDH1 R132H localized to the cytosol (Fig. 5D). Collectively, these data demonstrate that localization of IDH mutants to mitochondria rather than the cytosol results in greater 2HG accumulation in cells.
FIGURE 5.

Mitochondrial localization of cytosolic IDH1 mutant results in greater accumulation of 2HG in cells. A, An IDH1 construct designed to mislocalize to the mitochondria was engineered by placing the mitochondrial targeting sequence (MTS) of IDH2 at the N terminus of IDH1. Ten additional amino acids immediately following the MTS were also included to ensure proper processing, similar to that performed for previous IDH domain-swapping experiments in yeast (29). B, the desired localization of the mito-IDH1 construct was confirmed by Western blot of non-heavy and heavy membrane fractions of 293T cells 48 h following transfection of the indicated tagged constructs. C, Myc-tagged versions of the appropriately localized cytosolic IDH1 R132H or the mislocalized mito-IDH1 R132H were transfected into 293T cells at the doses indicated. Forty-eight hours post-transfection, cells were harvested and assessed for cellular 2HG accumulation by GC-MS (top) or IDH1 expression by Western blot (bottom). Data are representative of 3 independent experiments. D, FLAG-tagged versions of mitochondrial IDH2 R172K, cytosolic IDH1 R132H, and mito-IDH1 R132H were transfected into 293T cells. Forty-eight hours post-transfection, cells were harvested and assessed for 2HG accumulation by GC-MS (top) or protein expression by Western blot (bottom, images presented are panels from different areas of the same gel). Data are mean ± S.D. of 3 biological replicates from a representative experiment. *, p < 0.01.
DISCUSSION
The common feature of recurrent cancer-associated mutations in the isocitrate dehydrogenase enzymes is a neomorphic enzymatic activity converting α-ketoglutarate to the oncometabolite 2HG. In many functional studies to date, the various 2HG-producing IDH1/2 mutations have been considered equivalent. However, it is becoming increasingly clear that distinct neomorphic IDH1/2 alleles are not found with equal frequencies across different cancers and are not associated with the same prognosis (supplemental Table S1). For example, IDH2 Arg-140 mutations are rarely found in cancer outside of myeloid malignancies. But within myeloid malignancies, IDH2 Arg-140 is the most commonly mutated IDH residue in AML, and is uniquely associated with a better AML prognosis relative to IDH2 Arg-172 and IDH1 Arg-132 mutations (20–22). Moreover, little work has addressed the importance of differential subcellular localization of the IDH1 and IDH2 proteins.
The data presented here demonstrate that the particular residue that is mutated within the IDH active site has consequences for both the amount of 2HG that accumulates and the cellular consequences of IDH mutation. Both Arg-140 and Arg-172 mutations in mitochondrial IDH2 can produce increasing amounts of 2HG at increasing cellular expression levels, and both are insensitive to impairment of wild-type IDH activity. However, across multiple expression conditions and cell types, IDH2 Arg-140 mutations consistently result in less 2HG accumulation than IDH2 Arg-172 mutations. A recent report documenting similar differences when these mutations were knocked-in to an endogenous IDH2 allele in colon cancer cells via homologous recombination provides further support for this finding (30). Here, we demonstrate that the quantitative difference in 2HG accumulation between IDH2 Arg-140 and Arg-172 mutations is within a range that can variably alter gene expression in non-transformed cells and the ability of these lineage-specific progenitor cells to differentiate into terminally differentiated cells. In addition to cancer, these data are relevant to the inborn error of metabolism d-2HG aciduria. In this disease marked by widespread 2HG elevation in cells and body fluids, nearly 50% of cases harbor IDH2 Arg-140 mutations, but other IDH mutations are not found. We propose that only the lower 2HG levels resulting from IDH2 Arg-140 mutations are compatible with embryogenesis and that higher 2HG levels resulting from other germline IDH mutations may be lethal in utero.
It remains possible that the major downstream effects of 2HG that are critical to transformation vary between different cancers. 2HG can inhibit the catalytic activity of TET2, a DNA 5-methylcytosine hydroxylase. In AML, the mutual exclusivity of IDH mutations with TET2 mutations (16), as well as the emerging phenotypes of TET2 knock-out mice (31–33), provides evidence that inhibition of TET2 function can be a critical event sufficient to promote hematologic malignancy. In contrast, other cancers like glioma harbor IDH mutations but not TET2 mutations. We have proposed that in glioma, concerted inhibition of Jumonji histone demethylases by 2HG may assume an important tumorigenic role that cannot be recapitulated by simply inhibiting TET2 (17). 2HG can also inhibit prolyl hydroxylases that modify collagen (19, 34), which are likely to be of more importance in solid tumors such as glioma and chondrosarcoma than in liquid tumors like AML. Sustained inhibition of histone demethylases and/or collagen modifying enzymes may require higher levels of 2HG than that required for effective inhibition of TET2 function in myeloid cells. This requirement may account for why the lower 2HG-producing IDH2 Arg-140 mutations are not found in solid tumors. Further studies, including those with genetically engineered mice expressing the various IDH mutations in different cell lineages, will be required to test this hypothesis.
Left unexplored in the literature to date is examination of the impact that differential subcellular localization of IDH1 and IDH2 mutant proteins has on the ability of these mutants to result in 2HG accumulation in cells. IDH1 Arg-132 and IDH2 Arg-172 are structurally analogous residues and play similar roles in substrate binding in the enzyme active site, yet expression of IDH1 Arg-132 and IDH2 Arg-172 mutations in cells at comparable levels did not result in equivalent levels of 2HG accumulation unless wild-type IDH1 was co-expressed. This result is unlikely to be due solely to intrinsically weaker enzymatic activity of IDH1 R132H relative to IDH2 R172K for producing 2HG, because mitochondrial targeting of IDH1 R132H also resulted in increased 2HG accumulation relative to IDH1 R132H in the cytosol. The results presented here suggest that unlike the case with mitochondrial IDH2 mutations, 2HG production from cytosolic IDH1 mutations is particularly dependent on metabolic flux through a wild-type IDH1 partner to increase the local availability of substrate.
Although both mitochondrial IDH2 and cytosolic IDH1 mutations predominantly occur in monoallelic fashion, cases with reduction to homozygosity of mutant IDH2 have been reported (26, 35), whereas loss of the wild-type IDH1 allele in an IDH1 mutant cancer has not been described to date. We have previously proposed that more efficient generation of 2HG with co-expression of WT and mutant IDH1 may exist (5). One possible explanation for this, that the WT:R132H heterodimer could have intrinsically better activity to directly reduce α-ketoglutarate to 2HG, is not supported by the recent data of Leonardi et al. (36) who shows that the recombinant WT:R132H heterodimer is not as efficient as the R132H:R132H homodimer at converting α-ketoglutarate to 2HG in vitro. Although Pietrak et al. (37) concluded that the potential for substrate channeling from isocitrate → α-ketoglutarate → 2HG through WT:R132H heterodimers is present, they also concluded that the WT subunit was not necessary for the production of 2HG by the mutant recombinant protein. This finding was arrived at when α-ketoglutarate and NADPH were supplied in excess in vitro. But particularly in light of other reports suggesting that 2HG in cells is directly produced from the pathway of glutamine → glutamate → α-ketoglutarate → 2HG (5, 38), it has remained unclear how retained expression of wild-type IDH1 could be important for 2HG production by mutant IDH1.
In contrast to these biochemical studies of purified proteins, the present data suggest that in intact cells, cytosolic mutant IDH1 is substrate limited and that wild-type IDH1 contributes to the local substrate production required for mutant IDH1 to produce 2HG. This finding adds to the growing evidence that IDH1 mutations do not act via dominant inhibition of wild-type IDH1. By contrast, our findings suggest that targeting the provision of substrate by wild-type IDH1 to mutant IDH1 may provide an additional mechanism to mitigate the ultimate effects of IDH mutations by reducing the ability to produce 2HG. This would be especially attractive if increased flux through wild-type IDH1 can be shown to be dependent on the activation of growth factor signaling pathways. Prior evidence has linked IDH1 expression levels to the activity of the sterol regulatory element-binding protein transcription factor (39). Increasing IDH1 expression has been suggested to alter cellular and organismal physiology by increasing the production of cytosolic NADPH (40). Further studies in non-transformed cell lines will be needed to test the dependence of 2HG production from cytosolic mutant IDH1 on the prior induction of growth factor signaling and increased wild-type IDH1 activity. But this hypothesis may partially explain the failure (to date) of genetically engineered mouse models to demonstrate the initiation of tumorigenesis by IDH1 mutation in a stem/progenitor cell (34, 41, 42). It appears from the present data that 2HG production from cytosolic mutant IDH1 is a dynamic process that is acutely sensitive to the background metabolic state of the cell.
In contrast to 2HG production from mutant IDH1 being dependent on wild-type IDH1, these data show that 2HG production from mutant IDH2 is not dependent on wild-type mitochondrial IDH2 (or IDH3) activity. This is consistent with the report that IDH2 mutants do not heterodimerize with IDH2 WT, unlike IDH1 R132H mutants that do heterodimerize with a wild-type partner (43). It remains a technical challenge in the field to reliably measure metabolite levels in the subcellular compartments of intact cells. Our study does not determine the percentages of α-ketoglutarate and NADPH within each subcellular compartment that are dependent on wild-type IDH activity and/or the metabolism of anaplerotic substrates like glutamine. However, the lack of dependence of 2HG production from mitochondrial IDH2 mutations on wild-type IDH activity is consistent with mitochondrial α-ketoglutarate levels being maintained by a variety of anaplerotic substrates, and with a high NADPH/NADP+ ratio being maintained in the mitochondria through the interconversion of NADH to NADPH by the mitochondrial trans-hydrogenase (44). Recent work from multiple groups has implicated wild-type IDH “reverse flux,” the NADPH-dependent reductive carboxylation of α-ketoglutarate back to isocitrate, as playing an important role for cell growth and viability, particularly in hypoxic conditions or in cells with impaired TCA cycle function (27, 45–47). Although there is debate regarding whether this reductive carboxylation is primarily dependent upon cytosolic IDH1 or mitochondrial IDH2, the metabolic conditions that favor 2HG production from IDH mutations are also likely to favor reductive carboxylation flux from wild-type IDH proteins. The present results suggest that relative to the cytosol, mitochondria provide a more consistently favorable environment for the reductive metabolism of glutamine-derived α-ketoglutarate.
Supplementary Material
Acknowledgments
We thank members of the Thompson laboratory, especially Ji Zhang and Gaspare La Rocca, for technical assistance and critical discussions and feedback on the manuscript. We thank Ouathek Ouerfelli and Maria Spassova of the Organic Synthesis Core Facility and George Sukenick of the NMR Analytical Core Facility at Memorial Sloan-Kettering Cancer Center for the synthesis of the deuterated 2HG internal standard, Rich Terek of Brown University for the CS-1 cell line, and Tahir Sheikh.
This work was supported, in whole or in part, by the National Institutes of Health and National Cancer Institute. C. B. Thompson is a co-founder of Agios Pharmaceuticals and a member of the board directors of Merck.

This article contains supplemental “Experimental Procedures,” Figs. S1–S2, and Table S1.
- IDH
- isocitrate dehydrogenase
- 2HG
- 2-hydroxyglutarate
- AML
- acute myeloid leukemia
- GC-MS
- gas chromatography-mass spectrometry
- ACO2
- mitochondrial aconitase
- MTS
- mitochondrial targeting sequence
- c-kit
- stem cell factor receptor
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Mardis E. R., Ding L., Dooling D. J., Larson D. E., McLellan M. D., Chen K., Koboldt D. C., Fulton R. S., Delehaunty K. D., McGrath S. D., Fulton L. A., Locke D. P., Magrini V. J., Abbott R. M., Vickery T. L., Reed J. S., Robinson J. S., Wylie T., Smith S. M., Carmichael L., Eldred J. M., Harris C. C., Walker J., Peck J. B., Du F., Dukes A. F., Sanderson G. E., Brummett A. M., Clark E., McMichael J. F., Meyer R. J., Schindler J. K., Pohl C. S., Wallis J. W., Shi X., Lin L., Schmidt H., Tang Y., Haipek C., Wiechert M. E., Ivy J. V., Kalicki J., Elliott G., Ries R. E., Payton J. E., Westervelt P., Tomasson M. H., Watson M. A., Baty J., Heath S., Shannon W. D., Nagarajan R., Link D. C., Walter M. J., Graubert T. A., DiPersio J. F., Wilson R. K., Ley T. J. (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Parsons D. W., Jones S., Zhang X., Lin J. C., Leary R. J., Angenendt P., Mankoo P., Carter H., Siu I. M., Gallia G. L., Olivi A., McLendon R., Rasheed B. A., Keir S., Nikolskaya T., Nikolsky Y., Busam D. A., Tekleab H., Diaz L. A., Jr., Hartigan J., Smith D. R., Strausberg R. L., Marie S. K., Shinjo S. M., Yan H., Riggins G. J., Bigner D. D., Karchin R., Papadopoulos N., Parmigiani G., Vogelstein B., Velculescu V. E., Kinzler K. W. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yan H., Parsons D. W., Jin G., McLendon R., Rasheed B. A., Yuan W., Kos I., Batinic-Haberle I., Jones S., Riggins G. J., Friedman H., Friedman A., Reardon D., Herndon J., Kinzler K. W., Velculescu V. E., Vogelstein B., Bigner D. D. (2009) IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao S., Lin Y., Xu W., Jiang W., Zha Z., Wang P., Yu W., Li Z., Gong L., Peng Y., Ding J., Lei Q., Guan K. L., Xiong Y. (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 324, 261–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dang L., White D. W., Gross S., Bennett B. D., Bittinger M. A., Driggers E. M., Fantin V. R., Jang H. G., Jin S., Keenan M. C., Marks K. M., Prins R. M., Ward P. S., Yen K. E., Liau L. M., Rabinowitz J. D., Cantley L. C., Thompson C. B., Vander Heiden M. G., Su S. M. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gross S., Cairns R. A., Minden M. D., Driggers E. M., Bittinger M. A., Jang H. G., Sasaki M., Jin S., Schenkein D. P., Su S. M., Dang L., Fantin V. R., Mak T. W. (2010) Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 207, 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ward P. S., Patel J., Wise D. R., Abdel-Wahab O., Bennett B. D., Coller H. A., Cross J. R., Fantin V. R., Hedvat C. V., Perl A. E., Rabinowitz J. D., Carroll M., Su S. M., Sharp K. A., Levine R. L., Thompson C. B. (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Amary M. F., Bacsi K., Maggiani F., Damato S., Halai D., Berisha F., Pollock R., O'Donnell P., Grigoriadis A., Diss T., Eskandarpour M., Presneau N., Hogendoorn P. C., Futreal A., Tirabosco R., Flanagan A. M. (2011) IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 224, 334–343 [DOI] [PubMed] [Google Scholar]
- 9. Borger D. R., Tanabe K. K., Fan K. C., Lopez H. U., Fantin V. R., Straley K. S., Schenkein D. P., Hezel A. F., Ancukiewicz M., Liebman H. M., Kwak E. L., Clark J. W., Ryan D. P., Deshpande V., Dias-Santagata D., Ellisen L. W., Zhu A. X., Iafrate A. J. (2012) Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 17, 72–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cairns R. A., Iqbal J., Lemonnier F., Kucuk C., de Leval L., Jais J. P., Parrens M., Martin A., Xerri L., Brousset P., Chan L. C., Chan W. C., Gaulard P., Mak T. W. (2012) IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 119, 1901–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ghiam A. F., Cairns R. A., Thoms J., Dal Pra A., Ahmed O., Meng A., Mak T. W., Bristow R. G. (2012) IDH mutation status in prostate cancer. Oncogene 31, 3826. [DOI] [PubMed] [Google Scholar]
- 12. Marcucci G., Maharry K., Wu Y. Z., Radmacher M. D., Mrózek K., Margeson D., Holland K. B., Whitman S. P., Becker H., Schwind S., Metzeler K. H., Powell B. L., Carter T. H., Kolitz J. E., Wetzler M., Carroll A. J., Baer M. R., Caligiuri M. A., Larson R. A., Bloomfield C. D. (2010) IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia. A cancer and leukemia group B study. J. Clin. Oncol. 28, 2348–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sjöblom T., Jones S., Wood L. D., Parsons D. W., Lin J., Barber T. D., Mandelker D., Leary R. J., Ptak J., Silliman N., Szabo S., Buckhaults P., Farrell C., Meeh P., Markowitz S. D., Willis J., Dawson D., Willson J. K., Gazdar A. F., Hartigan J., Wu L., Liu C., Parmigiani G., Park B. H., Bachman K. E., Papadopoulos N., Vogelstein B., Kinzler K. W., Velculescu V. E. (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274 [DOI] [PubMed] [Google Scholar]
- 14. Ward P. S., Cross J. R., Lu C., Weigert O., Abel-Wahab O., Levine R. L., Weinstock D. M., Sharp K. A., Thompson C. B. (2012) Identification of additional IDH mutations associated with oncometabolite R(−)-2-hydroxyglutarate production. Oncogene 31, 2491–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chowdhury R., Yeoh K. K., Tian Y. M., Hillringhaus L., Bagg E. A., Rose N. R., Leung I. K., Li X. S., Woon E. C., Yang M., McDonough M. A., King O. N., Clifton I. J., Klose R. J., Claridge T. D., Ratcliffe P. J., Schofield C. J., Kawamura A. (2011) The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Figueroa M. E., Abdel-Wahab O., Lu C., Ward P. S., Patel J., Shih A., Li Y., Bhagwat N., Vasanthakumar A., Fernandez H. F., Tallman M. S., Sun Z., Wolniak K., Peeters J. K., Liu W., Choe S. E., Fantin V. R., Paietta E., Löwenberg B., Licht J. D., Godley L. A., Delwel R., Valk P. J., Thompson C. B., Levine R. L., Melnick A. (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lu C., Ward P. S., Kapoor G. S., Rohle D., Turcan S., Abdel-Wahab O., Edwards C. R., Khanin R., Figueroa M. E., Melnick A., Wellen K. E., O'Rourke D. M., Berger S. L., Chan T. A., Levine R. L., Mellinghoff I. K., Thompson C. B. (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Turcan S., Rohle D., Goenka A., Walsh L. A., Fang F., Yilmaz E., Campos C., Fabius A. W., Lu C., Ward P. S., Thompson C. B., Kaufman A., Guryanova O., Levine R., Heguy A., Viale A., Morris L. G., Huse J. T., Mellinghoff I. K., Chan T. A. (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S. H., Ito S., Yang C., Wang P., Xiao M. T., Liu L. X., Jiang W. Q., Liu J., Zhang J. Y., Wang B., Frye S., Zhang Y., Xu Y. H., Lei Q. Y., Guan K. L., Zhao S. M., Xiong Y. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Patel J. P., Gönen M., Figueroa M. E., Fernandez H., Sun Z., Racevskis J., Van Vlierberghe P., Dolgalev I., Thomas S., Aminova O., Huberman K., Cheng J., Viale A., Socci N. D., Heguy A., Cherry A., Vance G., Higgins R. R., Ketterling R. P., Gallagher R. E., Litzow M., van den Brink M. R., Lazarus H. M., Rowe J. M., Luger S., Ferrando A., Paietta E., Tallman M. S., Melnick A., Abdel-Wahab O., Levine R. L. (2012) Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 366, 1079–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Green C. L., Evans C. M., Zhao L., Hills R. K., Burnett A. K., Linch D. C., Gale R. E. (2011) The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood 118, 409–412 [DOI] [PubMed] [Google Scholar]
- 22. Boissel N., Nibourel O., Renneville A., Huchette P., Dombret H., Preudhomme C. (2011) Differential prognosis impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood 117, 3696–3697 [DOI] [PubMed] [Google Scholar]
- 23. Kranendijk M., Struys E. A., van Schaftingen E., Gibson K. M., Kanhai W. A., van der Knaap M. S., Amiel J., Buist N. R., Das A. M., de Klerk J. B., Feigenbaum A. S., Grange D. K., Hofstede F. C., Holme E., Kirk E. P., Korman S. H., Morava E., Morris A., Smeitink J., Sukhai R. N., Vallance H., Jakobs C., Salomons G. S. (2010) IDH2 mutations in patients with d-2-hydroxyglutaric aciduria. Science 330, 336. [DOI] [PubMed] [Google Scholar]
- 24. Scully S. P., Berend K. R., Toth A., Qi W. N., Qi Z., Block J. A. (2000) Marshall Urist Award. Interstitial collagenase gene expression correlates with in vitro invasion in human chondrosarcoma. Clin. Orthop. Relat. Res. 376, 291–303 [DOI] [PubMed] [Google Scholar]
- 25. Susa M., Morii T., Yabe H., Horiuchi K., Toyama Y., Weissbach L., Hornicek F. J., Morioka H. (2009) Alendronate inhibits growth of high-grade chondrosarcoma cells. Anticancer Res. 29, 1879–1888 [PubMed] [Google Scholar]
- 26. Pansuriya T. C., van Eijk R., d'Adamo P., van Ruler M. A., Kuijjer M. L., Oosting J., Cleton-Jansen A. M., van Oosterwijk J. G., Verbeke S. L., Meijer D., van Wezel T., Nord K. H., Sangiorgi L., Toker B., Liegl-Atzwanger B., San-Julian M., Sciot R., Limaye N., Kindblom L. G., Daugaard S., Godfraind C., Boon L. M., Vikkula M., Kurek K. C., Szuhai K., French P. J., Bovée J. V. (2011) Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat. Genet. 43, 1256–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wise D. R., Ward P. S., Shay J. E., Cross J. R., Gruber J. J., Sachdeva U. M., Platt J. M., DeMatteo R. G., Simon M. C., Thompson C. B. (2011) Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. U.S.A. 108, 19611–19616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Geiser M., Cèbe R., Drewello D., Schmitz R. (2001) Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. BioTechniques 31, 88–90, 92 [DOI] [PubMed] [Google Scholar]
- 29. Contreras-Shannon V., McAlister-Henn L. (2004) Influence of compartmental localization on the function of yeast NADP+-specific isocitrate dehydrogenases. Arch. Biochem. Biophys. 423, 235–246 [DOI] [PubMed] [Google Scholar]
- 30. Grassian A. R., Lin F., Barrett R., Liu Y., Jiang W., Korpal M., Astley H., Gitterman D., Henley T., Howes R., Levell J., Korn J. M., Pagliarini R. (2012) Isocitrate dehydrogenase (IDH) mutations promote a reversible ZEB1/mir-200-dependent epithelial-mesenchymal (EMT) transition. J. Biol. Chem. 287, 42180–42194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Z., Cai X., Cai C. L., Wang J., Zhang W., Petersen B. E., Yang F. C., Xu M. (2011) Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118, 4509–4518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moran-Crusio K., Reavie L., Shih A., Abdel-Wahab O., Ndiaye-Lobry D., Lobry C., Figueroa M. E., Vasanthakumar A., Patel J., Zhao X., Perna F., Pandey S., Madzo J., Song C., Dai Q., He C., Ibrahim S., Beran M., Zavadil J., Nimer S. D., Melnick A., Godley L. A., Aifantis I., Levine R. L. (2011) Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20, 11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quivoron C., Couronné L., Della Valle V., Lopez C. K., Plo I., Wagner-Ballon O., Do Cruzeiro M., Delhommeau F., Arnulf B., Stern M. H., Godley L., Opolon P., Tilly H., Solary E., Duffourd Y., Dessen P., Merle-Beral H., Nguyen-Khac F., Fontenay M., Vainchenker W., Bastard C., Mercher T., Bernard O. A. (2011) TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20, 25–38 [DOI] [PubMed] [Google Scholar]
- 34. Sasaki M., Knobbe C. B., Itsumi M., Elia A. J., Harris I. S., Chio I. I., Cairns R. A., McCracken S., Wakeham A., Haight J., Ten A. Y., Snow B., Ueda T., Inoue S., Yamamoto K., Ko M., Rao A., Yen K. E., Su S. M., Mak T. W. (2012) d-2-Hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 26, 2038–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pichler M. M., Bodner C., Fischer C., Deutsch A. J., Hiden K., Beham-Schmid C., Linkesch W., Guelly C., Sill H., Wölfler A. (2011) Evaluation of mutations in the isocitrate dehydrogenase genes in therapy-related and secondary acute myeloid leukaemia identifies a patient with clonal evolution to IDH2 R172K homozygosity due to uniparental disomy. Br. J. Haematol. 152, 669–672 [DOI] [PubMed] [Google Scholar]
- 36. Leonardi R., Subramanian C., Jackowski S., Rock C. O. (2012) Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J. Biol. Chem. 287, 14615–14620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pietrak B., Zhao H., Qi H., Quinn C., Gao E., Boyer J. G., Concha N., Brown K., Duraiswami C., Wooster R., Sweitzer S., Schwartz B. (2011) A tale of two subunits. How the neomorphic R132H IDH1 mutation enhances production of αHG. Biochemistry 50, 4804–4812 [DOI] [PubMed] [Google Scholar]
- 38. Seltzer M. J., Bennett B. D., Joshi A. D., Gao P., Thomas A. G., Ferraris D. V., Tsukamoto T., Rojas C. J., Slusher B. S., Rabinowitz J. D., Dang C. V., Riggins G. J. (2010) Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 70, 8981–8987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shechter I., Dai P., Huo L., Guan G. (2003) IDH1 gene transcription is sterol regulated and activated by SREBP-1a and SREBP-2 in human hepatoma HepG2 cells. Evidence that IDH1 may regulate lipogenesis in hepatic cells. J. Lipid Res. 44, 2169–2180 [DOI] [PubMed] [Google Scholar]
- 40. Koh H. J., Lee S. M., Son B. G., Lee S. H., Ryoo Z. Y., Chang K. T., Park J. W., Park D. C., Song B. J., Veech R. L., Song H., Huh T. L. (2004) Cytosolic NADP+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. J. Biol. Chem. 279, 39968–39974 [DOI] [PubMed] [Google Scholar]
- 41. Atai N. A., Renkema-Mills N. A., Bosman J., Schmidt N., Rijkeboer D., Tigchelaar W., Bosch K. S., Troost D., Jonker A., Bleeker F. E., Miletic H., Bjerkvig R., De Witt Hamer P. C., Van Noorden C. J. (2011) Differential activity of NADPH-producing dehydrogenases renders rodents unsuitable models to study IDH1R132 mutation effects in human glioblastoma. J. Histochem. Cytochem. 59, 489–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sasaki M., Knobbe C. B., Munger J. C., Lind E. F., Brenner D., Brüstle A., Harris I. S., Holmes R., Wakeham A., Haight J., You-Ten A., Li W. Y., Schalm S., Su S. M., Virtanen C., Reifenberger G., Ohashi P. S., Barber D. L., Figueroa M. E., Melnick A., Zúñiga-Pflücker J. C., Mak T. W. (2012) IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 488, 656–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jin G., Reitman Z. J., Spasojevic I., Batinic-Haberle I., Yang J., Schmidt-Kittler O., Bigner D. D., Yan H. (2011) 2-hydroxyglutarate production, but not dominant negative function, is conferred by glioma-derived NADP-dependent isocitrate dehydrogenase mutations. PLoS One 6, e16812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rydström J. (2006) Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta 1757, 721–726 [DOI] [PubMed] [Google Scholar]
- 45. Filipp F. V., Scott D. A., Ronai Z. A., Osterman A. L., Smith J. W. (2012) Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res. 25, 375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Metallo C. M., Gameiro P. A., Bell E. L., Mattaini K. R., Yang J., Hiller K., Jewell C. M., Johnson Z. R., Irvine D. J., Guarente L., Kelleher J. K., Vander Heiden M. G., Iliopoulos O., Stephanopoulos G. (2012) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mullen A. R., Wheaton W. W., Jin E. S., Chen P. H., Sullivan L. B., Cheng T., Yang Y., Linehan W. M., Chandel N. S., DeBerardinis R. J. (2012) Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.