

Background: CNTF plays an important role in brain development and injury.
Results: S-Nitrosoglutathione induces CNTF expression in astrocytes via NO signaling dependent PPAR-γ transactivation.
Conclusion: NO signaling in astrocytes favors the beneficial outcomes of reactive astrogliosis.
Significance: Endogenously produced NO or its exogenous source has potential to modulate the outcomes of reactive astrogliosis in vivo.
Keywords: Astrocytes, Brain, Multiple Sclerosis, Neurochemistry, Neuroinflammation, Neuroprotection, Nitric Oxide, p38 MAPK, Protein Kinase G (PKG), STAT3
Abstract
Accumulating evidence suggests that reactive astrogliosis has beneficial and detrimental outcomes in various CNS disorders, but the mechanism behind this dichotomy is unclear. Recent advances in this direction suggested that NO signaling is critical to regulate the outcomes of reactive astrogliosis in vivo. Using biochemical and genetic approaches, we here investigated the effect of S-nitrosoglutathione (GSNO; a physiological NO donor) in astrocytes in vitro settings. GSNO enhanced the expressions of glial fibrillary acidic protein and neurotrophic factors including ciliary neurotrophic factor (CNTF) in astrocytes in a dose-dependent manner. The enhanced CNTF expression in GSNO-treated astrocytes was ascribed to NO-mediated sGC/cGMP/PKG signaling. It was associated with p38 MAPK-dependent increased peroxisome proliferator-activated receptor-γ transactivation. In addition, the chromatin accessibility of peroxisome proliferator-activated receptor-γ accompanied with ATF2 and CREB (cAMP-response element-binding protein) was enhanced across the CNTF gene promoter in GSNO treated astrocytes. Interestingly, secreted CNTF was responsible for increased expression of glial fibrillary acidic protein in GSNO-treated astrocytes in an autocrine manner via a JAK2- and STAT3-dependent mechanism. In addition, CNTF secreted by GSNO-treated astrocytes enhanced the differentiation of immature oligodendrocytes in vitro. These effects of GSNO were consistent with an endogenously produced NO in astrocytes stimulated with proinflammatory cytokines in vitro. We conclude that NO signaling induces CNTF expression in astrocytes that favors the beneficial outcomes of reactive astrogliosis in vivo. Our data suggest that the endogenously produced NO or its exogenous source has potential to modulate the outcomes of reactive astrogliosis to protect CNS under pathological conditions.
Introduction
Evidence is emerging to suggest that astrocytes interact with the vasculature, neurons, and other astrocytes via signaling biological mediators and transport processes that in turn regulate blood flow and modulate brain impulse transmission (1). Cross-talk between astrocytes and neurons via neurotrophic factor release is considered to be the primary event in the maintenance of CNS homeostasis (2). In CNS, astrocytes are known to be immunocompetent cells that rapidly respond to various stimuli leading to reactive astrogliosis, which is the pathological hallmark of various CNS disorders (2, 3). Reactive astrogliosis is characterized by progressive cellular hypertrophy in astrocytes and enhanced cellular proliferation in response to CNS insult (2, 4). Previously, reactive astrogliosis was considered to be a uniformly negative and maladaptive event, and inhibition of this process was a therapeutic strategy (5). Surprisingly, recent studies provided compelling evidence that reactive astrogliosis has both detrimental and beneficial outcomes that are context-dependent and determined by complex and combinatorial inter/intracellular signaling mechanisms (6–8). Dichotomy behind these outcomes of reactive astrogliosis, however, remains to be understood.
The increased expression of glial fibrillary acidic protein (GFAP)3 in astrocytes is considered as a marker for reactive astrogliosis in CNS insult (9, 10). In addition, reactive astrocytes express an increased level of inducible nitric-oxide synthase that produces NO, which is considered to be detrimental to axons and oligodendrocytes (OLs) as documented in multiple sclerosis (MS) brain lesions (11, 12). S-Nitrosoglutathione (GSNO), a physiological NO donor, is reported to induce GFAP expression in astrocytes by inducing NO/soluble guanylyl cyclase (sGC)/cGMP/PKG signaling (13). Likewise, ciliary neurotrophic factor (CNTF) has also been documented to induce GFAP expression in astrocytes via activation of the JAK2 and STAT3 signaling (14, 15). Importantly, CNTF is reported to enhance metabolic plasticity in astrocytes that contribute to develop resistance against metabolic insults in brain (16). In addition, CNTF is reported to inhibit inducible inducible nitric-oxide synthase expression and thus NO production in reactive astrocytes (17). These findings suggest that NO- and CNTF-induced inter-/intracellular mechanisms are critical to modulate the outcomes of reactive astrogliosis.
Under physiological conditions, NO has been reported to participate in the neurotransmission and cell differentiation processes (18–20). The excessive production of NO, however, is considered to be detrimental to CNS under pathological conditions (21, 22). Over the past decade and a half, S-nitrosothiols (i.e. GSNO) has emerged as a regulator of various intracellular signaling mechanisms via protein S-nitrosylation (23, 24). We recently documented that GSNO treatment attenuates experimental autoimmune encephalomyelitis in a murine model of MS via inhibition of mononuclear cell infiltration into the CNS and restoration of blood brain barrier integrity (25, 26). GSNO treatment was also protective in other disease models such as stroke (27, 28) and traumatic spine and brain injury (29–31). Considering the biphasic role of NO in brain injury models, we sought to determine whether and how NO signaling modulates the outcomes of reactive astrogliosis under physiological and pathological conditions using in vitro settings.
MATERIALS AND METHODS
Chemicals and Reagents
Unless otherwise stated, all chemicals were purchased from Sigma. DMEM (4.5 g/liter glucose), FBS, mouse IgG, and rabbit polyclonal IgG (control primary antibodies) and secondary antibodies, i.e. Alexa Fluor conjugated with anti-rabbit IgG or anti-mouse IgG, were purchased from Invitrogen. Antibodies against phospho-JAK2, phospho-activating transcription factor 2 (ATF2), -phosphorylated ERK1/2, -phosphorylated p38 MAPK, -PPAR-γ, -histone H3, and -β-actin were purchased from Cell Signaling Technology (Danvers, MA). Rat recombinant CNTF, PDGF-AA, TNF-α, basic FGF2, IFN-γ, and anti-rat CNTF antibodies were purchased from R&D Systems Inc. (Minneapolis, MN). GSNO (100 mm in DMSO) was purchased from World Precision Instruments (Sarasota, FL). GW9662 (10 μm in DMSO), cell-permeable STAT3 inhibitor peptide (2 mg/ml in water), JAK2 inhibitor II (5 mm in DMSO), SB203580 (2.5 mm in DMSO), and apocynin (5 mm in DMSO) including anti-A2B5 antibodies were purchased from EMD Millipore (Billerica, MA). S-Nitroso-N-acetyl-d-l-penicillamine (SNAP; 100 mm in DMSO), U0126 (2.5 mm in DMSO), NS-2028 (10 μm in DMSO), MY-5445 (5 mm in DMSO), KT-5823 (100 μm in DMSO), 8Br-cGMP (10 mm in water), Rp-8Br-cGMP (10 mm in water), and L-NAME hydrochloride (10 mm in DMSO) were purchased from EnzoLife Sciences (Farmingdale, NY). Antibodies against GFAP, CNTF (immunostaining) and CREB-binding protein (CBP) were purchased from Abcam (Cambridge, MA). Precast SDS-PAGE gels and SYBR Green super mix were purchased from Bio-Rad. ECL-detecting reagents and nitrocellulose membranes were purchased from GE Healthcare.
Generation of Cortical Astrocytes and OL Cultures
Cortical glial cell cultures were generated from the brains of P1-P2 Sprague-Dawley rats obtained from pregnant mothers purchased from Harlan Laboratories (Dublin, VA) as described earlier (32) using purification methods described by McCarthy and de Vellis (33). Briefly, pups were euthanized by decapitation in accordance with the Medical University of South Carolina Institutional Animal Care and Use Committee and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The dissociated rat cortices were cultured on poly-lysine-coated cultures flasks in DMEM containing 10% FBS and 4 mm l-glutamine. After 10 days flasks were shaken at 250 revolutions per min for 2 h to remove microglia. To ensure complete removal of the microglia and OLs/OL progenitor cells, culture shaking was repeated twice after 1 or 2 days. Next, astrocytes cultures were exposed to 5 mm l-leucine methyl ester for 2 h to kill microglia contamination (34) followed by culturing for 24 h in fresh media. This method provided >99.0% GFAP-positive cells determined by FACS analysis. Purified cortical astrocytes (2000 cells/cm2) were plated in 6-well cell culture plates or glass slide chambers for 24 h. 70–80% confluent cultures were treated with GSNO or various pharmacological agents. For OL differentiation studies, conditioned media with GSNO (GSNO-ACM) or vehicle (Control-ACM) treatments were generated by treating astrocytes with GSNO (0.2 mm) or vehicle for 6 h followed by replacement of GSNO free DMEM and incubation for another 48 h. Likewise, conditioned media were generated from cytokine-treated astrocytes in the presence/or absence of apocynin or cytokine-treated astrocytes with knockdown of p47-phox for experimental studies.
OL Differentiation Studies
OLs/OL progenitor cells dislodged from the astrocyte layer were replated on laminin-2 coated glass chamber slides in high glucose Sato-based medium after checking their purity as described earlier (35). Purified OLs/OL progenitor cells (2000 cells/cm2) were plated in glass chamber slides in Sato medium supplemented with PDGF-AA and bovine FGF-2 (10 ng/ml each). After 48 h, fresh Sato medium supplemented with 10 ng/ml CNTF was added to generate immature OLs and incubated for another 48 h before culture them in GSNO-ACM, Control-ACM, and/or cytokine-treated astrocyte-conditioned media.
RNA Preparation, cDNA Synthesis, and Quantitative Real-time PCR Analysis
Cells were carefully processed for RNA isolation using TRIzol reagent followed by cDNA synthesis and real-time PCR analysis using iCycler iQ Real-time PCR Detection System (Bio-Rad) as described previously (32). Gene-specific primer for rat gene sequences (β-actin, forward primer (FP) 5′-agagaagctgtgctatgttgccct-3′ and reverse primer (RP) 5′-accgctcattgccgatagtgatga-3′; GFAP, FP 5′-agaaggccacctcaagaggaacat-3′ and RP 5′-ttaggccctcacactgtatggcaa-3′; myelin basic protein (MBP), FP 5′-ctctggcaaggactcacacac-3′ and RP 5′-tctgctgagggacaggcctctc-3; CNTF, FP cttcaagagctctcacagtg-3′ and RP tgcttatctttggccccataat-3′; insulin-like growth factor-1 (IGF-1), FP 5′-tgacatgcccaagactcagaagga-3′ and RP 5′-ggttgctcaagcagcaaaggatct-3′; leukemia inhibitory factor (LIF); FP 5′-tcacggcaacctcatgaaccagatwere-3′ and RP 5′-ccattggcatggaaaggtgggaaa-3′; COX-2; FP 5′-atcgatgccatggaactgtatccc-3′ and RP, 5′-ctcttacagctcagttgaacgcct-3′) were designed using Primer Quest software and purchased from Integrated DNA Technologies (Coralville, IA). Thermal cycling conditions were as follows: activation of iTaqTM DNA polymerase in SYBR Green super mix at 95 °C for 10 min followed by 40 cycles of amplification at 95 °C for 30 s and 59–60 °C for 45–60 s. The specificity and detection methods for data analysis are as described earlier (36).
Immunofluorescence and Phase Contrast Microscopy
For single-labeling, standard methodology was used. Briefly, slides were blocked by using an Image-iT® fixation and permeabilization kit (Invitrogen) and incubated with appropriately diluted primary antibody (1:100) at 4 °C overnight followed by washing and further incubation with secondary antibodies and Alexa Fluor-488 goat anti-rabbit IgG antibodies (1:500) for 1 h. For double-labeling, slides were incubated simultaneously with both types of primary antibodies after blocking with a serum-PBS solution at 4 °C overnight as described above. Secondary antibodies, Alexa Fluor-488-conjugated goat anti-rabbit IgG (CNTF) or Alexa Fluor-594-conjugated rabbit anti-mouse IgG (GFAP) antibodies, were used. Slides were also incubated with Alexa Fluor-conjugated IgG antibodies without primary antibody as negative controls and an appropriate mouse IgG or rabbit polyclonal IgG as isotype controls. After thorough washings, slides were mounted with Fluoromount-G (Electron Microscopy Sciences) containing Hoechst. Slides were examined with fluorescence microscope (Olympus BX-60), and images were captured at magnifications ×400 with an Olympus digital camera (Optronics; Goleta, CA) using a dual-band pass filter. The contrast and brightness of images were processed using Adobe Photoshop CS 5 software.
Numbers of processes and their length in OLs were determined by photographing random fields of cells with Olympus CKX41 inverse microscope at an amplification of ×200. Q Capture Pro 7 imaging software (Q Imaging, Surrey BC, Canada) was used for image processing, counting of cell soma and processes, and measuring of the OL process length. Depending on cell density, 15–25 cells/photograph were analyzed to count cell processes to the major cell body. Likewise, process length was measured in OLs from the major cell body to the end of process and calculated for statistical analysis.
Western Blot Analysis
Cells were lysed in ice-cold lysis buffer (radioimmune precipitation assay buffer, Thermo Scientific) and processed for Western blot analysis as described previously (32). Autoradiographs were scanned and quantified the band intensity by using ImageJ software from the NIH (free download at rsb.info.nih.gov).
Collection of Cellular Cytosolic and Nuclear Fractions
Cytosolic and nuclear fractions of cells were collected as described earlier (36). Then cytosolic and nuclear fractions were subjected to Western blot analysis and the ratio of protein band intensity of nuclear and cytosol fraction was calculated accordingly as described for Western blot analysis.
Co-immunoprecipitation and ELISA Methods
Nuclear proteins (150–200 μg/ml) were incubated with anti-CBP antibodies (20 μg/ml) in PBS with 0.01% Tween 20 overnight at 4 °C on an orbital shaker. After 24 h, 50 μl (1.5 mg) of Dynabeads protein A (Enzo life Technologies, Grass Island, NY) were added and incubated for 10 min followed by collection of beads using magnet. Beads for washed twice with PBS/w Tween 20, and samples were analyzed by Western blot analysis. CNTF levels in the culture supernatants were measured by using ELISA (R&D Systems Inc., Minneapolis, MN) as per the instructions in the product manual.
Transfection Studies
Lipofectamine was used for transient transfection of cells according to the manufacturer's instructions. Briefly, 70–80% confluent astrocyte cultures in 6-well cell culture plates after 24 h in DMEM with 10% FBS were transfected with 3.3 nmol of CNTF, STAT3, JAK2, p47-phox, and/or control (scramble) siRNA duplexes using a protocol described by Santa Cruz Biotechnology (Santa Cruz, CA). After 24 h, cells were cultured in fresh DMEM with 10% FBS for another 48 h followed by treatment with agents for 24 h and harvesting. Knockdown of the targeted protein in cells was examined by Western blot analysis that showed reduction of 40–60%.
ChIP Assay
Cells were cross-linked with 1% formaldehyde for 10 min at 37 °C and washed 3 times with ice-cold PBS containing 1 mm phenylmethylsulfonyl fluoride and 1% aprotinin. Soluble chromatin was prepared using a ChIP assay kit purchased from Active Motif (Carlsbad, CA) according to the manufacturer's recommendations and immunoprecipitated without (control) or with anti-CBP antibody and normal goat IgG. After washes and elution, precipitates were heated overnight at 65 °C to reverse cross-linking of DNA and protein. DNA fragments were purified by phenol-chloroform extraction and ethanol precipitation. The purified DNA was subjected to real-time PCR amplification using the primers specific for rat CNTF (NM_013166.1 (−) 04 kb: GPR1068780 (−) 04A containing CREB-binding domain present in the CNTF promoter purchased from Qiagen (Valencia, CA). Likewise, RNA polymerase II Chip-Grade Antibody kit was purchased from Qiagen containing GAPDH promoter primers as positive controls and non-coding region IGX1a primers as negative controls. Real-time PCR amplification conditions were 95 °C for 10 min and 40 cycles of 95 °C, 15 s and 60 °C for 60 s. Data were analyzed as described for real-time PCR analysis.
Cell Viability Testing
Cell viability of astrocytes treated with pharmacological agents was determined by a lactate dehydrogenase release assay kit (Roche Diagnostics) or trypan blue exclusion assay by mixing 10 μl of 0.4% trypan blue solution with 10 μl of cell suspension and reading on TC10 automated cell counter (Bio-Rad).
Detection of Nitrite Levels
Nitrite levels in the culture supernatants were measured by using Greiss reagent as described earlier (37).
Statistical Analysis
Data are given as the composite mean ± S.E. and analyzed by using Student's t test or one-way multiple range analysis of variance followed by a Bonferroni post-test; p values were determined for three to four separate samples in each experiment using GraphPad Prism 5.0 software (GraphPad Software Inc. San Diego, CA). p values <0.05 were considered significant.
RESULTS
S-Nitrosothiols Induce Hypertrophic Astrogliosis in Treated Astrocytes under Physiological Conditions
In experiments reported here we used cortical astrocyte cultures treated with S-nitrosothiols to investigate whether and how NO signaling modulates the outcomes of reactive astrogliosis in vivo. GSNO has recently been reported to regulate various cellular functions (23, 38). In addition, earlier we documented that exogenously administered GSNO restores blood brain barrier integrity and attenuates disease severity in experimental autoimmune encephalomyelitis (25, 26) and traumatic brain injury (30, 31) models. These studies provide evidence that regardless of the excessive expression of inducible nitric-oxide synthase/NO production by activated CNS glial cells in response to inflammatory insult, the exogenously administered GSNO has a tendency to limit the generation of circulating reactive nitrogen species NO/ONOO in vivo (39). It was reinforced by decrease in the level of nitrosylated proteins in a traumatic brain injury model (31) as well as NO-mediated inhibition of Th17 cell-mediated CNS inflammation (40). These findings suggest that the reduced bioavailability of GSNO at the tissue level is the limiting factor in brain disorders. Our findings could be explained by the enhanced GSNO reductase activity that degrades cellular GSNO as documented in severe respiratory failure and asthmatic disease conditions (41) or the conversion of NO into reactive nitrogen species as reported in brain disorders (39, 42).
We first sought to determine the effect of GSNO in astrocytes in vitro conditions. Consistent with previous report (13), we observed hypertrophic astrogliosis in GSNO-treated astrocytes as evident from significant increases in GFAP mRNA transcripts in astrocytes treated with graded concentrations of GSNO compared with controls (Fig. 1A). It was accompanied with a change in the morphology of GFAP-positive astrocytes treated with GSNO than controls (Fig. 1B). Next, to investigate whether GSNO promotes the beneficial function of astrocytes in vivo, we measured the level of mRNA transcripts for neurotrophic factors, i.e. CNTF, IGF-1, and LIF, in similarly treated astrocytes. GSNO significantly enhanced transcripts for CNTF, IGF-1, and LIF mRNA in treated astrocytes compared with controls (Fig. 1C). Interestingly, CNTF mRNA transcripts were significantly higher than IGF-1 or LIF mRNA transcripts in GSNO-treated astrocytes (Fig. 1C).
FIGURE 1.

Effect of GSNO treatment in astrocytes. Cortical astrocytes were plated (2000 × cm2) in 6-well cell culture plates or glass slide chambers. After 24 h, cells were treated with GSNO for another 24 h. A, the composite mean ± S.E. of four experiments depicts the ratio of GFAP to β-actin mRNA transcripts in treated astrocytes. B, representative fields of the slides (n = 5) depict the morphology of GFAP positive astrocytes determined by immunocytochemistry and fluorescence microscopy (magnifications ×400). C, the composite mean ± S.E. of three experiments depicts the ratio of IGF-1, LIF, and CNTF to β-actin mRNA transcripts in astrocytes treated with GSNO (0.2 mm) for 24 h. Statistical significance as indicated **, p < 0.01; ***, p < 0.001; NS, not significant.
Further comprehensive analysis revealed that GSNO enhances CNTF mRNA transcripts in a dose-dependent manner (Fig. 2A). It was true with similarly treated astrocytes with another S-nitrosothiol, SNAP (Fig. 2B). Consistent with RNA data, CNTF protein level was elevated significantly in the culture supernatants of astrocytes treated with GSNO or SNAP compared with controls (Fig. 2C) including in the cell lysate (Fig. 2, D and E). These findings were further confirmed by immunocytochemistry studies showing the increased accumulation of CNTF in the cytoplasm of astrocytes treated with GSNO or SNAP (Fig. 2F). To distinguish whether these observed effects of S-nitrosothiols in astrocytes are mediated by NO signaling or GSH, we treated astrocytes with GSH (5 mm) in parallel studies. As expected, no induction of CNTF expression or change in the morphology was observed in astrocytes treated with glutathione (data not shown). Notably, GSNO or SNAP concentrations used to treat astrocytes were not toxic as confirmed by cell viability with lactate dehydrogenase release (supplemental Fig. S1A) or trypan blue exclusion (data not shown) assays. Together, these data imply that NO signaling induces hypertrophic astrogliosis in astrocytes under physiological conditions.
FIGURE 2.
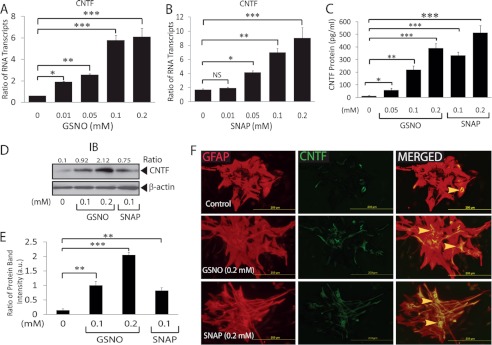
S-Nitrosothiols induce CNTF expression in astrocytes. Cortical astrocytes were cultured as detailed in Fig. 1 legend and treated with GSNO or SNAP. A and B, the composite mean ± S.E. of four experiments of treated astrocytes for 24 h depicts the ratio of CNTF to β-actin mRNA transcripts. C, the composite mean ± S.E. of three experiments depicts CNTF levels in the culture supernatants of treated astrocytes for 48 h measured by ELISA. A representative autoradiograph (D) and composite mean ± S.E. of three experiments depict the ratio of CNTF to β-actin band intensity (arbitrary units (a.u.)) in treated astrocytes (E) for 48 h. F, a representative field of slides (n = 5) of treated astrocytes for 72 h depicts the immunofluorescence for GFAP (left panel), CNTF (middle panel), and their co-localization (right panel) examined with fluorescence microscope (magnification ×400). The arrowhead depicts the localization of CNTF in the cytoplasm of astrocytes. Statistical significance as indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not significant; IB, immunoblot.
GSNO-induced sGC/cGMP/PKG Signaling Enhances CNTF and GFAP Expressions in Astrocytes
NO signaling is well known to induce the activation of sGC, thus elevating cGMP, a well established NO effector pathway (43). GSNO has recently been documented to increase GFAP expression in astrocytes via the sGC/cGMP/PKG-dependent pathway (13). Therefore, we then examined the possibility that GSNO may exert its influence on astrocytes to express CNTF via activation of sGC/cGMP/PKG pathway. To address this, we used specific inhibitors or agonists of NO signaling pathway to determine their effect on CNTF or GFAP expression in astrocytes simultaneously (Fig. 3A). Interestingly, GSNO-mediated increased CNTF mRNA transcripts in astrocytes were abolished by co-treatment with NS-2028 (a specific inhibitor of sGC) and/or KT-5823 (a specific inhibitor of PKG) (Fig. 3B). In contrast, MY-5445 (a specific cGMP phosphodiesterase inhibitor) or 8Br-cGMP (PKG activator) mimicked the effect of GSNO in astrocytes (Fig. 3B). Consistent with previous report (13), GSNO-mediated increased GFAP mRNA transcripts were reversed by co-treatment with NS-2028 and/or KT-5823 (Fig. 3C). This effect of GSNO was consistent with astrocytes treated with MY-5445 or 8Br-cGMP (Fig. 3C). Consistent with RNA data, CNTF and GFAP proteins levels were also elevated accordingly in treated astrocytes with GSNO or 8Br-cGMP (Fig. 3, D and E). In contrast, this effect of GSNO was reversed by Rp-8Br-cGMP (competitive analog of cGMP that blocks PKG activity) in treated astrocytes (Fig. 3, D and E). Concentrations of the inhibitors and/or agonists of this NO signaling cascade used in the study were not toxic to cells as confirmed by lactate dehydrogenase release (supplemental Fig. S1B) or trypan blue exclusion (data not shown) assays. These findings suggest that sGC/cGMP/PKG signaling participates in the induction of both CNTF and GFAP genes in GSNO-treated astrocytes.
FIGURE 3.

NO effector sGC/cGMP/PKG pathway enhances the expressions of CNTF and GFAP in astrocytes. Cortical astrocytes were cultured in 6-well cell culture plates as detailed in the Fig. 1 legend and treated with various agents. A, the schematic depicts the NO signaling effector sGC/cGMC/PKG pathway. The pharmacological inhibitors or agonists of this pathway used in the study are in bold. The composite mean ± S.E. of four experiments depicts the ratio of CNTF (B) and GFAP (C) to β-actin mRNA transcripts in treated astrocytes for 24 h. D, a representative autoradiograph depicts CNTF and GFAP protein levels in treated astrocytes for 48 h. E, the composite mean ± S.E. of three experiments depicts the % change in protein band intensity normalized with β-actin in treated astrocytes in D. Statistical significance is as indicated: $, p < 0.05; $$, p < 0.01; $$$, p < 0.001. NS, not significant versus no treatment. *, p < 0.05; ***, p < 0.001 versus GSNO treatment.
GSNO-mediated Induction of CNTF Gene Is Regulated by CREB/PPAR-γ/ATF-2 via Activation of MAPKs in Astrocytes
Earlier CNTF was documented to induce GFAP expression in astrocytes (14, 15). Likewise, NO signaling is reported to induce GFAP expression in astrocytes (13). We then asked whether the induction of CNTF and GFAP genes was linked in GSNO-treated astrocytes. Importantly, NO/sGC/PKG signaling has been documented to activate MAPK in different cell types (44, 45). Therefore, we first determined the activation status of MAPKs in GSNO-treated astrocytes. As expected, the enhanced phosphorylation of both ERK1/2 and p38 MAPK was observed in GSNO-treated astrocytes in a dose-dependent manner (Fig. 4, A and B). GSNO-mediated enhanced phosphorylation of ERK1/2 and p38 MAPK was reversed by co-treatment with NS-2028 and/or KT-5823 (Fig. 4, A and B). MY-5445 mimicked the effect of GSNO in treated astrocytes (Fig. 4, A and B). Because p38 MAPK activity is involved in the activation of PPAR-γ in endothelial cells (46), we next examined whether GSNO induces PPAR-γ activity in astrocytes. Interestingly, an enhanced nuclear translocation of PPAR-γ from the cytosol was observed as an indicator of its transcriptional activation in GSNO-treated astrocytes in a dose-dependent manner (Fig. 4C). PPAR-γ agonist ciglitazone was used as positive control (data not shown). This GSNO-mediated induction of PPAR-γ transcriptional activity in astrocytes was reversed by SB203580 (p38 MAPK inhibitor) but not by U0126, a MEK inhibitor (Fig. 4, D–E). This was true with astrocytes co-treated with GSNO and GW9662 (PPAR-γ antagonist) or Rp-8Br-cGMP (Fig. 4, D and E). In contrast, 8Br-cGMP mimicked the effect of GSNO on PPAR-γ transactivation in astrocytes (Fig. 4, D and E). Together, these data suggest that NO signaling induces PPAR-γ transactivation in astrocytes via p38 MAPK-dependent mechanism.
FIGURE 4.

GSNO-mediated activation of CREB/PPAR-γ/ATF2 induces CNTF gene in astrocytes. Cortical astrocytes were cultured in 6-well cell culture plates as detailed in the Fig. 1 legend. A, a representative autoradiograph depicts the phosphorylation of ERK1/2 and p38 MAPK in astrocytes treated with graded concentrations of GSNO and/or in the presence of NS-2028 (100 nm), KT-5823 (50 nm), and/or MY-5445 (5 μm) for 2 h. IB, immunoblot. B, the composite mean ± S.E. of three experiments depicts the ratio of phosphorylated to β-actin protein band intensity in treated astrocytes (A). a.u., arbitrary units. C and D, the representative autoradiograph depicts the distribution of PPAR-γ in the nucleus and cytosol of astrocytes treated with graded concentrations of GSNO and/or in the presence of GW9662 (100 nm), 8Br-cGMP (0.5 mm), Rp-8Br-cGMP (5 μm), U0126 (5 μm), and SB203580 (5 μm) for 24 h. E, the composite mean ± S.E. of three experiments depicts the ratio of PPAR-γ protein bands intensity in nucleus to cytosol in treated astrocytes (C and D). F, a representative autoradiograph depicts the level of PPAR-γ and ATF2 protein in the nucleus of treated astrocytes for 24 h analyzed by co-immunoprecipitation (IP) and Western blot analysis. G, the composite mean ± S.E. of four experiments depicts CNTF gene promoter activity normalized with GAPDH gene promoter activity in astrocytes treated with different concentrations of GSNO for 24 h analyzed by ChIP assay. H, the composite mean ± S.E. of four experiments depicts the ratio of CNTF to β-actin mRNA transcripts in treated astrocytes for 24 h. Statistical significance as indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001. NS, no treatment.
Next, to investigate whether GSNO promotes the chromatin accessibility of PPAR-γ in astrocytes, we performed co-immunoprecipitation of nuclear proteins using anti-CBP antibodies. Fig. 4F depicts that GSNO enhances interactions of CBP (transcriptional co-activator) with PPAR-γ and ATF2, which reinforce the observed NO-mediated induction of PPAR-γ transactivation in astrocytes. We next examined whether GSNO-mediated enhanced interaction among CBP, PPAR-γ, and ATF2 is responsible for the induction of CNTF gene. To do this, a ChIP assay was performed using anti-CBP antibodies that showed the enhanced chromatin accessibility of PPAR-γ/ATF2 across the CNTF gene promoter in astrocytes treated with GSNO or 8Br-cGMP (Fig. 4G). However, GSNO-mediated induction of CNTF gene promoter activity was reversed by co-treatment with Rp-8Br-cGMP in astrocytes (Fig. 4G).
Further comprehensive analysis revealed that GSNO-mediated induction of CNTF gene transcription was ascribed to the activation of PPAR-γ and MAPK in astrocytes. This was evident from the reversal of GSNO-mediated enhanced CNTF mRNA transcripts in the presence of GW9662 or SB203580 including in the presence of U0126 in astrocytes (Fig. 4H). Concentrations of the inhibitors for MAPK or PPAR-γ were not toxic to astrocytes as confirmed by cell viability with lactate dehydrogenase release (supplemental Fig. S1C) or trypan blue exclusion (data not shown) assays. Together, these findings imply that NO signaling induces the CNTF gene in GSNO-treated astrocytes by enhancing the chromatin accessibility of CREB/PPAR-γ/ATF2 across the CNTF gene promoter via activation of both p38 MAPK and ERK1/2.
CNTF Is Responsible for Increased GFAP Expression in GSNO-treated Astrocytes
Earlier CNTF was reported to induce GFAP expression in astrocytes via induction of JAK2/STAT3 signaling (14, 15). We observed a parallel increase in the expressions of CNTF and GFAP in GSNO-treated astrocytes (Figs. 1 and 2). We reasoned that the enhanced CNTF expression may be responsible for the induction of GFAP gene in GSNO-treated astrocytes. Thus, we treated astrocytes with GSNO in the presence of CNTF neutralizing antibodies or used astrocytes transiently transfected with CNTF siRNAs. The observed enhanced GFAP mRNA transcripts in GSNO-treated astrocytes were significantly reversed in the presence of rat CNTF neutralizing antibodies (Fig. 5A). Consistent with RNA data, the GSNO-mediated increased GFAP level in astrocytes was reversed in the presence of CNTF neutralizing antibodies (Fig. 5B). This was true with the knockdown of CNTF gene expression in astrocytes that demonstrated the reversal of GSNO-mediated increased CNTF mRNA transcripts in control astrocytes (Fig. 5C). The knockdown of CNTF protein in transiently transfected astrocytes was confirmed by Western blot analysis (supplemental Fig. S2A).
FIGURE 5.

Secreted CNTF induces GFAP expression in GSNO-treated astrocytes. Cortical astrocytes were plated in 6-well cell culture plates as detailed in the Fig. 1 legend. A, the composite mean ± S.E. of four experiments depicts the ratio of GFAP to β-actin mRNA transcripts in treated astrocytes for 24 h. Abs, antibodies. B, a representative autoradiograph (n = 4) depicts GFAP levels in treated astrocytes for 48 h. C, the Composite mean ± S.E. of three experiments depicts the ratio of GFAP to β-actin mRNA transcripts in treated astrocytes transiently transfected with control (scramble) or CNTF siRNAs. D, the composite mean ± S.E. of three experiments depicts the ratio of GFAP to β-actin mRNA transcripts in treated astrocytes with GSNO (0.2 mm) or CNTF (30 ng/ml) in the presence or absence of JAK2 inhibitor II (50 μm) or STAT3 inhibitor peptide (5 μg/ml) for 24 h. E, the composite mean ± S.E. of three experiments depicts the ratio of GFAP to β-actin mRNA transcripts in treated astrocytes after transient transfection with JAK2 or STAT3 siRNAs. F, a representative autoradiograph (n = 3) depicts % change in the protein level of JAK2 and STAT3 compared with controls in transiently transfected astrocytes (E). Statistical significance is as indicated. **, p < 0.01; ***, p < 0.001; NS, not significant.
Further analysis revealed that GSNO-mediated increased GFAP mRNA transcripts in astrocytes was reversed by inhibitors of JAK2 or STAT3 (Fig. 5D). This data were consistent with the inhibition of CNTF-mediated increased GFAP mRNA transcripts in astrocytes treated with inhibitors of JAK2 and STAT3 (Fig. 5D). Likewise, GSNO-mediated increased GFAP mRNA transcripts in astrocytes were abolished by knockdown of JAK2 or STAT3 expressions (Fig. 5E). This was also true for CNTF-mediated increased GFAP mRNA transcripts in astrocytes that were reversed by knockdown of JAK2 or STAT3 expressions (Fig. 5E). The reduction of JAK2 and STAT3 proteins in transiently transfected astrocytes with siRNAs was examined by Western blot analysis (Fig. 5F). Together, these data suggest that CNTF induced signaling is responsible for enhanced GFAP expression in GSNO-treated astrocytes in an autocrine manner.
CNTF Secreted by GSNO-treated Astrocytes Enhances OL Differentiation
Next, to test whether CNTF secreted by GSNO-treated astrocytes has implications to protect CNS under pathological conditions, we used conditioned media of astrocytes treated with GSNO (GSNO-ACM) or vehicle (Control-ACM) to treat immature OLs in vitro settings. Interestingly, immature OLs cultured in GSNO-ACM demonstrated the enhanced arborization in OLs compared with those treated with Control-ACM as revealed by counting of OL processes in images captured by phase-contrast microscopy (Fig. 6, A and B). Consistent with this, the length of OL processes was enhanced in OLs treated with GSNO-ACM more so than Control-ACM (Fig. 6C). These results were further supported by increased MBP mRNA transcripts (a signature gene of matured OLs) in GSNO-ACM-treated OLs than Control-ACM (Fig. 6D). As expected, GSNO-ACM effect on OLs was abolished by co-treatment with CNTF neutralizing antibodies (Fig. 6, A–D). To further determine whether CNTF is the sole neurotrophic factor secreted by astrocytes that contributes to OL differentiation, we treated immature OLs with different concentrations of recombinant CNTF protein. As expected, CNTF increased MBP mRNA transcripts in immature OLs significantly at its concentration 15 ng/ml and peaked at 30 ng/ml compared with controls (supplemental Fig. S3A). This effect of CNTF was reversed by CNTF neutralizing antibodies (supplemental Fig. S3A). Together, these data imply that CNTF secreted by GSNO-treated astrocytes contributes to OL differentiation.
FIGURE 6.

CNTF secreted by GSNO treated astrocytes enhances OL differentiation. Immature OLs (2000 cells/cm2) plated in glass slide chambers or 6-well culture plates were cultured for 96 h in culture supernatants (astrocyte conditioned media; 100%) of astrocytes treated with GSNO (GSNO-ACM) or vehicle (Control-ACM). A, a representative phase contrast micrograph (n = 5) of immature OLs cultured in control-ACM or GSNO-ACM in the presence or absence of CNTF neutralizing antibodies (α-CNTF-Abs) depicts OL morphology photographed by light microscopy at magnification ×200. B, the composite mean ± S.E. of four experiments depicts the number of processes per cell determined by counting of the phase contrast micrographs. C, the composite mean ± S.E. of four experiments depicts the length of process per cell measured in the phase contrast micrographs. D, the composite mean ± S.E. of four experiments depicts the ratio of MBP to β-actin mRNA transcripts in treated immature OLs. Statistical significance as indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Endogenously Produced NO Mimics the Effect of GSNO in Astrocytes
We next asked whether the endogenously produced NO in astrocytes can mimic the effect of GSNO. To do this we treated astrocytes with proinflammatory cytokines i.e. IFN-γ plus TNF-α as an inducer of inducible nitric-oxide synthase (47). Importantly, proinflammatory cytokines are documented to induce reactive oxygen species generation in glial cells via induction of NADPH oxidase activity (48). Reactive oxygen species tend to convert NO into its highly reactive peroxynitrite species that are detrimental in several CNS disorders (39, 42). Therefore, we treated astrocytes with cytokines in the presence of apocynin (NADPH oxidase inhibitor) or used astrocytes transiently transfected with p47-phox (a subunit of NADPH oxidase) siRNAs to increase cellular level of endogenous NO. The reduction of p47-phox protein level in transiently transfected astrocytes was confirmed by Western blot analysis (supplemental Fig. S2B). Interestingly, astrocytes treated with cytokines in the presence of apocynin showed increased CNTF and GFAP mRNA transcripts compared with vehicle treatment or controls (Fig. 7, A and B). This was true with similarly treated astrocyte knockdown for p47-phox expression than controls (Fig. 7, A and B). As expected, this effect of cytokines in astrocytes with reduced levels of p47-phox expression was abolished by co-treatment with L-NAME (Fig. 7, A and B). Consistent with RNA data, CNTF protein levels were elevated significantly in culture supernatants of astrocytes treated with cytokines in the presence of apocynin or those astrocytes knockdown for p47-phox expression compared with vehicle treatment or controls (supplemental Fig. S3B). As expected, cytokine-mediated increased CNTF protein in the culture supernatants of astrocytes with reduced p47-phox level was reversed by co-treatment with L-NAME (supplemental Fig. S3B). However, nitrite levels in the culture supernatants were elevated in cytokine-treated astrocytes with apocynin or vehicle including astrocytes with reduced p47-phox expression or controls and that were reduced by co-treatment with L-NAME (Fig. 7D). Together, these data imply that endogenously produced NO in cytokine-treated astrocytes induces CNTF expression thus hypertrophic astrogliosis under reduced oxidative stress conditions.
FIGURE 7.

Endogenously produced NO mimics GSNO effect in astrocytes. Cortical astrocytes were plated in 6-well cell culture plates as detailed in the Fig. 1 legend. A and B, the composite mean ± S.E. of three experiments depicts the ratio of CNTF and GFAP to β-actin mRNA transcripts in astrocytes treated with TNF-α (15 ng/ml) plus IFN-γ (25 ng/ml) in the presence of apocynin or vehicle as well as in transiently transfected astrocytes with p47-phox or control (scramble) siRNAs and treated with L-NAME for 24 h. C, the composite mean ± S.E. of three experiments depicts the ratio of MBP to β-actin mRNA transcripts in immature OLs plated as detailed in Fig. 6 legend and cultured for 96 h in culture supernatants (astrocyte-conditioned media; 50%) of cytokine-treated astrocytes (A and B). D, the composite mean ± S.E. of three experiments depicts nitrite levels in the culture supernatants of cytokine treated astrocytes for 24 h. Statistical significance as indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001. NS, not significant.
To further validate these findings, we cultured immature OLs in the culture supernatants of treated astrocytes (Fig. 7, A and B). The enhanced MBP mRNA transcripts were observed in treated immature OLs with conditioned media of cytokine-treated astrocytes in the presence of apocynin compared with vehicle or controls (Fig. 7C). This was true with cytokine-treated astrocytes with knockdown of p47-phox expression than controls (Fig. 7C). As expected, the observed increased MBP mRNA transcripts in OLs treated with conditioned media of cytokine-treated astrocytes having reduced p47-phox expression were reversed by conditioned media of similarly treated astrocytes in the presence of L-NAME (Fig. 7C). Together, these data suggest that the endogenously produced NO mimics the effect of GSNO, albeit produced by regulation of cellular redox environment.
DISCUSSION
Astrocytes, constituting the majority of resident glial cells, are almost five times more in number than neurons in normal adult brain (49). As endfeet around brain capillaries, astrocytes control the entry of metabolites to induce energy substrates into brain parenchyma from the vasculature (50). Increasing evidence suggests that astrocytes play a central role in CNS functioning by regulating the immune responses under pathological conditions (3). Astrocytes behave differently in terms of the secretion of neurotrophic factors or inflammatory mediators under physiological and pathological conditions (51, 52). Activation of astrocytes following CNS injury may have beneficial effects that include the monitoring and controlling of the extracellular water, pH, and ion homeostasis to promote the repair process. In contrast, the increased production of inflammatory mediators by activated astrocytes following CNS injury favors their detrimental effects surpassing beneficial effects (53, 54). Therefore, an understanding of the mechanism(s) that regulates the outcome of reactive astrogliosis is an active area of investigation to develop potential therapy for CNS disorders.
To begin to understand that NO signaling modulates the outcome of reactive astrogliosis, we evaluated the effect of GSNO in astrocytes. NO, a short-lived and diffusible free radical, has various roles as a signaling and effector molecule in diverse biological systems including neuronal messaging, vasodilation, and inhibition of platelet aggregation (55). In contrast, free NO is implicated with toxic effects under oxidative conditions because of its conversion into highly reactive peroxynitrite in several CNS disorders (18). Our findings demonstrated that NO signaling favors the beneficial outcomes of reactive astrogliosis as evident from the increased CNTF and GFAP expression in GSNO treated astrocytes (Figs. 1 and 2). Importantly, the increased CNTF expression in GSNO-treated astrocytes was ascribed to the NO effector sGC/cGMP/PKG pathway (Fig. 3). It was associated with p38 MAPK-dependent increased transactivation of PPAR-γ and its increased chromatin accessibility accompanied with ATF2 and CREB across the CNTF gene promoter (Fig. 4). However, ERK1/2 phosphorylation was also involved in the induction of CNTF expression in GSNO-treated astrocytes (Fig. 4H). We further demonstrated that the increased GFAP expression in GSNO-treated astrocytes was ascribed to CNTF-mediated induction of JAK2/STAT3 signaling in an autocrine manner (Fig. 5). In addition, CNTF secreted by GSNO-treated astrocytes enhanced the differentiation of immature OLs (Fig. 6). Finally, we showed that the endogenously produced NO by astrocytes stimulated with proinflammatory cytokines under reduced oxidative stress conditions mimics the effect of GSNO (Fig. 7). Together, these findings provide evidence that NO signaling induces CNTF expression in astrocytes that favors the beneficial outcome of reactive astrogliosis to protect CNS under physiological and pathological conditions (Fig. 8). Understanding of this NO signaling mechanism has implications to search for better therapeutics for CNS disorders.
FIGURE 8.

The schematic depicts how NO signaling does induce CNTF and GFAP expression in astrocytes. NO induces sGC activation that produces intracellular cGMP from GTP hydrolysis resulting in the activation of PKG and CREB. Activated PKG induces the phosphorylation of p38 MAPK and ERK1/2 that consequently leads to ATF2 phosphorylation and the activation of COX-2 that provides ligands for PPAR-γ translocation. The chromatin accessibility of activated CREB induces interactions of PPAR-γ and ATF2 to the CNTF gene promoter. Subsequently, secreted CNTF induces JAK2-dependent phosphorylation of STAT3, and its interaction with activated CREB and ATF2 induces GFAP expression in astrocytes in an autocrine manner. Likewise, cytokine induced inducible nitric-oxide synthase (NOS-II) expression thus NO production mimics the effect of GSNO in astrocytes. Pol II, polymerase II.
Previously, NO signaling has been reported to induce CREB activation as the survival mechanism in BDNF-expressing neurons in brain (56, 57). Our findings showed that NO-mediated induction of CREB activation increases the chromatin accessibility of PPAR-γ/ATF2 across the CNTF gene promoter in astrocytes. This effect of NO signaling was ascribed to the PKG-mediated phosphorylation of p38 MAPK and ERK1/2 (Fig. 4). In agreement with this, NO signaling has been documented to increase the phosphorylation of p38 MAPK that in turn induces PPAR-γ transactivation (46, 58). In addition, ERK1/2 and p38 MAPK are reported to increase the phosphorylation of ATF2 and its nuclear translocation in cells (60, 61). Importantly, PPAR transactivation in cells depends upon the bioavailability of their natural ligands synthesized by COX2 via p38 MAPK-dependent mechanism in cells (37, 62, 63). In line with this, we observed the increased level of COX-2 in astrocytes treated with GSNO (supplemental Fig. S2C). In addition, GSNO-induced PPAR-γ transactivation was inhibited by the inhibitors of p38 MAPK or PPAR-γ, suggesting that NO signaling increases the bioavailability of PPAR-γ endogenous ligands. Importantly, PPAR-γ activators are documented to enhance anti-inflammatory activities in astrocytes and other cell types (64, 65). In light of these studies, we conclude that NO signaling in astrocytes is crucial to provide anti-inflammatory and neuroprotective activities in experimental autoimmune encephalomyelitis and traumatic brain injury models (25, 26, 30, 31).
GFAP is the filamentous protein rapidly expressed by activated astrocytes in response to inflammatory cytokines or neurotrophic factors (9, 10, 14, 15). Earlier GFAP expression in GSNO-treated astrocytes was documented to be independent of NF-κB activation (13). We here demonstrated that CNTF secreted by GSNO-treated astrocytes is responsible for increased GFAP expression that was ascribed to JAK2/STAT3 signaling (Fig. 5). Consistent with this, CNTF has previously been reported to induce GFAP expression in astrocytes via activation of JAK2/STAT3 signaling (14, 15). Our findings provide evidence that CNTF-induced GFAP expression and NO-mediated CREB activation are linked in GSNO-treated astrocytes. These conclusions are supported by the observed increased phosphorylation of CREB and STAT3 in reactive astrocytes (66) that support our hypothesis that NO signaling in astrocytes is crucial to maintain CNS homeostasis.
CNTF has previously been documented to enhance the differentiation of astrocytes (67) that eventually enhances their potential to protect them against metabolic insults (16). In addition, CNTF-mediated induction of hypertrophic astrogliosis has been documented to protect neurons against various CNS insults (16). Our data demonstrated that CNTF secreted by GSNO-treated astrocytes enhances immature OLs differentiation. This effect of GSNO was evident with the endogenously produced NO in astrocytes treated with cytokines under reduced oxidative stress conditions (Fig. 7). Of note, a higher concentration of CNTF (15 ng/ml) was required to induce MBP expression in OLs than was observed with lower levels of CNTF (∼350 pg/ml) in the culture supernatant of GSNO-treated astrocytes (supplemental Fig. S3A and Fig. 2C). These data indicate that CNTF may exhibit synergistic activity with other neurotrophic factors (i.e. IGF-1 or LIF) secreted by GSNO-treated astrocytes to influence OLs. Moreover, CNTF was reported to provide CNS protection against various insults and the protection of differentiating OLs to promote CNS remyelination (59, 68, 69). Together, these findings suggest that NO-mediated induction of CNTF in astrocytes favors the beneficial outcomes of reactive astrogliosis.
In summary, our findings suggest that NO signaling induces CNTF expression in astrocytes. The secreted CNTF by GSNO-treated astrocytes eventually induces hypertrophic astrogliosis and enhances the differentiation of OLs that has implications to protect CNS under physiological and pathological conditions. Future studies using GSNO or other NO donors hold promise as therapeutic candidates (individually or in combination with PPAR-γ agonists) for incurable diseases such as MS or traumatic brain injury.
Supplementary Material
Acknowledgments
We thank all members of our laboratory for valuable comments and help during the course of this study. We thank especially Joyce Brian and Chara William for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants NS-22576, NS-37766, VA-1BX001072, VA-BX001999, C06 RR018823, and C06 RR015455.

This article contains supplemental Figs. S1—S3.
- GFAP
- glial fibrillary acidic protein
- OL
- oligodendrocyte
- MS
- multiple sclerosis
- GSNO
- S-nitrosoglutathione
- sGC
- soluble guanylyl cyclases
- CNTF
- ciliary neurotrophic factor
- ATF2
- activating transcription factor 2
- SNAP
- S-nitroso-N-acetyl-d-l-penicillamine
- CBP
- cAMP-response element-binding protein (CREB)-binding protein
- MBP
- myelin basic protein
- LIF
- leukemia inhibitory factor
- PKG
- protein kinase G
- L-NAME
- NG-nitro-l-arginine methyl ester
- ACM
- astrocyte-conditioned media
- Rp-8Br-cGMP 8-Bromoguanosine-3′
- 5′-cyclic monophosphorothioate Rp- isomer.
REFERENCES
- 1. Nedergaard M., Ransom B., Goldman S. A. (2003) New roles for astrocytes. Redefining the functional architecture of the brain. Trends Neurosci. 26, 523–530 [DOI] [PubMed] [Google Scholar]
- 2. Seifert G., Schilling K., Steinhäuser C. (2006) Astrocyte dysfunction in neurological disorders. A molecular perspective. Nat. Rev. Neurosci. 7, 194–206 [DOI] [PubMed] [Google Scholar]
- 3. Dong Y., Benveniste E. N. (2001) Immune function of astrocytes. Glia 36, 180–190 [DOI] [PubMed] [Google Scholar]
- 4. Vega-Avelaira D., Moss A., Fitzgerald M. (2007) Age-related changes in the spinal cord microglial and astrocytic response profile to nerve injury. Brain Behav. Immun. 21, 617–623 [DOI] [PubMed] [Google Scholar]
- 5. Rostworowski M., Balasingam V., Chabot S., Owens T., Yong V. W. (1997) Astrogliosis in the neonatal and adult murine brain post-trauma. Elevation of inflammatory cytokines and the lack of requirement for endogenous interferon-γ. J. Neurosci. 17, 3664–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sofroniew M. V. (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Herx L. M., Yong V. W. (2001) Interleukin-1 β is required for the early evolution of reactive astrogliosis following CNS lesion. J. Neuropathol. Exp. Neurol. 60, 961–971 [DOI] [PubMed] [Google Scholar]
- 8. Myer D. J., Gurkoff G. G., Lee S. M., Hovda D. A., Sofroniew M. V. (2006) Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 129, 2761–2772 [DOI] [PubMed] [Google Scholar]
- 9. Sriram K., Benkovic S. A., Hebert M. A., Miller D. B., O'Callaghan J. P. (2004) Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration. Key signaling pathway for astrogliosis in vivo? J. Biol. Chem. 279, 19936–19947 [DOI] [PubMed] [Google Scholar]
- 10. Eng L. F., Ghirnikar R. S. (1994) GFAP and astrogliosis. Brain Pathol. 4, 229–237 [DOI] [PubMed] [Google Scholar]
- 11. Liu B., Neufeld A. H. (2001) Nitric-oxide synthase-2 in human optic nerve head astrocytes induced by elevated pressure in vitro. Arch. Ophthal. 119, 240–245 [PubMed] [Google Scholar]
- 12. Haider L., Fischer M. T., Frischer J. M., Bauer J., Höftberger R., Botond G., Esterbauer H., Binder C. J., Witztum J. L., Lassmann H. (2011) Oxidative damage in multiple sclerosis lesions. Brain 134, 1914–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brahmachari S., Fung Y. K., Pahan K. (2006) Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J. Neurosci. 26, 4930–4939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Na Y. J., Jin J. K., Kim J. I., Choi E. K., Carp R. I., Kim Y. S. (2007) JAK-STAT signaling pathway mediates astrogliosis in brains of scrapie-infected mice. J. Neurochem. 103, 637–649 [DOI] [PubMed] [Google Scholar]
- 15. Kahn M. A., Huang C. J., Caruso A., Barresi V., Nazarian R., Condorelli D. F., de Vellis J. (1997) Ciliary neurotrophic factor activates JAK/Stat signal transduction cascade and induces transcriptional expression of glial fibrillary acidic protein in glial cells. J. Neurochem. 68, 1413–1423 [DOI] [PubMed] [Google Scholar]
- 16. Escartin C., Pierre K., Colin A., Brouillet E., Delzescaux T., Guillermier M., Dhenain M., Déglon N., Hantraye P., Pellerin L., Bonvento G. (2007) Activation of astrocytes by CNTF induces metabolic plasticity and increases resistance to metabolic insults. J. Neurosci. 27, 7094–7104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilkins A., Compston A. (2005) Trophic factors attenuate nitric oxide-mediated neuronal and axonal injury in vitro. Roles and interactions of mitogen-activated protein kinase signalling pathways. J. Neurochem. 92, 1487–1496 [DOI] [PubMed] [Google Scholar]
- 18. Bruhwyler J., Chleide E., Liégeois J. F., Carreer F. (1993) Nitric oxide. A new messenger in the brain. Neurosci. Biobehav. Rev. 17, 373–384 [DOI] [PubMed] [Google Scholar]
- 19. Magrinat G., Mason S. N., Shami P. J., Weinberg J. B. (1992) Nitric oxide modulation of human leukemia cell differentiation and gene expression. Blood 80, 1880–1884 [PubMed] [Google Scholar]
- 20. Bishop A., Anderson J. E. (2005) NO signaling in the CNS. from the physiological to the pathological. Toxicology 208, 193–205 [DOI] [PubMed] [Google Scholar]
- 21. Bayir H., Kagan V. E., Clark R. S., Janesko-Feldman K., Rafikov R., Huang Z., Zhang X., Vagni V., Billiar T. R., Kochanek P. M. (2007) Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J. Neurochem. 101, 168–181 [DOI] [PubMed] [Google Scholar]
- 22. Sharma H. S., Drieu K., Alm P., Westman J. (2000) Role of nitric oxide in blood-brain barrier permeability, brain edema, and cell damage following hyperthermic brain injury. An experimental study using EGB-761 and Gingkolide B pretreatment in the rat. Acta Neurochir. Suppl. 76, 81–86 [DOI] [PubMed] [Google Scholar]
- 23. Hess D. T., Matsumoto A., Kim S. O., Marshall H. E., Stamler J. S. (2005) Protein S-nitrosylation. Purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166 [DOI] [PubMed] [Google Scholar]
- 24. Hess D. T., Stamler J. S. (2012) Regulation by S-nitrosylation of protein post-translational modification. J. Biol. Chem. 287, 4411–4418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nath N., Morinaga O., Singh I. (2010) S-nitrosoglutathione a physiologic nitric oxide carrier attenuates experimental autoimmune encephalomyelitis. J. Neuroimmune Pharmacol. 5, 240–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Prasad R., Giri S., Nath N., Singh I., Singh A. K. (2007) GSNO attenuates EAE disease by S-nitrosylation-mediated modulation of endothelial-monocyte interactions. Glia 55, 65–77 [DOI] [PubMed] [Google Scholar]
- 27. Khan M., Sekhon B., Giri S., Jatana M., Gilg A. G., Ayasolla K., Elango C., Singh A. K., Singh I. (2005) S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J. Cereb. Blood Flow Metab. 25, 177–192 [DOI] [PubMed] [Google Scholar]
- 28. Khan M., Jatana M., Elango C., Paintlia A. S., Singh A. K., Singh I. (2006) Cerebrovascular protection by various nitric oxide donors in rats after experimental stroke. Nitric oxide 15, 114–124 [DOI] [PubMed] [Google Scholar]
- 29. Chou P. C., Shunmugavel A., El Sayed H., Desouki M. M., Nguyen S. A., Khan M., Singh I., Bilgen M. (2011) Preclinical use of longitudinal MRI for screening the efficacy of S-nitrosoglutathione in treating spinal cord injury. J. Magn. Reson. Imaging 33, 1301–1311 [DOI] [PubMed] [Google Scholar]
- 30. Khan M., Im Y. B., Shunmugavel A., Gilg A. G., Dhindsa R. K., Singh A. K., Singh I. (2009) Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J. Neuroinflammation 6, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khan M., Sakakima H., Dhammu T. S., Shunmugavel A., Im Y. B., Gilg A. G., Singh A. K., Singh I. (2011) S-Nitrosoglutathione reduces oxidative injury and promotes mechanisms of neurorepair following traumatic brain injury in rats. J. Neuroinflammation 8, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paintlia A. S., Paintlia M. K., Khan M., Vollmer T., Singh A. K., Singh I. (2005) HMG-CoA reductase inhibitor augments survival and differentiation of oligodendrocyte progenitors in animal model of multiple sclerosis. FASEB J. 19, 1407–1421 [DOI] [PubMed] [Google Scholar]
- 33. McCarthy K. D., de Vellis J. (1980) Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J. Cell Biol. 85, 890–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Giulian D., Baker T. J. (1986) Characterization of ameboid microglia isolated from developing mammalian brain. J. Neurosci. 6, 2163–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paintlia A. S., Paintlia M. K., Singh A. K., Orak J. K., Singh I. (2010) Activation of PPAR-γ and PTEN cascade participates in lovastatin-mediated accelerated differentiation of oligodendrocyte progenitor cells. Glia 58, 1669–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paintlia A. S., Paintlia M. K., Singh A. K., Singh I. (2008) Inhibition of rho family functions by lovastatin promotes myelin repair in ameliorating experimental autoimmune encephalomyelitis. Mol. Pharmacol. 73, 1381–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paintlia A. S., Paintlia M. K., Singh I., Singh A. K. (2006) IL-4-induced peroxisome proliferator-activated receptor γ activation inhibits NF-κB transactivation in central nervous system (CNS) glial cells and protects oligodendrocyte progenitors under neuroinflammatory disease conditions. Implication for CNS-demyelinating diseases. J. Immunol. 176, 4385–4398 [DOI] [PubMed] [Google Scholar]
- 38. Lipton S. A., Choi Y. B., Pan Z. H., Lei S. Z., Chen H. S., Sucher N. J., Loscalzo J., Singel D. J., Stamler J. S. (1993) A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364, 626–632 [DOI] [PubMed] [Google Scholar]
- 39. Murphy S. (2000) Production of nitric oxide by glial cells. Regulation and potential roles in the CNS. Glia 29, 1–13 [DOI] [PubMed] [Google Scholar]
- 40. Niedbala W., Alves-Filho J. C., Fukada S. Y., Vieira S. M., Mitani A., Sonego F., Mirchandani A., Nascimento D. C., Cunha F. Q., Liew F. Y. (2011) Regulation of type 17 helper T-cell function by nitric oxide during inflammation. Proc. Natl. Acad. Sci. U.S.A. 108, 9220–9225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Que L. G., Liu L., Yan Y., Whitehead G. S., Gavett S. H., Schwartz D. A., Stamler J. S. (2005) Protection from experimental asthma by an endogenous bronchodilator. Science 308, 1618–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abramov A. Y., Duchen M. R. (2005) The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid β peptides. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 360, 2309–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arnold W. P., Mittal C. K., Katsuki S., Murad F. (1977) Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. U.S.A. 74, 3203–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Doronzo G., Viretto M., Russo I., Mattiello L., Di Martino L., Cavalot F., Anfossi G., Trovati M. (2011) Nitric oxide activates PI3-K and MAPK signalling pathways in human and rat vascular smooth muscle cells. Influence of insulin resistance and oxidative stress. Atherosclerosis 216, 44–53 [DOI] [PubMed] [Google Scholar]
- 45. Browning D. D., McShane M. P., Marty C., Ye R. D. (2000) Nitric oxide activation of p38 mitogen-activated protein kinase in 293T fibroblasts requires cGMP-dependent protein kinase. J. Biol. Chem. 275, 2811–2816 [DOI] [PubMed] [Google Scholar]
- 46. Ptasinska A., Wang S., Zhang J., Wesley R. A., Danner R. L. (2007) Nitric oxide activation of peroxisome proliferator-activated receptor γ through a p38 MAPK signaling pathway. FASEB J. 21, 950–961 [DOI] [PubMed] [Google Scholar]
- 47. Singh I., Pahan K., Khan M., Singh A. K. (1998) Cytokine-mediated induction of ceramide production is redox-sensitive. Implications to proinflammatory cytokine-mediated apoptosis in demyelinating diseases. J. Biol. Chem. 273, 20354–20362 [DOI] [PubMed] [Google Scholar]
- 48. Mander P. K., Jekabsone A., Brown G. C. (2006) Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J. Immunol. 176, 1046–1052 [DOI] [PubMed] [Google Scholar]
- 49. Sofroniew M. V., Vinters H. V. (2010) Astrocytes. Biology and pathology. Acta Neuropathol. 119, 7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kacem K., Lacombe P., Seylaz J., Bonvento G. (1998) Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter. A confocal microscopy study. Glia 23, 1–10 [PubMed] [Google Scholar]
- 51. Morganti-Kossmann M. C., Satgunaseelan L., Bye N., Kossmann T. (2007) Modulation of immune response by head injury. Injury 38, 1392–1400 [DOI] [PubMed] [Google Scholar]
- 52. Makwana M., Jones L. L., Cuthill D., Heuer H., Bohatschek M., Hristova M., Friedrichsen S., Ormsby I., Bueringer D., Koppius A., Bauer K., Doetschman T., Raivich G. (2007) Endogenous transforming growth factor β 1 suppresses inflammation and promotes survival in adult CNS. J. Neurosci. 27, 11201–11213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ghirnikar R. S., Yu A. C., Eng L. F. (1994) Astrogliosis in culture. III. Effect of recombinant retrovirus expressing antisense glial fibrillary acidic protein RNA. J. Neurosci. Res. 38, 376–385 [DOI] [PubMed] [Google Scholar]
- 54. Yu A. C., Lee Y. L., Eng L. F. (1993) Astrogliosis in culture. I. The model and the effect of antisense oligonucleotides on glial fibrillary acidic protein synthesis. J. Neurosci. Res. 34, 295–303 [DOI] [PubMed] [Google Scholar]
- 55. Nathan C. (1992) Nitric oxide as a secretory product of mammalian cells. FASEB J. 6, 3051–3064 [PubMed] [Google Scholar]
- 56. Chien W. L., Liang K. C., Fu W. M. (2008) Enhancement of active shuttle avoidance response by the NO-cGMP-PKG activator YC-1. Eur. J. Pharmacol. 590, 233–240 [DOI] [PubMed] [Google Scholar]
- 57. Ciani E., Guidi S., Bartesaghi R., Contestabile A. (2002) Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons. Implication for a survival role of nitric oxide. J. Neurochem. 82, 1282–1289 [DOI] [PubMed] [Google Scholar]
- 58. Browning D. D., Windes N. D., Ye R. D. (1999) Activation of p38 mitogen-activated protein kinase by lipopolysaccharide in human neutrophils requires nitric oxide-dependent cGMP accumulation. J. Biol. Chem. 274, 537–542 [DOI] [PubMed] [Google Scholar]
- 59. Albrecht P. J., Murtie J. C., Ness J. K., Redwine J. M., Enterline J. R., Armstrong R. C., Levison S. W. (2003) Astrocytes produce CNTF during the remyelination phase of viral-induced spinal cord demyelination to stimulate FGF-2 production. Neurobiol. Dis. 13, 89–101 [DOI] [PubMed] [Google Scholar]
- 60. Han J., Jiang Y., Li Z., Kravchenko V. V., Ulevitch R. J. (1997) Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature 386, 296–299 [DOI] [PubMed] [Google Scholar]
- 61. Crowe D. L., Shemirani B. (2000) The transcription factor ATF-2 inhibits extracellular signal regulated kinase expression and proliferation of human cancer cells. Anticancer Res. 20, 2945–2949 [PubMed] [Google Scholar]
- 62. Puigserver P., Rhee J., Lin J., Wu Z., Yoon J. C., Zhang C. Y., Krauss S., Mootha V. K., Lowell B. B., Spiegelman B. M. (2001) Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol. Cell 8, 971–982 [DOI] [PubMed] [Google Scholar]
- 63. Li Z., Jansen M., Ogburn K., Salvatierra L., Hunter L., Mathew S., Figueiredo-Pereira M. E. (2004) Neurotoxic prostaglandin J2 enhances cyclooxygenase-2 expression in neuronal cells through the p38MAPK pathway. A death wish? J. Neurosci. Res. 78, 824–836 [DOI] [PubMed] [Google Scholar]
- 64. Janabi N. (2002) Selective inhibition of cyclooxygenase-2 expression by 15-deoxy-Δ(12,14)(12,14)-prostaglandin J(2) in activated human astrocytes but not in human brain macrophages. J. Immunol. 168, 4747–4755 [DOI] [PubMed] [Google Scholar]
- 65. Luna-Medina R., Cortes-Canteli M., Alonso M., Santos A., Martínez A., Perez-Castillo A. (2005) Regulation of inflammatory response in neural cells in vitro by thiadiazolidinones derivatives through peroxisome proliferator-activated receptor γ activation. J. Biol. Chem. 280, 21453–21462 [DOI] [PubMed] [Google Scholar]
- 66. Hebert M. A., O'Callaghan J. P. (2000) Protein phosphorylation cascades associated with methamphetamine-induced glial activation. Ann. N.Y. Acad. Sci. 914, 238–262 [DOI] [PubMed] [Google Scholar]
- 67. Hughes S. M., Lillien L. E., Raff M. C., Rohrer H., Sendtner M. (1988) Ciliary neurotrophic factor induces type-2 astrocyte differentiation in culture. Nature 335, 70–73 [DOI] [PubMed] [Google Scholar]
- 68. Louis J. C., Magal E., Takayama S., Varon S. (1993) CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death. Science 259, 689–692 [DOI] [PubMed] [Google Scholar]
- 69. Mayer M., Bhakoo K., Noble M. (1994) Ciliary neurotrophic factor and leukemia inhibitory factor promote the generation, maturation, and survival of oligodendrocytes in vitro. Development 120, 143–153 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.