

Background: Calcium-independent phospholipase A2γ (iPLA2γ) is a mediator of complement-induced glomerular injury.
Results: Complement stimulated iPLA2γ through activation of mitogen-activated protein kinases.
Conclusion: Phosphorylation of iPLA2γ plays a role in activation and signaling.
Significance: Understanding the regulation of iPLA2γ activity is essential for developing novel therapeutic approaches to glomerular injury and proteinuria.
Keywords: Arachidonic Acid, Cell Signaling, Eicosanoids Biosynthesis, Kidney, MAP Kinases (MAPKs), Glomerulonephritis
Abstract
In experimental membranous nephropathy, complement C5b-9-induces glomerular epithelial cell (GEC) injury and proteinuria. The effects of C5b-9 are mediated via signaling pathways, including calcium-independent phospholipase A2γ (iPLA2γ), and mitogen-activated protein kinases (MAPKs) such as extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38. The iPLA2γ pathway is cytoprotective. This study addresses the mechanisms of iPLA2γ activation. iPLA2γ activity was monitored by quantifying prostaglandin E2 (PGE2) production. In GECs, iPLA2γ localized at the endoplasmic reticulum and mitochondria. Complement-mediated production of PGE2 was amplified in GECs that overexpress iPLA2γ, compared with control cells, and was blocked by the iPLA2γ inhibitor bromoenol lactone in both iPLA2γ-overexpressing and control GECs. In GECs that overexpress iPLA2γ, complement-mediated PGE2 production was reduced by inhibitors of MAP/ERK kinase 1 (MEK1) and p38 but not JNK. In COS-1 cells that overexpress iPLA2γ and cyclooxygenase-1, PGE2 production was induced by co-expression of constitutively active MEK1 or MAPK-interacting kinase 1 (MNK1) as well as by stimulation with epidermal growth factor (EGF) + ionomycin. Complement- and EGF + ionomycin-stimulated iPLA2γ activity was attenuated by the S511A/S515A double mutation. Moreover, complement and EGF + ionomycin enhanced phosphorylation of Ser-511. Thus, complement-mediated activation of iPLA2γ is mediated via ERK and p38 pathways, and phosphorylation of Ser-511 and/or Ser-515 plays a key role in the catalytic activity and signaling of iPLA2γ. Defining the mechanisms by which complement activates iPLA2γ provides opportunities for development of novel therapeutic approaches to GEC injury and proteinuria.
Introduction
Phospholipases A2 (PLA2s)3 comprise a family of enzymes that hydrolyze the acyl bond at the sn-2 position of phospholipids to yield free fatty acids such as arachidonic acid (AA), and lysophospholipids (1, 2). Both products represent precursors for signaling molecules that can exert multiple biological functions. AA can be converted into bioactive eicosanoids by the effect of cyclooxygenases (COX), lipoxygenases, and cytochrome P450 (3). Schaloske and Dennis (4) classified PLA2 enzymes into 15 groups based on their sequence homologies, and from a functional point of view these groups can be recombined into five principal types of PLA2s, secreted PLA2, cytosolic PLA2 (cPLA2), calcium-independent PLA2 (iPLA2), platelet-activating factor acetylhydrolases, and lysosomal PLA2. iPLA2s are members of group VI family of PLA2 enzymes (4). The first and most extensively studied enzyme in this group is VIA, which has two isoforms, iPLA2-VIA-1 and -2 (iPLA2β short and iPLA2β long). The second iPLA2 isoform, Group VIB (iPLA2γ), is homologous to iPLA2β in the C-terminal catalytic domain but shows no similarities in N-terminal region; accordingly, iPLA2γ may have distinct regulatory properties from those of iPLA2β (5, 6).
Various PLA2 enzymes have been shown to mediate pathways of cell injury in experimental disease models (7–11). For example, cPLA2α and its products are important mediators of complement-induced glomerular epithelial cell (GEC; podocyte) injury in the passive Heymann nephritis model of membranous nephropathy (12, 13). In passive Heymann nephritis, GECs (an important component of the glomerular permselectivity barrier) (14, 15) are targeted by the complement C5b-9 membrane attack complex, which leads to noncytolytic GEC injury (12, 13). Injury is associated with activation of diverse signaling pathways, which include phospholipases as well as protein kinases, proteinases, COX2, endoplasmic reticulum (ER) stress, reactive oxygen species, and others. These pathways contribute to changes in GEC lipid structure and function, actin cytoskeleton reorganization, and displacement of filtration slit diaphragm proteins, ultimately resulting in proteinuria (12, 13, 16). Recently, we demonstrated that C5b-9 stimulated a Ca2+-independent PLA2 activity in GECs, and complement-induced release of [3H]AA and prostaglandin E2 (PGE2) was amplified in GECs that overexpress iPLA2γ (13, 16). Furthermore, overexpression of iPLA2γ attenuated complement-induced GEC injury, and this effect was reversed by the iPLA2-directed inhibitor, bromoenol lactone (BEL) as well as indomethacin, suggesting that the cytoprotective effect of iPLA2γ was at least in part mediated by generation of prostanoids (16).
Although we and others have shown functional coupling of iPLA2γ with COX1, leading to prostanoid production (17), the functions of iPLA2γ have not been fully delineated (18). In resting cells, iPLA2s, including iPLA2β, are involved in the maintenance of membrane phospholipids by generating lysophospholipid acceptors that are reacylated with fatty acids. Therefore, iPLA2 plays a housekeeping role by facilitating phospholipid remodeling (19–21). Inhibition of iPLA2 in HEK293 and INS-1 cells altered the amounts of several phospholipids and resulted in decreased cell growth and p53 activation (22–24). During oxidative stress, an ER-associated iPLA2 in renal proximal tubular cells (iPLA2γ) recognizes, cleaves, and removes oxidized phospholipids from the ER membrane (25); thus, iPLA2γ may act to repair or prevent lipid peroxidation during oxidative stress (25). Another study demonstrated that expression of shRNA against iPLA2γ increased lipid peroxidation and induced apoptosis in renal cells (26). iPLA2 is involved in signal transduction pathways that include mitogen-activated protein kinase (MAPK) p38, epidermal growth factor (EGF) receptor, the tumor suppressor gene, p53, and cell cycle-regulator, p21 (27). A diverse array of cellular processes has been proposed to be regulated by iPLA2γ, including cellular proliferation (28), assembly of very low density lipoprotein (29), apoptosis (30), endothelial cell platelet activating factor synthesis (31), tumorigenesis, cell injury, and chemotaxis (7).
iPLA2γ is a membrane-bound enzyme that is reported to localize at the ER, peroxisomes, and mitochondria (25). These distinct sites of localization may be a result of specific domains in the structure of the enzyme (32). iPLA2γ gene transcription and translation appear complex, as distinct translation initiation sites, resulting in the production of 88-, 77-, 74-, and 63-kDa forms of the enzyme were reported (33). iPLA2γ contains a consensus site for nucleotide binding and a lipase consensus motif in its C-terminal half as well as potential cAMP-dependent protein kinase, protein kinase C, and extracellular signal-regulated kinase (ERK) phosphorylation sites (32). The lipase consensus motif GVSTG (amino acids 481–485 in the C-terminal region) is essential for Ca2+-independent PLA2 catalytic activity, and substitution of Ala for Ser-483 or Asp-627 results in loss of PLA2 activity (34). To date, it is not known if/how phosphorylation would affect iPLA2γ activity.
The goal of the present study was to further characterize the activation of iPLA2γ in complement-induced GEC injury. Specifically, we addressed the role of various kinases known to be activated by complement. In GECs, we demonstrate the subcellular localization of iPLA2γ at the ER and mitochondria, which was dependent on the N-terminal region of iPLA2γ. Complement-induced activation of iPLA2γ was mediated via ERK and p38 pathways. Stimulation of iPLA2γ was dependent on phosphorylation of Ser-511 and/or Ser-515 via MAPK-interacting kinase 1 (MNK1).
EXPERIMENTAL PROCEDURES
Materials
Tissue culture media, G418 (Geneticin), plasmid pRc/RSV, and Lipofectamine 2000 were from Invitrogen. Electrophoresis reagents were from Bio-Rad. Mouse monoclonal anti-green fluorescent protein (GFP), sheep anti-COX1, rabbit anti-MEK1 (C-18), and rabbit anti-MAP/ERK kinase kinase 1 (MEKK1) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-calnexin was from Assay Designs and Stressgen (Ann Arbor, MI). Mouse monoclonal anti-α-tubulin was from Sigma. Rabbit phospho-p44/42 MAPK (Thr-202/Tyr-204), rabbit anti-phospho-p38 (Thr-180/Tyr-182), rabbit anti-phospho-JNK (Thr-183/Tyr-185), and rabbit anti-phospho-Ser/Thr Akt substrate antibody (which recognizes the sequence (R/K)XX(pS/T)) were from Cell Signaling Technology (Danvers, MA). MitoTracker Red CMXRos was from Molecular Probes (Eugene, OR). Enhanced chemiluminescence (ECL) reagents were from GE Healthcare. BEL, CGP57380, PLA2 assay kits and PGE2 enzyme immunoassay kits were from Cayman Chemical (Ann Arbor, MI). PD98059 was from Calbiochem. Human iPLA2γ wild type (WT) in pcDNA 1.1 was kindly provided by Drs. Richard Gross and David Mancuso (Washington University School of Medicine, St. Louis, MO) (16). R4F-MEK cDNA was provided by Dr. Natalie Ahn (University of Colorado, Boulder, CO) (35). pFC-MEKK, the constitutively active form of MEKK1, and pFC-MKK3, the constitutively active form of MKK3, were from Stratagene (La Jolla, CA). C8 and C8-deficient serum were from Complement Technology (Tyler, TX). pcDNA3-myc-MAPK-activated protein kinase-2 (MK2) WT and constitutively active mutant cDNA3-myc-MK2-EE were kindly provided by Professor Matthias Gaestel (Institute of Biochemistry, Medical School, Hannover, Germany) (36). Constructs encoding glutathione S-transferase (GST)-tagged MNK1 (pEBG-MNK1), a constitutively active form of MNK1 (pEBG-T332D), dominant-interfering MNK1 mutant (pEBG-T2A2), and pEBG empty vector were kindly provided by Dr. Jonathan Cooper (Fred Hutchinson Cancer Research Center, Seattle, WA) (37). Other reagents were from Sigma.
Cloning and Construction of iPLA2γ Mutants
WT, full-length (782 amino acid; M1) human iPLA2γ in pcDNA 1.1 was digested with SalI and SacII and was subcloned into pEGFP-C1 vector (Clontech) at SalI and SacII restriction sites to produce M1 GFP-iPLA2γ WT (M1 denotes that the iPLA2γ cDNA sequence begins at the codon for the first methionine). Mutations in potential ERK phosphorylation sites (Ser-271 and Ser-168 to Ala as well as the double mutation) were constructed by PCR-based mutagenesis (primers are presented in Table 1). A double mutation in two other potential phosphorylation sites (Ser-511 and Ser-515 to Ala) was constructed using an analogous approach (Table 1). For construction of N-terminally truncated (M4) GFP-iPLA2γ (i.e. iPLA2γ cDNA sequence beginning at the codon for the 4th methionine, amino acid 221) PCR reactions were performed with primers M4-F1 in combination with R1 (Table 1). All GFP-iPLA2γ mutant cDNAs were verified by DNA sequencing.
TABLE 1.
PCR primers employed to construct iPLA2γ mutants
Bold letters denote base substitutions encoding for mutated amino acids.
| GFP-iPLA2γ | Primer | Primer sequence (5′ to 3′) |
|---|---|---|
| GFP-iPLA2γ S271A | F1 | CCGGAGCTCCTGCAGTCGACATGTCTATTAATCT |
| R2 | CGCAGAAGGAGCTGTAGGCTTG | |
| F2 | CAAGCCTACAGCTCCTTCTGCG | |
| R1 | CCTTGTTCCTCCACCATCAA | |
| GFP-iPLA2γ S168A | F1 | CCGGAGCTCCTGCAGTCGACATGTCTATTAATCT |
| R3 | GGAAAAGGAGCCTTTTCTGCTGAT | |
| F3 | GCAGAAAAGGCTCCTTTTCCAGAA | |
| GFP-iPLA2γ S511A/S515A | R1 | CCTTGTTCCTCCACCATCAA |
| F1 | GCGAGAAAAGATTATCGCAAG | |
| R4 | TTGTGCAAATACATCTGCTCCTAATTTTC | |
| F4 | AATTAGGAGCAGATGTATTTGCACAAAATGT | |
| R1 | GGGGTTTCTTGCTGTTTCAA | |
| M4 GFP-iPLA2γ | M4-F1 | CCGGAGCTCAAATGAAATGTCTCAACAAAAGGAAAATG |
| M4-R1 | CCTTGTTCCTCCACCATCAA |
Cell Culture and Transfection
Rat GEC culture and characterization have been described previously (38). GECs were maintained in K1 medium on plastic substratum. Cells were stably transfected with M1 or M4 GFP-iPLA2γ WT plasmids using Lipofectamine 2000 reagent according to the manufacturer's instructions. After selection with G418 and expansion, cells were sorted by flow cytometry to obtain cells with the highest expression of GFP-iPLA2γ WT. Fluorescence microscopy and immunoblotting were used to confirm GFP- iPLA2γ overexpression. A clone of GECs containing the neomycin-resistance gene was used as a control (GEC-Neo). COS-1 cells were cultured in DMEM, 10% fetal bovine serum and were transfected transiently with GFP-iPLA2γ and/or COX1 cDNAs using Lipofectamine 2000.
Incubation of GECs with Complement
GECs in monolayer culture were washed twice and incubated with rabbit anti-GEC antiserum (5% v/v) in modified Krebs-Henseleit buffer containing 145 mm NaCl, 5 mm KCl, 0.5 mm MgSO4, 1 mm Na2HPO4, 0.5 mm CaCl2, 5 mm glucose, and 20 mm Hepes, pH 7.4, for 30 min at 22 °C. The cells were then incubated for 40 min at 37 °C with normal human serum (NS, 2% v/v; with full complement activity) or heat-inactivated (decomplemented) human serum (HIS, 2% v/v; incubated at 56 °C for 60 min) in controls (39, 40).
PGE2 Assay
Stimulated iPLA2 enzymatic activity was monitored by measuring PGE2 production. After incubation, supernatants were collected to quantify PGE2. The amount of PGE2 released into supernatants was equivalent to that from cells plus supernatants, indicating that most PGE2 was released from cells into supernatants. PGE2 was quantified using an enzyme immunoassay kit according to the manufacturer's instructions. The range of the standard curve in the assay was 4–1000 pg of PGE2/100 μl of sample (41). PGE2 concentration was calculated according to standard formulas.
PLA2 Assay
PLA2 activity was measured in COS-1 cell extracts using a PLA2 activity assay kit according to the manufacturer's instructions and as described previously (42). In this assay, hydrolysis of arachidonoyl thiophosphatidylcholine at the sn-2 position by PLA2 releases a free thiol that is detected by 5,5′-dithio-bis-2-nitrobenzoic acid. Briefly, cells were homogenized in 50 mm Hepes, pH 7.4, containing 1 mm EDTA. Cell homogenates were cleared by centrifugation at 8500 × g for 10 min at 4 °C. The reaction was initiated by the addition of 2-arachidonoyl thiophosphatidylcholine to cell extracts in buffer containing 80 mm Hepes, pH 7.4, 150 mm NaCl, 4 mm Triton X-100, 30% glycerol, and 1 mg/ml BSA. Duplicate samples were incubated with and without 10 μm BEL. After 60 min at 22 °C, the reaction was terminated by the addition of 1 mm 5,5′-dithio-bis-2-nitrobenzoic acid, and the absorbance was measured at 450 nm. To determine iPLA2 activity, the optical density obtained in the presence of BEL was subtracted from the total optical density (42) (in control cells, ∼20% of PLA2 activity was inhibited by BEL). The value of the group with maximum iPLA2 activity was set to 1.0, and the iPLA2 activities of the other groups were calculated as percent of maximum.
Immunoblotting
Cells were lysed in ice-cold buffer containing 1% Triton X-100, 125 mm NaCl, 10 mm Tris, pH 7.4, 1 mm EGTA, 2 mm Na3VO4, 10 mm sodium pyrophosphate, 25 mm NaF, and protease inhibitor mixture (Roche Diagnostics). Equal amounts of lysate proteins were dissolved in Laemmli buffer and subjected to SDS-PAGE under reducing conditions. Proteins were then electrophoretically transferred onto a nitrocellulose membrane and blocked at room temperature for 60 min with 5% dry milk in buffer containing 10 mm Tris, pH 7.5, 50 mm NaCl, 2.5 mm EDTA, and 0.05% Tween 20. The membrane was then incubated with primary and secondary antibodies and developed with ECL.
Immunofluorescence Microscopy
GECs expressing GFP-iPLA2γ WT (M1 or M4) and GEC-Neo (control) were cultured on glass coverslips for 24 h. All reactions were carried out at 22 °C. To examine the localization of GFP-iPLA2γ WT at the ER, cells were fixed with 3% paraformaldehyde in PBS for 30 min and permeabilized with 0.1% Triton-X 100 in PBS for 30 min. After washing with PBS, GECs were incubated with rabbit anti-calnexin antiserum or normal rabbit serum (negative control) diluted in 3% BSA for 30 min. Cells were washed and incubated with rhodamine-conjugated goat anti-rabbit IgG in 3% BSA for 30 min. Nuclei were counter-stained with 4′6-diamindino 2-phenylindole (DAPI, 30 nm) in PBS for 4–5 min just before mounting the coverslips onto glass slides. Staining was visualized with a Zeiss AxioObserver fluorescence microscope with visual output connected to an AxioCam digital camera. To visualize mitochondria, GECs expressing GFP-iPLA2γ WT (M1 or M4) and GEC-Neo (control), on coverslips were incubated for 15 min at 37 °C with MitoTracker Red CMXRos (25 nm). Cells were then fixed with 3% (w/v) paraformaldehyde in PBS for 30 min. After washing, coverslips were mounted onto glass slides and visualized with a fluorescence microscope.
Statistics
Data are presented as the mean ± S.E. One-way analysis of variance was used to determine significant differences among groups. Where significant differences were found, individual comparisons were made between groups using the t-statistic and by adjusting the critical value according to Tukey's or Bonferroni's method. Statistical significance was considered to be p < 0.05.
RESULTS
M1 GFP-iPLA2γ WT Is Enzymatically Active
To study the effect of complement on iPLA2γ activation, we first established a subclone of GECs that stably overexpresses M1 GFP-iPLA2γ WT (M1 GEC-iPLA2γ). By immunoblotting, M1 GEC-iPLA2γ was expressed as a 115-kDa protein, consistent with GFP (27 kDa) fused with the 88-kDa isoform of iPLA2γ (Fig. 1A). When M1 GFP-iPLA2γ WT or untagged iPLA2γ was transfected in COS-1 cells together with COX1, PGE2 production was markedly increased compared with untransfected control (Fig. 1B) or cells transfected with COX1 alone (Fig. 1C). This increase was attenuated by the addition of BEL (Fig. 1C). Together the results indicate that M1 GFP-iPLA2γ WT is enzymatically active.
FIGURE 1.
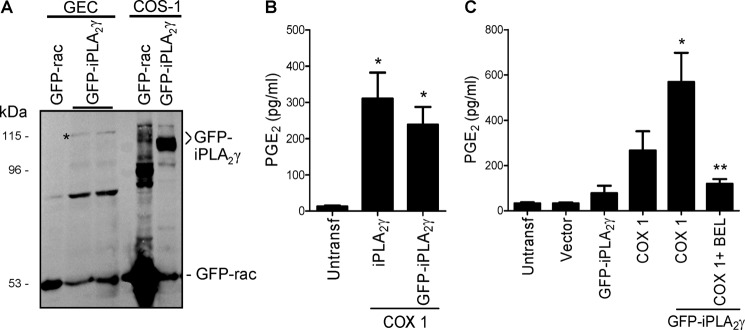
Expression and activity of M1 GFP-iPLA2γ WT. A, GECs were stably transfected, and COS-1 cells were transiently transfected with M1 GFP-iPLA2γ WT or a GFP-rac fusion protein for comparison. Lysates were immunoblotted with antibody to GFP. * denotes M1 GFP-iPLA2γ in GECs, which migrated slower than M1 GFP-iPLA2γ in COS-1 cells, possibly because of differential post-translational modifications in the two cell lines. B, COS-1 cells were transiently transfected with untagged full-length iPLA2γ, M1 GFP-iPLA2γ WT, and COX1. PGE2 production was measured in cell supernatants 24 h after transfection. Both GFP-tagged and untagged enzymes increased PGE2 release. *, p < 0.001 iPLA2γ + COX1 and GFP-iPLA2γ + COX1 versus untransfected (Untransf) cells, three experiments. C, COS-1 cells were untransfected or were transiently transfected with vector, M1 GFP-iPLA2γ WT, COX1, or M1 GFP-iPLA2γ WT + COX1. Expression of COX1 alone increased PGE2 release compared with control, whereas GFP-iPLA2γ + COX1 substantially amplified the increase in PGE2 production. This increase was inhibited by coincubation with 30 μm BEL (6 h). *, p < 0.001 GFP-iPLA2γ + COX1 versus untransfected or vector and p < 0.001 GFP-iPLA2γ + COX1 versus GFP-iPLA2γ, p < 0.05 GFP-iPLA2γ + COX1 versus COX1; **, p < 0.001 GFP-iPLA2γ + COX1 + BEL versus GFP-iPLA2γ + COX1, 7 experiments.
iPLA2γ Localizes at the ER and Mitochondria
The expression and localization of iPLA2 isoforms varies considerably among different cell types, and the role of iPLA2 may be determined by the isoform present in a specific cell or organelle (43). We examined the localization of M1 GFP-iPLA2γ WT in stably transfected GECs. By fluorescence microscopy, confluent monolayers of resting GECs demonstrated green fluorescent staining mainly in the perinuclear region (Fig. 2, A and C). A significant portion of the M1 GFP-iPLA2γ WT co-localized with calnexin (Fig. 2, B and D), indicating localization at the ER. In resting cells not expressing M1 GFP-iPLA2γ WT, calnexin staining showed a similar distribution in the perinuclear region, indicating that expression of M1 GFP-iPLA2γ WT did not affect the structure of the ER (data not shown).
FIGURE 2.

Subcellular localization of M1 GFP-iPLA2γ WT and N-terminally truncated GFP-iPLA2γ (M4). GECs stably transfected with M1 GFP-iPLA2γ WT show predominantly perinuclear green fluorescent staining (A). Cells were labeled with antibody to calnexin (red staining; B) to localize the ER, whereas nuclei were counterstained with DAPI (blue fluorescence; C). Calnexin staining was perinuclear (B). Panel D shows co-localization of M1 GFP-iPLA2γ WT and calnexin (yellow-orange staining). GECs that express M1 GFP-iPLA2γ WT (E) were stained with Mitotracker red, a marker of the mitochondria (F). Panel G shows co-localization of GFP-iPLA2γ with Mitotracker red. GECs expressing M1 GFP-iPLA2γ WT were treated with digitonin to permeabilize the plasma membranes (H). Digitonin treatment did not affect the localization of M1 GFP-iPLA2γ WT. GECs stably transfected with M4 GFP-iPLA2γ mainly show cytoplasmic green fluorescent staining (I and M). There is some minor perinuclear accentuation, and there appear to be aggregates in occasional cells. GECs were stained with anti-calnexin antibody (red staining; J), Mitotracker red (N), and DAPI (blue fluorescence; K and O). Panel L (merge of I–K) shows only minor co-localization of M4 GFP-iPLA2γ with calnexin. Panel P (merge of M–O) shows an absence of co-localization of M4 GFP-iPLA2γ with Mitotracker red.
A portion of the M1 GFP-iPLA2γ WT did not appear to co-localize with calnexin but localized at the mitochondria (Fig. 2, E–G). The appearance of the mitochondria was unaffected by the expression of M1 GFP-iPLA2γ WT (data not shown). Finally, we treated the GECs expressing M1 GFP-iPLA2γ WT with digitonin to permeabilize the plasma membranes and release cytosolic proteins into cell supernatants. Distribution of the GFP fluorescence in the digitonin-treated cells was similar to that seen in untreated (intact) cells (Fig. 2H), further supporting the association of iPLA2γ with intracellular organelles. Together the results shown in Fig. 2 indicate that in GECs M1 GFP-iPLA2γ WT is associated with at least two organelles, the ER and mitochondria.
N-terminally Truncated iPLA2γ (M4 GFP-iPLA2γ) Is Mislocalized
The catalytic domain of iPLA2γ is located in the C-terminal region of the protein, whereas the role of the N-terminal region is poorly defined. Indeed, a previous study showed that iPLA2γ contains four N-terminal methionine residues that may act as translation initiation sites, resulting in 88-, 77-, 74-, and 63-kDa forms of iPLA2γ in SF9 insect cells (33). To determine if the N-terminal region may be involved in localization and/or regulation of iPLA2γ catalytic activity, we deleted the 220 N-terminal amino acids (spanning between the 1st and 4th methionine) to generate a short form of iPLA2γ in which the GFP-iPLA2γ fusion would be at the 4th methionine (M4 GFP- iPLA2γ). Expression of M4 GFP-iPLA2γ in COS-1 cells showed a prominent band at ∼92 kDa (Fig. 3A) that represents GFP (27 kDa) fused with the 63-kDa form of iPLA2γ. When both M1 and M4 isoforms were expressed at serially increasing concentrations in COS-1 cells (together with COX1), PGE2 production by M1 GFP-iPLA2γ WT was markedly greater compared with M4 GFP-iPLA2γ despite weaker expression (Fig. 3, A–C). PGE2 production by M4 GFP-iPLA2γ was trivial, as it was not significantly greater compared with control cells.
FIGURE 3.
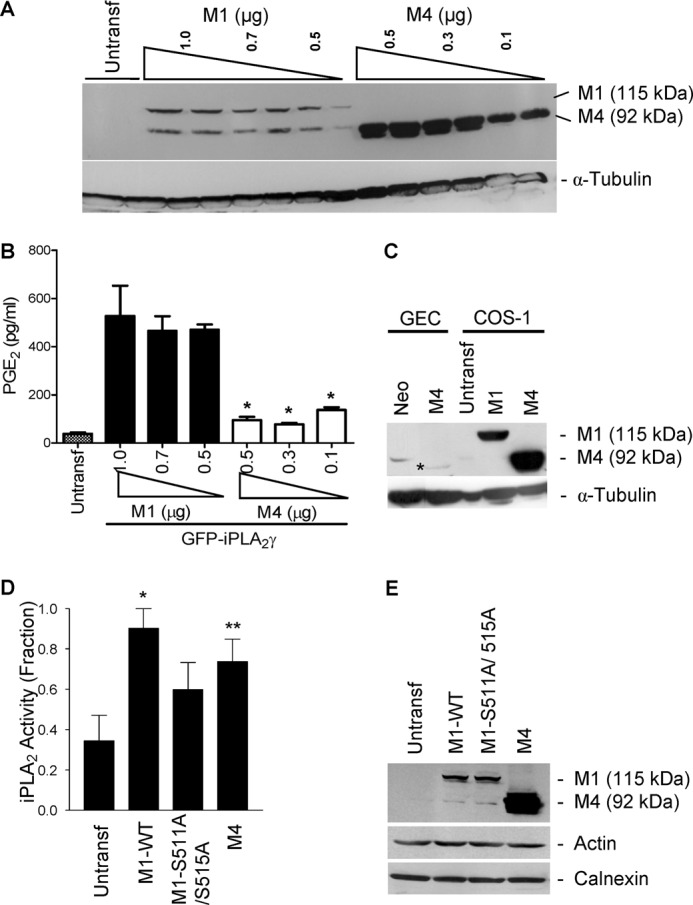
Expression and activity of GFP-iPLA2γ WT and mutants. COS-1 cells were co-transfected with N-terminally truncated (M4) GFP-iPLA2γ (0.1–0.5 μg of plasmid DNA) and for comparison with M1 GFP-iPLA2γ WT (0.5–1.0 μg of plasmid DNA), both with COX1. A, anti-GFP antibody immunoblot shows greater expression of M4 GFP-iPLA2γ compared with M1 GFP-iPLA2γ WT. Expression tended to increase with increasing doses of plasmid DNA. The lower band in the M1 lanes is nonspecific. B, PGE2 release in COS-1 cells expressing M4 GFP-iPLA2γ or M1 GFP-iPLA2γ WT (both with COX1) was normalized for corresponding protein expression. In these experiments basal PGE2 release (untransfected cells) was 38.2 pg/ml. PGE2 production by M1 GFP-iPLA2γ WT was markedly greater compared with M4 GFP-iPLA2γ. *, p < 0.0001 M4 + COX1 (0.5, 0.3, and 0.1 μg) versus M1 + COX1 (1, 0.7, and 0.5 μg), three experiments. C, GECs were stably transfected, and COS-1 cells were transiently transfected with M4 GFP-iPLA2γ. GEC-Neo and COS-1 cells transiently transfected with M1 GFP-iPLA2γ WT are presented for comparison. Lysates were immunoblotted with antibody to GFP. * denotes M4 GFP-iPLA2γ in GECs. The bands in GEC-Neo and untransfected COS-1 cells are nonspecific. D, PLA2 activity in untransfected (control) COS-1 cells and COS-1 cells expressing M1 GFP-iPLA2γ WT, M1 GFP-iPLA2γ S511A/S515A double mutant, and M4 GFP-iPLA2γ is shown. Cell extracts were prepared 24 h after transfection, and iPLA2 activity was monitored by release of AA from 2-arachidonoyl-phosphatidylcholine (“Experimental Procedures”). iPLA2 activities of M1 GFP-iPLA2γ WT and M4 GFP-iPLA2γ were significantly greater compared with control. iPLA2 activity of M1 GFP-iPLA2γ S511A/S515A tended to be greater than control. *, p < 0.01 M1 GFP-iPLA2γ WT versus control, **, p < 0.05 M4 GFP-iPLA2γ WT versus control, four experiments. In these experiments basal iPLA2 activity (control cells) was 0.69 nmol/min/ml. E, cell lysates were immunoblotted with antibodies to GFP (iPLA2γ), actin, or calnexin (marker of ER).
We also employed an in vitro assay to measure iPLA2 enzymatic activity in extracts of control COS-1 cells and COS-1 cells expressing M1 GFP-iPLA2γ WT or M4 GFP-iPLA2γ. Using 2-arachidonoyl phosphatidylcholine as substrate, M1 GFP-iPLA2γ WT activity was significantly greater than control. M4 GFP-iPLA2γ also demonstrated significant activity (Fig. 3D). Thus, M4 GFP-iPLA2γ is active in vitro but not in intact cells.
In the next set of experiments, we employed GECs stably transfected with M4 GFP-iPLA2γ (Fig. 3C) to examine the localization. M4 GFP-iPLA2γ was found mainly in the cytosol, and only a minor portion was co-localized with calnexin (Fig. 2, I–L). Some cells contained what appeared to be aggregates of M4 GFP-iPLA2γ (Fig. 2I). M4 GFP-iPLA2γ did not co-localize with Mitotracker red (Fig. 2, M–P). Thus, deletion of the N-terminal region of iPLA2γ leads to mislocalization of the enzyme away from the ER and mitochondria. Taken together, M4 GFP-iPLA2γ is enzymatically active but may not be functional in releasing AA and PGE2 in intact cells due to the mislocalization of the enzyme from the membrane containing the substrate.
Complement Induces Release of PGE2 in GECs That Overexpress iPLA2γ
A previous study demonstrated that in GECs, complement can induce release of [3H]AA via activation of iPLA2γ (16). Consistent with the previous results, when GEC-Neo cells were incubated with antibody and sublytic NS (to form C5b-9) or HIS in controls, PGE2 production increased significantly, which was inhibited by R-BEL, a specific inhibitor of iPLA2γ (Fig. 4A). Next, we compared PGE2 release in M1 GEC-iPLA2γ and GEC-Neo. Overexpression of M1 GFP-iPLA2γ WT did not affect the basal PGE2 production (during incubation with HIS). However, after incubation with complement, PGE2 release was significantly amplified in M1 GEC-iPLA2γ compared with GEC-Neo (Fig. 4B), and the complement-mediated PGE2 release in M1 GEC-iPLA2γ was almost completely inhibited by BEL (racemic mixture, non-selective inhibitor of iPLA2β and -γ) (Fig. 4B), in keeping with earlier results (16). Therefore, activation of complement is coupled with stimulation of iPLA2γ activity. To verify that the PGE2 release produced by antibody and NS was actually due to formation of C5b-9, antibody-sensitized M1 GEC-iPLA2γ were exposed to C8-deficient serum or C8-deficient serum reconstituted with C8 (40). C8-deficient serum alone had no significant effect on PGE2 production, whereas C8-deficient serum reconstituted with C8 increased PGE2 release significantly (Fig. 4C).
FIGURE 4.
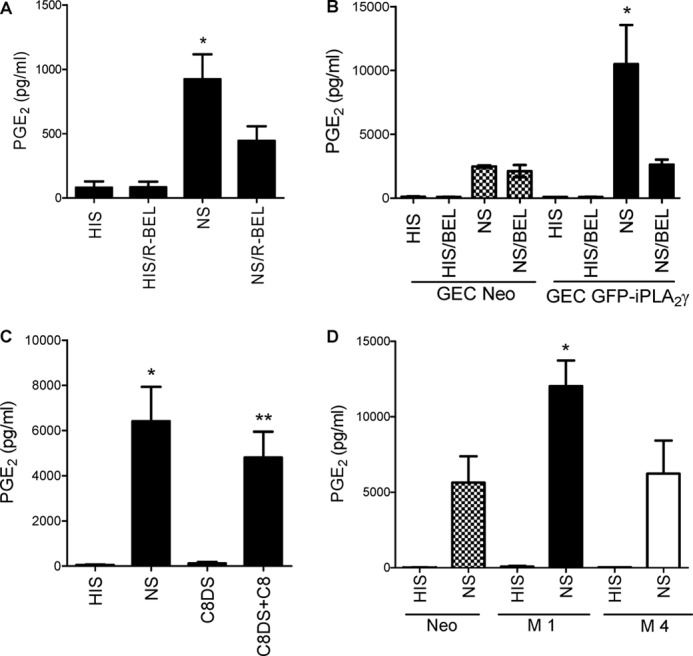
Complement induces production of PGE2 via iPLA2γ. A, shown is the role of endogenous iPLA2γ. Neo GECs were incubated with anti-GEC antiserum for 30 min at 22 °C in the presence or absence of the iPLA2γ-directed inhibitor R-BEL (10 μm). Cells were then incubated at 37 °C with 2% NS (to form C5b-9) or HIS in controls with or without R-BEL for 40 min. Then PGE2 production was measured in cell supernatants. Complement stimulated PGE2 production, and the increase was significantly attenuated by R-BEL. *, p < 0.0001 NS versus HIS and p < 0.001 NS versus NS/R-BEL, three experiments. B, complement-induced production of PGE2 is amplified in GECs that overexpress M1 GFP-iPLA2γ WT (GEC GFP-iPLA2γ). GECs that express M1 GFP-iPLA2γ WT were incubated with antibody and complement with or without BEL as above. M1 GFP-iPLA2γ WT markedly amplified complement-induced PGE2 production, and the increase was attenuated by BEL (30 μm). *, p < 0.001 GEC-GFP-iPLA2γ (NS) versus GEC-Neo (NS), p < 0.001 GEC-GFP-iPLA2γ (NS/BEL) versus GEC-GFP-iPLA2γ (NS), three experiments. C, PGE2 production is dependent on C5b-9 assembly. GECs that express M1 GFP-iPLA2γ WT were incubated with antibody and C8-deficient serum (C8DS) with or without purified C8. When C8DS was reconstituted with C8, PGE2 production amplified significantly. *, p < 0.0001 NS versus HIS; **, p < 0.001 C8DS+C8 versus C8DS, 3 experiments. D, M4 GFP-iPLA2γ is inactive in intact GECs. Neo GECs or GECs that stably express M1 GFP-iPLA2γ WT or M4 GFP-iPLA2γ were incubated with antibody and complement as above. *, p < 0.001 GEC-M1 GFP-iPLA2γ WT (NS) versus GEC-Neo (NS) and p < 0.001 GEC-M1 GFP-iPLA2γ WT (NS) versus GEC-M4 GFP-iPLA2γ (NS), three experiments.
We also tested the effect of complement on PGE2 release in the GECs stably expressing M4 GFP-iPLA2γ. In contrast to M1 GEC-iPLA2γ, the effect of complement on PGE2 production in the M4-expressing cells was not significantly different from GEC-Neo (Fig. 4D). The result indicates that M4 GFP-iPLA2γ is less active in the cell and is in keeping with the experiments involving transient transfection of the M1 and M4 forms of iPLA2γ in COS-1 cells (Fig. 3B).
Role of MAPKs in iPLA2γ Activation
The C5b-9 complex is assembled in the plasma membranes of cells, whereas iPLA2γ is localized at the ER and mitochondria, i.e. at organelles generally separated from the plasma membrane. It is, therefore, unlikely that C5b-9 interacts with iPLA2γ directly. Instead, it is reasonable to propose that C5b-9 may activate iPLA2γ via intermediary signals. C5b-9 has been shown to increase the cytosolic Ca2+ concentration and activate MAPKs, including ERK, JNK, and p38 (39). In the next series of experiments we examined if complement-induced activation of iPLA2γ was mediated through these MAPK pathways. We employed several MAPK pathway inhibitors including the MEK1 inhibitor, PD98059, the p38 inhibitor SB203580, and the JNK inhibitor SP600125 (44). M1 GEC-iPLA2γ were preincubated with each inhibitor and were then incubated with antibody and complement (Fig. 5A). The complement-induced release of PGE2 was inhibited significantly by SB203580 and PD98059. SP600125 tended to decrease the complement-mediated production of PGE2, but the effect was not significant. PD98059 and SB203580 were reported to cross-react and inhibit COX1 and COX2 activities (45). We, therefore, tested FR167653 and U0126, inhibitors of p38 and MEK1, respectively, which do not inhibit COX1 and COX2 (46) and are structurally distinct from SB203580 and PD98059. Both FR167653 and U0126 inhibited the complement-mediated production of PGE2 (Fig. 5B), confirming a role for p38 and ERK pathways in iPLA2γ activation by complement.
FIGURE 5.
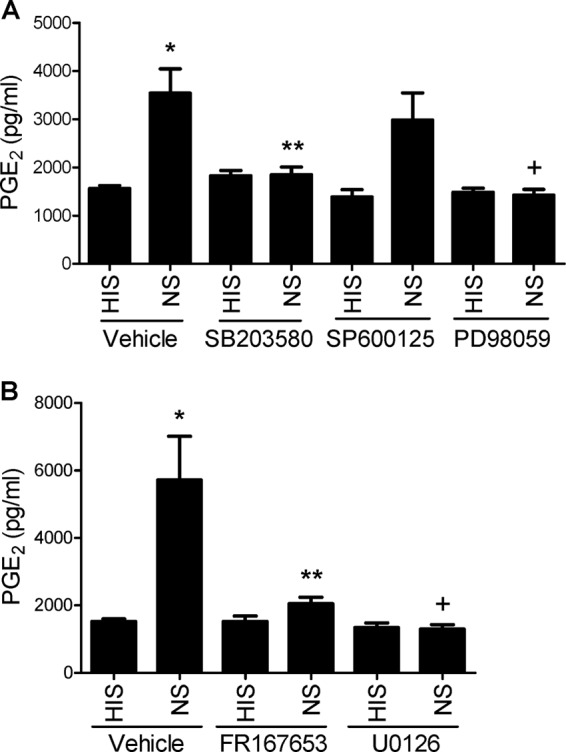
Complement-induced PGE2 production in GECs that stably express M1 GFP-iPLA2γ WT is mediated by ERK and p38. A, GECs were incubated with antibody and complement (as in Fig. 4) in the presence of inhibitors of p38 (SB203580; 10 μm), MEK1 (PD98059; 50 μm), or JNK (SP600125; 10 μm). Complement-induced PGE2 production was reduced significantly by the p38 and MEK1 inhibitors. *, p < 0.0001 NS versus HIS (vehicle); **p < 0.01 SB203580 versus vehicle (NS); +, p < 0.001 PD98059 versus vehicle (NS), six experiments. B, GECs were incubated with antibody and complement in the presence of inhibitors of p38 (FR167653; 10 μm) and MEK1 (U0126; 50 μm). Complement-induced PGE2 production was reduced significantly by both drugs. *, p < 0.0001 NS versus HIS (vehicle); **, p < 0.001 FR167653 versus vehicle (NS); +, p < 0.0001 U0126 versus vehicle (NS), four experiments.
MAPKs Enhance iPLA2γ Activity in COS-1 Cells
To confirm the role of MAPK pathways in the regulation of iPLA2γ activity, we transiently co-transfected COS-1 cells with M1 GFP-iPLA2γ WT, COX1, and constitutively active mutants of MEK1 (kinase upstream of ERK), MEKK1 (kinase upstream of JNK and possibly p38), and MKK3 (kinase upstream of p38). All three constitutively active mutants enhanced PGE2 production compared with control (vector) (Fig. 6).
FIGURE 6.
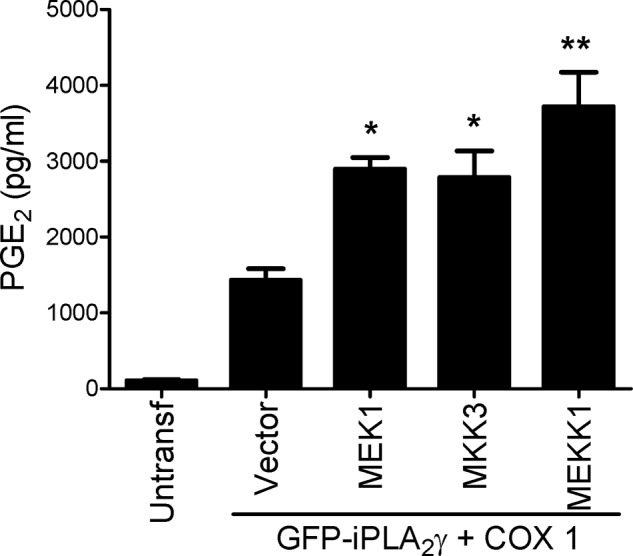
Activation of MAPK pathways stimulates iPLA2γ-mediated PGE2 production. COS-1 cells were co-transfected with M1 GFP-iPLA2γ WT, COX1, and constitutively active mutants of MEK1, MKK3, or MEKK1 or with empty vector. Untransfected cells are additional controls. PGE2 release was measured 48 h after transfection. *, p < 0.01; **, p < 0.0001 versus vector, three experiments.
The expression and function of the constitutively active mutants were evaluated in the same sets of experiments. Constitutively active MEK1 migrated slightly faster than the endogenous MEK1 (45 kDa) (Fig. 7A). ERK phosphorylation (which reflects ERK activation) was increased 1.7-fold by the constitutively active MEK1 (Fig. 7, A and B), confirming functional activity. Constitutively active MEKK1 was expressed as a protein of ∼35 kDa (Fig. 7C), and it stimulated phosphorylation of JNK as expected by ∼3-fold (Fig. 7, C and D). In addition, the constitutively active MEKK1 activated the ERK pathway by 2.5-fold (Fig. 7B) and showed modest but significant activation of the p38 pathway (Fig. 7, E and F), consistent with earlier studies showing that when overexpressed, the MEKK1 mutant can activate ERK and p38 (47). Unexpectedly, we could not detect phosphorylation of p38 after transfection of the constitutively active mutant of MKK3 (Fig. 7, E and F) even though MKK3 increased PGE2 release (Fig. 6). Possibly, phosphorylation of p38 was very transient, limiting its detectability. In summary, the results support a role for ERK in the activation of iPLA2γ, as constitutively active protein kinases that stimulated ERK phosphorylation also stimulated iPLA2γ activity, and complement-stimulated iPLA2γ activity was blocked by ERK pathway-directed inhibitors. By analogy, p38 may also stimulate iPLA2γ, as constitutively active MEKK1 stimulated iPLA2γ and p38 phosphorylation, and complement-stimulated iPLA2γ activity was blocked by p38 inhibitors. However, the role of MKK3 could not be established definitively and will require additional study. Finally, the results do not support a role for JNK in the activation of iPLA2γ.
FIGURE 7.

Effects of constitutively active kinases on ERK, JNK, and p38 phosphorylation. COS-1 cells were co-transfected with M1 GFP-iPLA2γ WT, COX1, and constitutively active mutants of MEK1, MKK3, and MEKK1 or with empty vector. A, lysates were immunoblotted with antibodies to MEK1 (upper panel) or phospho-ERK (pERK; lower panel). Endogenous MEK1 is present in all samples; constitutively active MEK1 (R4F-MEK1) migrates ahead of endogenous MEK1 (MEK1; 45 kDa). Both constitutively active MEK1 and MEKK1 stimulated ERK phosphorylation. B, densitometric quantification of pERK is shown. *, p < 0.01 MEK1 versus vector; **, p < 0.0001 MEKK1 versus vector, three experiments. C, lysates were immunoblotted with antibodies to MEKK1 (upper and middle panels) or phospho-JNK (pJNK; lower panel). Endogenous MEKK1 is present in all samples; constitutively active MEKK1 migrates at 35 kDa. Constitutively active MEKK1 stimulated JNK phosphorylation. D, densitometric quantification of pJNK is shown. *, p < 0.0001 MEKK1 versus vector, three experiments. E, lysates were immunoblotted with antibodies to phospho-p38 (pp38; upper panel) or tubulin (lower panel). Constitutively active MEKK1 stimulated p38 phosphorylation. F, densitometric quantification of pp38. *, p < 0.001 MEKK1 versus vector, three experiments.
EGF Together with Ionomycin Enhances iPLA2γ Activity in COS-1 Cells
EGF is a well known activator of the ERK pathway, and COS-1 cells express abundant EGF receptor. To further substantiate the role of the ERK pathway in the activation of iPLA2γ, we investigated the effect of EGF on PGE2 release in COS-1 cells that were transiently transfected with M1 GFP-iPLA2γ WT (and COX1). Treatment of COS-1 cells with EGF alone did not affect PGE2 release (Fig. 8A). Although iPLA2 is Ca2+-independent (does not require Ca2+ for its catalytic activity), iPLA2 activity may nonetheless be regulated by Ca2+ or a Ca2+-dependent factor (48). For this reason, we used the Ca2+ ionophore ionomycin to induce a Ca2+ influx. Interestingly, stimulation with EGF in the presence of ionomycin increased PGE2 release by more than 4-fold compared with EGF alone, ionomycin alone, or untreated (Fig. 8A). Furthermore, BEL inhibited iPLA2γ activity induced by EGF + ionomycin significantly (Fig. 8B). Finally, we confirmed that incubation of COS-1 cells with EGF + ionomycin induced ERK phosphorylation (Fig. 8C). Thus, the effect of ionomycin + EGF on iPLA2γ activation is analogous to the effect of C5b-9, which also induces a Ca2+ influx and activation of ERK.
FIGURE 8.

EGF together with ionomycin enhances iPLA2γ activity. COS-1 cells were co-transfected with M1 GFP-iPLA2γ WT and COX1. After 24 h COS-1 cells were incubated with EGF (100 ng/ml) and/or ionomycin (Iono, 1.0–1.5 μm) for 40 min. A, stimulation by EGF and ionomycin significantly increased PGE2 release compared with unstimulated cells or cells stimulated with each agonist alone. *, p < 0.001 EGF + ionomycin versus untreated, p < 0.05 EGF + ionomycin versus ionomycin (alone), p < 0.001 EGF + ionomycin versus EGF (alone), four experiments. B, COS-1 cells were co-transfected with M1 GFP-iPLA2γ WT and COX1. After 24 h 1 group of cells was preincubated with BEL (30 μm) for 6.5 h. Then cells were incubated for 30 min with EGF and ionomycin with or without BEL. The increase of PGE2 release was inhibited almost completely in presence of BEL. *, p < 0.01 EGF + ionomycin versus untreated. *, p < 0.01 EGF + ionomycin versus EGF + ionomycin + BEL, 3 experiments. C, cell lysates were immunoblotted with antibodies to phospho-ERK (pERK) or tubulin. EGF and ionomycin enhanced phosphorylation of ERK.
Mutations in Putative ERK Phosphorylation Sites Do Not Affect iPLA2γ Activity
Analysis of the iPLA2γ protein sequence by the Scansite program (49) suggested that Ser-168 (EKSP amino acid motif) and Ser-271 (PTSP motif) may be ERK phosphorylation sites. Given the substantial evidence for the activation of iPLA2γ via the ERK pathway, in the next series of the studies we examined if iPLA2γ may be a direct target of ERK. We constructed three mutant forms of M1 GFP-iPLA2γ, including S168A, S271A, and S168A/S271A double mutation, and tested their activities in COS-1 cells. PGE2 production stimulated by EGF + ionomycin with the iPLA2γ mutants did not differ from the WT (Fig. 9A), whereas the expression levels of all constructs were comparable (Fig. 9B). Thus, single or double mutations of putative ERK phosphorylation sites did not affect iPLA2γ activation by EGF + ionomycin, suggesting that the effect of ERK on the stimulation of iPLA2γ is indirect.
FIGURE 9.
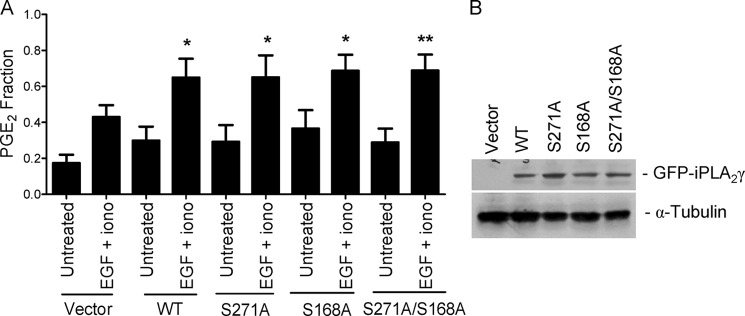
Mutations in putative ERK phosphorylation sites do not affect iPLA2γ activity. COS-1 cells were transiently transfected with M1-GFP-iPLA2γ WT, the S168A and S271A mutants, or the S168A/S271A double mutant together with COX1. A, after 24 h, cells were untreated or were incubated with EGF (100 ng/ml) + ionomycin (Iono, 1.5 μm) for 40 min. PGE2 release was stimulated significantly by EGF + ionomycin in the cells expressing M1 GFP-iPLA2γ WT and all mutants. *, p < 0.05; **, p < 0.01 versus corresponding untreated groups, seven experiments. In these experiments basal PGE2 release (vector + COX1-transfected, untreated cells) was 827 pg/ml. B, cell lysates were immunoblotted with antibodies to GFP or tubulin. The blot shows comparable levels of expression.
Mutations in Ser-511 and Ser-515 Inhibit iPLA2γ Activation
A recent phosphoproteomic analysis of mitochondrial proteins in murine heart revealed two iPLA2γ phosphorylation sites, Ser-505 and Thr-509, corresponding to Ser-511 and Ser-515 in human iPLA2γ (50). The authors suggested that the two phosphorylation sites (RKLGpSDVFpSQNV) may be in the context of MK2 or casein kinase I substrate motifs (50). Based on these results, we constructed a S511A/S515A double mutant form of M1 GFP-iPLA2γ and tested its activity in COS-1 cells. The in vitro enzymatic activity of M1 GFP-iPLA2γ S511A/S515A in COS-1 cells tended to be greater compared with control but was lower compared with M1 GFP-iPLA2γ WT (Fig. 3D). In addition, PGE2 production stimulated by EGF + ionomycin was attenuated significantly in the COS-1 cells expressing the S511A/S515A double mutant compared with WT (Fig. 10A), whereas the expression levels of WT and double mutant were comparable (Fig. 10B). Basal PGE2 levels in iPLA2γ WT and S511A/S515A-expressing cells appeared comparable (Fig. 10A).
FIGURE 10.

The S511A/S515A double mutation inhibits stimulated iPLA2γ activity. COS-1 cells were transiently transfected with M1-GFP-iPLA2γ WT, GFP-iPLA2γ S511A/S515A mutant, or empty vector together with COX1. A, after 24 h cells were untreated or were incubated with EGF (100 ng/ml) + ionomycin (Iono, 1.5 μm) for 40 min. PGE2 release was stimulated significantly by EGF + ionomycin in the cells expressing M1 GFP-iPLA2γ WT. *, p < 0.0001 versus corresponding untreated group. The increase in PGE2 release was smaller in cells expressing GFP-iPLA2γ S511A/S515A mutant. **, p < 0.01 versus cells expressing M1 GFP-iPLA2γ WT and treated with EGF + ionomycin, four experiments. In these experiments basal PGE2 release (vector + COX1-transfected, untreated cells) was 191 pg/ml. B, cell lysates were immunoblotted with antibodies to GFP or tubulin. The blot shows comparable levels of expression. C, cell lysates were immunoprecipitated with anti-GFP antibody (+) and were immunoblotted with anti-(R/K)XX(pS/T) or anti-GFP antibodies. The blot shows enhanced phosphorylation of Ser-511 (pS511) in the M1-GFP-iPLA2γ WT in EGF + ionomycin-stimulated cells. Phosphorylation of Ser-511 is faint in unstimulated WT-expressing cells and is absent in the mutant. D, shown are total lysates of the above immunoprecipitation experiments blotted with anti-GFP antibody.
Next, we tested if Ser-511 in iPLA2γ WT was phosphorylated by EGF + ionomycin stimulation using an antibody that identifies the (R/K)XX(pS/T) motif, corresponding to KLGpS in iPLA2γ. Phosphorylation of Ser-511 was evident in stimulated cells; in some experiments, faint phosphorylation was detected in unstimulated cells (Fig. 10C). In contrast, the S511A/S515A mutant iPLA2γ was not phosphorylated by EGF + ionomycin (Fig 10C), confirming that in the WT enzyme Ser-511 is the relevant phosphorylation site. These results suggest that the iPLA2γ activation by EGF + ionomycin is mediated by the direct phosphorylation of iPLA2γ by a kinase, which is downstream of ERK. Phosphorylation of Ser-515 in iPLA2γ WT was not monitored due to the unavailability of an antibody that identifies the DVFpSQ motif.
In a previous study we demonstrated that complement induced a robust activation-specific phosphorylation of MK2 (∼4-fold above control) and that activation of MK2 was mediated by both ERK and p38 kinase pathways (51). Indeed, MK2 is regarded primarily as a substrate of p38 kinase but is also reported to be a substrate of ERK (44, 51). Given that Ser-511 and Ser-515 may represent a MK2 phosphorylation motif, we investigated if expression of MK2 would stimulate the activity of iPLA2γ WT. By analogy to constitutively active MEK1 and MEKK1 (as shown in Fig. 6), WT and constitutively active MK2 were expressed with iPLA2γ WT in COS-1 cells. Despite robust expression, neither WT nor constitutively active MK2 stimulated PGE2 production consistently (data not shown).
Another protein kinase that is activated by ERK and p38 is MNK1 (44). To determine if the effect of the ERK pathway on iPLA2γ activation was mediated by MNK1, we expressed GFP-iPLA2γ in COS-1 cells and examined PGE2 production after stimulation with EGF + ionomycin in the presence or absence of the MNK1-directed inhibitor CGP57380 (44). Stimulated PGE2 production was blocked completely by CGP57380 (Table 2). CGP57380 was reported to have some inhibitory activity against MEK1 (44); however, we verified that EGF-induced phosphorylation of ERK was not reduced in the presence of CGP57380 (result not shown). Therefore, inhibition of the EGF + ionomycin-induced activation of iPLA2γ was most likely due to the inhibition of MNK1.
TABLE 2.
Effect of the MNK1 inhibitor, CGP57380, on PGE2 production
COS-1 cells were co-transfected with M1 GFP-iPLA2γ WT and COX1. After 24 h some cells were treated with CGP57380 (20 μm) for 16 h. Then cells were incubated with EGF + ionomycin (see the legend to Fig. 8) for 40 min. Stimulated PGE2 production was inhibited by CGP57380. Basal PGE2 release (untransfected, untreated cells) was 51 pg/ml.
| Transfection/treatment | PGE2 fraction |
|---|---|
| Untransfected, untreated | 0.13 ± 0.02 |
| Untransfected + CGP57380 | 0.17 ± 0.04 |
| GFP-iPLA2γ + COX, untreated | 0.65 ± 0.04 |
| GFP-iPLA2γ + COX, EGF + ionomycin | 0.91 ± 0.04 |
| GFP-iPLA2γ + COX, EGF + ionomycin + CGP57380 | 0.56 ± 0.07a |
a p < 0.001 EGF + ionomycin + CGP57380 vs. EGF + ionomycin, three experiments.
To confirm that MNK1 can activate and phosphorylate iPLA2γ, COS-1 cells were transfected with WT GFP-iPLA2γ and COX1 in the presence of WT, constitutively active (T332D), or dominant interfering (T2A2) forms of MNK1 or with empty vector. Both MNK1 WT and T332D enhanced PGE2 production compared with control (vector), whereas MNK1 T2A2 did not show a significant effect (Fig. 11, A and B). We tested Ser-511 phosphorylation by constitutively active MNK1 in iPLA2γ WT using the anti-(R/K)XX(pS/T) antibody. Phosphorylation of Ser-511 was enhanced when COS-1 cells were cotransfected with MNK1 T332D compared with empty vector (Fig. 11, C and D). Together these results support the view that phosphorylation of iPLA2γ on Ser-511 in response to EGF stimulation is mediated by MNK1.
FIGURE 11.

Constitutively active MNK1 activates and phosphorylates iPLA2γ. COS-1 cells were transiently co-transfected with M1 GFP-iPLA2γ WT, COX1 and GST-MNK1 WT, GST-MNK1 T332D, and GST-MNK1 T2A2 or with empty vector. A, PGE2 release was measured 48 h after transfection. *, p < 0.05 MNK1 WT versus vector; *, p < 0.01 MNK1 T332D versus vector and **, p < 0.05 MNK1 T332D versus MNK1 T2A2, five experiments. In these experiments basal PGE2 release (vector + M1 GFP-iPLA2γ WT + COX1-transfected cells) was 164 pg/ml. B, lysates were immunoblotted with antibodies to GFP, GST, or actin. The blot shows comparable levels of expression. C, COS-1 cells were transiently co-transfected with M1 GFP-iPLA2γ WT and GST-MNK1 T332D or vector. After 48 h cells were treated with ionomycin (10 μm, 40 min) (ionomycin was included in these experiments to enhance the phosphorylation signal, although ionomycin did not independently induce phosphorylation; see Fig. 12). Cell lysates were immunoprecipitated with anti-GFP antibody (+) or nonimmune IgG in controls (−) and were immunoblotted with anti-(R/K)XX(pS/T) or anti-GFP antibodies. The blot shows enhanced phosphorylation of iPLA2γ Ser-511 (pS511) in MNK1 T332D transfected cells. D, total lysates of the above immunoprecipitation experiments blotted with anti-GFP or anti-GST (MNK1) antibodies are shown.
Complement Induces Phosphorylation of iPLA2γ on Ser-511
In these experiments we assessed if complement-mediated activation of iPLA2γ involves phosphorylation. First, we tested if Ser-511 in iPLA2γ WT is phosphorylated by complement in GECs using the anti-(R/K)XX(pS/T) antibody. Phosphorylation of Ser-511 was stimulated in complement-treated GECs (NS) compared with control (HIS; Fig. 12A). In some experiments ionomycin was included together with NS and HIS incubations; however, ionomycin did not modulate Ser-511 phosphorylation either in complement-treated cells or control (Fig. 12A). Because complement increases the cytosolic Ca2+ concentration (16, 52), ionomycin would not be expected to provide any additional stimulatory effect. Second, we compared complement-induced PGE2 release in GECs expressing M1 GFP-iPLA2γ WT or GFP-iPLA2γ S511A/S515A and GEC-Neo. Overexpression of M1 GFP-iPLA2γ WT or the S511A/S515A mutant did not affect the basal PGE2 production (during incubation with HIS). After incubation with complement (NS), PGE2 release was significantly amplified in cells expressing M1 GFP-iPLA2γ compared with GEC-Neo (Fig. 12B), in keeping with previous experiments (Fig. 4, B and D). PGE2 release in complement-treated cells expressing the S511A/S515A mutant was significantly lower compared with WT (Fig. 12B) despite comparable levels of expression (Fig. 12C). Together, these results indicate that complement at least in part activates iPLA2γ via direct phosphorylation on Ser-511, most likely by MNK1.
FIGURE 12.
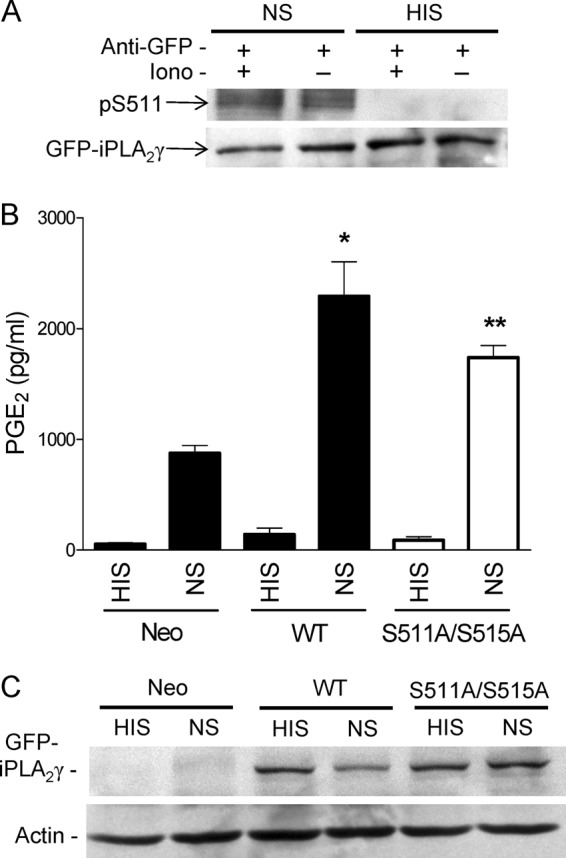
Complement induces phosphorylation of iPLA2γ on Ser-511. GECs were transiently transfected with M1 GFP-iPLA2γ WT. After 24 h cells were incubated with ionomycin (Iono, 5 μm) for 30 min (+) and then incubated with antibody and NS (3%, 40 min) or HIS in controls. A, cell lysates were immunoprecipitated with anti-GFP antibody (+) and were immunoblotted with anti-(R/K)XX(pS/T) or anti-GFP antibodies. The blot shows phosphorylation of Ser-511 (pS511) in NS-stimulated cells (with or without ionomycin). Phosphorylation is absent in the HIS-stimulated cells. B, GEC neo and GECs that express M1 GFP-iPLA2γ WT or GFP-iPLA2γ S511A/S515A (transient transfection) were incubated with antibody and complement as above. PGE2 production was amplified in NS-stimulated M1 GFP-iPLA2γ WT-expressing cells, whereas the amplification was smaller in GECs expressing the double mutant. *, p < 0.001 M1 GFP-iPLA2γ WT (NS) versus GEC-Neo (NS) and **, p < 0.05 M1 GFP-iPLA2γ WT (NS) versus GFP-iPLA2γ S511A/S515A (NS), three experiments. C, cell lysates were immunoblotted with antibodies to GFP or actin. The blot shows comparable levels of GFP-iPLA2γ expression.
DISCUSSION
The present study demonstrates that complement C5b-9 activates endogenous and ectopic iPLA2γ (Fig. 4). Activation of iPLA2γ occurs via ERK and p38 pathways (Fig. 5–9) and is dependent on an increase in cytosolic Ca2+ concentration and phosphorylation of the enzyme on Ser-511 and/or Ser-515 most likely via MNK1 (Fig. 11, Table 2). To our knowledge this is the first demonstration of phosphorylation-dependent activation of iPLA2γ. Stimulation of iPLA2γ by complement was coupled with production of PGE2. Similar to this result in the present study, iPLA2γ was shown to augment AA release and PGE2 production, which was associated with increased cell growth in a human colorectal adenocarcinoma cell line (HAC-7) (17). Coupling of iPLA2γ with COX1 was also shown in HEK293 cells (17).
In the present study we examined the cellular localization of iPLA2γ to better understand the mechanisms by which iPLA2γ hydrolyzes membrane phospholipids. M1 GFP-iPLA2γ WT was found mainly in the perinuclear region and co-localized with markers of the ER and mitochondria (Fig. 2). Deletion of the 220-amino acid N-terminal region (M4 GFP-iPLA2γ) altered the localization of the enzyme such that a significant portion of M4 GFP-iPLA2γ shifted to the cytosol (Fig. 2). Our result is in keeping with the study of Tanaka et al. (34), which showed that deletion of a 362-amino acid N-terminal region of iPLA2γ caused a portion of the enzyme to shift from the membrane to the cytosol in a cell fractionation study. Moreover, the N-terminal fragment localized solely in the membrane but not in the cytosolic fraction (34). Deletion of the N-terminal region of iPLA2γ did not abolish iPLA2γ enzymatic activity in vitro (Fig. 3D), consistent with the study by Tanaka et al. (34). However, the N-terminal truncated form of iPLA2γ was unable to induce significant PGE2 production when expressed in cells (Fig. 3). Taken together, these results indicate that the N-terminal region is involved in the membrane association of iPLA2γ, thereby allowing the enzyme access to phospholipid substrate intracellularly. In addition, the N-terminal region may have positive regulatory elements that could enhance iPLA2γ activity. Mislocalization of M4 GFP-iPLA2γ from the membrane may be expected to reduce PGE2 production, as COX1 is localized in the ER membrane and would couple with iPLA2γ-mediated AA release (17).
In previous studies it was demonstrated that C5b-9 can activate cPLA2α to augment production of [3H]AA and prostanoids (12, 13). Complement did not stimulate group IIA-secreted PLA2, and in contrast to iPLA2γ, overexpression of iPLA2β in GECs did not amplify complement-dependent release of [3H]AA (16, 52). Thus, both cPLA2α and iPLA2γ can contribute to complement-dependent release of AA. Previous studies in GECs demonstrated that complement induced an increase in cPLA2α catalytic activity, in association with Ser-505 phosphorylation, although this phosphorylation was not essential for cPLA2α activation (12, 13). In addition, glomerular cPLA2α was phosphorylated in vivo in passive Heymann nephritis (51).
By analogy to cPLA2α, complement activated iPLA2γ through intermediate signals, including protein kinases. C5b-9 can activate MAPK pathways in GECs (12, 13). In GECs overexpressing GFP-iPLA2γ WT, the complement-induced release of PGE2 was blocked by two distinct chemical inhibitors of both the ERK and p38 pathways but not JNK (Fig. 5). Conversely, constitutively active MAPK pathway mutants (in particular MEK and MEKK1) increased iPLA2γ-dependent PGE2 production (Fig. 6). Both MEK and MEKK1 induced activation-specific phosphorylation of ERK (Fig. 7). Taken together, the ERK and p38 pathways can mediate the activation of iPLA2γ by complement. A role for MAPKs in the activation of iPLA2 has been reported previously. Thrombin stimulated both ERK and p38 and iPLA2 activity in vascular smooth muscle cells and ventricular myocytes (53, 54). In mouse neural cells, p38 facilitated iPLA2 activity during hypoxia (55). These studies did not, however, differentiate between iPLA2β and iPLA2γ. To further substantiate the role of the ERK pathway in the activation of iPLA2γ, we showed that EGF + ionomycin stimulated PGE2 release in COS-1 cells expressing GFP-iPLA2γ WT in association with ERK activation (Figs. 8 and 9). Interestingly, unlike the constitutively active mutants of MEK1, MKK3, or MEKK1, the stimulatory effect of EGF in cells required the addition of ionomycin (to increase the cytosolic Ca2+ concentration) despite the Ca2+-independent catalytic properties of iPLA2γ in vitro. In keeping with previous reports, this finding suggests that activation of iPLA2γ in agonist-stimulated cells may involve a Ca2+-regulated process (5, 6), possibly activation of Ca2+-dependent protein kinases, such as calmodulin (56). Alternatively, Ca2+ may enhance activation of iPLA2γ directly. A recent study showed that divalent cations (Ca2+ or Mg2+) can activate iPLA2γ in heart mitochondria, leading to release of eicosanoids and lysolipids, possibly by facilitating mitochondrial phospholipid hydrolysis by iPLA2γ (57).
Tanaka et al. (6) suggested that iPLA2γ may have multiple potential phosphorylation sites. We carried out a mutagenesis analysis of iPLA2γ to determine the regulation of iPLA2γ activity by the ERK pathway. Mutation of two putative ERK phosphorylation sites, i.e. S168A and S271A, and S168A/S271A double mutation did not abrogate the stimulated activity of iPLA2γ WT (Fig. 9), implying that iPLA2γ was not a direct target of ERK. Another report suggested that the MAPK pathway might be involved in iPLA2γ activation indirectly, but no supporting data were presented (17). Based on a phosphoproteomic analysis of murine cardiac mitochondrial proteins (50), we then mutated Ser-511 and Ser-515 to Ala and showed that this double mutation significantly attenuated EGF + ionomycin- as well as complement-stimulated iPLA2γ-dependent PGE2 production (Figs. 10 and 12). Moreover, phosphorylation of Ser-511 was induced by EGF + ionomycin and by complement (Figs. 10 and 12). Phosphorylation of Ser-511 and/or Ser-515 could induce a conformational change in the enzyme, leading to an increase in catalytic activity. Further studies will be required to define the mechanism more precisely.
The Ser-511 and Ser-515 phosphorylation sites were proposed to be a phosphorylation motif for MK2 (50), and given that complement was shown to activate MK2 via the ERK or p38 pathway (51), it was reasonable to examine if MK2 may be the kinase downstream of ERK that phosphorylates iPLA2γ. However, we were not able to show stimulation of iPLA2γ after MK2 overexpression. The protein kinase MNK1 was also activated by ERK and p38, and the amino acid sequence preceding Ser-511 in iPLA2γ (KLGS) resembles the MNK1 phosphorylation motif in eukaryotic translation initiation factor 4E (KSGS) (44). In the present study the EGF + ionomycin-stimulated activity of iPLA2γ was blocked by a MNK1-directed inhibitor (Table 2). Expression of WT and constitutively active MNK1 stimulated PGE2 release via iPLA2γ, and constitutively active MNK1 enhanced Ser-511 phosphorylation (Fig. 11). Thus, activation of iPLA2γ by complement most likely involves an ERK-MNK1 pathway, although an additional role of another kinase downstream of ERK remains a possibility. Interestingly, a previous study has shown that phosphorylation and activation of cPLA2α can occur via p38 and MNK1 (58).
Various PLA2 enzymes have been shown to regulate pathways leading to cell injury in experimental disease models. These effects may be associated with generation of prostanoids (59), p38 activation (60), and induction of ER stress (40). There are both cytoprotective and injurious consequences related to the complement-mediated activation of PLA2 enzymes and production of prostanoids (13, 16). Overexpression of iPLA2γ attenuated complement-mediated injury in cultured GECs, and the cytoprotective effect was in part mediated through prostaglandin production (16). Further studies will be required to determine if activation of iPLA2γ is cytoprotective in C5b-9-mediated GECs in vivo, i.e. in attenuating development of proteinuria in experimental membranous nephropathy. Another potential mechanism of iPLA2γ cytoprotection may be related to the localization and action of iPLA2γ at the ER (Fig. 2). Such actions could include changes in ER membrane lipid composition, alterations in ER Ca2+ transporters, or modification of ER Ca2+ stores. Moreover, iPLA2γ could perturb the ER membrane to initiate an adaptive ER stress response as a feedback mechanism to limit complement-induced cell injury.
In GECs, a portion of iPLA2γ was localized at the mitochondria (Fig. 2). iPLA2γ may protect renal cortical mitochondria from oxidant-induce lipid peroxidation and dysfunction (43). Thus, in the presence of oxidized phospholipid acyl chains, iPLA2γ may hydrolyze damaged acyl chains and allow for reesterification with normal fatty acids, thereby repairing mitochondrial membrane phospholipids. So far, we have not conclusively defined the site of phospholipid hydrolysis by complement-stimulated iPLA2γ. Both ER and mitochondrial lipids in GECs contain AA (61); however, because COX isoenzymes are localized at the ER and the nuclear membranes but not at the mitochondria, the production of PGE2 suggests a coupling of AA release with COX at the ER. Definition of the subcellular sites of phospholipid hydrolysis and the functional role, including the cytoprotective mechanisms of iPLA2γ, will require further investigation.
This work was supported by Canadian Institutes of Health Research Grants MOP-53264 (to A. V. C.) and MOP-53335 (to T. T.). This work was also supported by the Kidney Foundation of Canada (to A. V. C.).
- PLA2
- phospholipase A2
- cPLA2
- cytosolic PLA2
- iPLA2
- calcium-independent PLA2
- AA
- arachidonic acid
- BEL
- bromoenol lactone
- COX
- cyclooxygenase
- ER
- endoplasmic reticulum
- GEC
- glomerular epithelial cell
- HIS
- heat-inactivated human serum
- MEK1
- MAP/ERK kinase 1
- MEKK1
- MAP/ERK kinase kinase 1
- MK2
- MAPK-activated protein kinase-2
- MKK3
- MAPK kinase 3
- MNK1
- MAPK-interacting kinase 1
- NS
- normal human serum
- PG
- prostaglandin.
REFERENCES
- 1. Murakami M., Kudo I. (2002) Phospholipase A2. J. Biochem. 131, 285–292 [DOI] [PubMed] [Google Scholar]
- 2. Six D. A., Dennis E. A. (2000) The expanding superfamily of phospholipase A2 enzymes. Classification and characterization. Biochim. Biophys. Acta 1488, 1–19 [DOI] [PubMed] [Google Scholar]
- 3. Funk C. D. (2001) Prostaglandins and leukotrienes. Advances in eicosanoid biology. Science 294, 1871–1875 [DOI] [PubMed] [Google Scholar]
- 4. Schaloske R. H., Dennis E. A. (2006) The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta 1761, 1246–1259 [DOI] [PubMed] [Google Scholar]
- 5. Mancuso D. J., Jenkins C. M., Gross R. W. (2000) The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A2. J. Biol. Chem. 275, 9937–9945 [DOI] [PubMed] [Google Scholar]
- 6. Tanaka H., Takeya R., Sumimoto H. (2000) A novel intracellular membrane-bound calcium-independent phospholipase A2. Biochem. Biophys. Res. Commun. 272, 320–326 [DOI] [PubMed] [Google Scholar]
- 7. Balsinde J., Balboa M. A. (2005) Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells. Cell. Signal. 17, 1052–1062 [DOI] [PubMed] [Google Scholar]
- 8. Bonventre J. V. (1999) The 85-kDa cytosolic phospholipase A2 knockout mouse. A new tool for physiology and cell biology. J. Am. Soc. Nephrol. 10, 404–412 [DOI] [PubMed] [Google Scholar]
- 9. Chilton F. (1996) Would the real role(s) for secretory PLA2s please stand up. J. Clin. Invest. 97, 2161–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cummings B. S., McHowat J., Schnellmann R. G. (2000) Phospholipase A2s in cell injury and death. J. Pharmacol. Exp. Ther. 294, 793–799 [PubMed] [Google Scholar]
- 11. Portilla D. (1999) Role of fatty acid β-oxidation and calcium-independent phospholipase A2 in ischemic acute renal failure. Curr. Opin. Nephrol. Hypertens. 8, 473–477 [DOI] [PubMed] [Google Scholar]
- 12. Cybulsky A. V., Quigg R. J., Salant D. J. (2005) Experimental membranous nephropathy redux. Am. J. Physiol. Renal Physiol. 289, F660–F671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cybulsky A. V. (2011) Membranous nephropathy. Contrib. Nephrol. 169, 107–125 [DOI] [PubMed] [Google Scholar]
- 14. Mundel P., Shankland S. J. (2002) Podocyte biology and response to injury. J. Am. Soc. Nephrol. 13, 3005–3015 [DOI] [PubMed] [Google Scholar]
- 15. Pavenstädt H., Kriz W., Kretzler M. (2003) Cell biology of the glomerular podocyte. Physiol. Rev. 83, 253–307 [DOI] [PubMed] [Google Scholar]
- 16. Cohen D., Papillon J., Aoudjit L., Li H., Cybulsky A. V., Takano T. (2008) Role of calcium-independent phospholipase A2 in complement-mediated glomerular epithelial cell injury. Am. J. Physiol. Renal Physiol. 294, F469–F479 [DOI] [PubMed] [Google Scholar]
- 17. Murakami M., Masuda S., Ueda-Semmyo K., Yoda E., Kuwata H., Takanezawa Y., Aoki J., Arai H., Sumimoto H., Ishikawa Y., Ishii T., Nakatani Y., Kudo I. (2005) Group VIB Ca2+-independent phospholipase A2γ promotes cellular membrane hydrolysis and prostaglandin production in a manner distinct from other intracellular phospholipases A2. J. Biol. Chem. 280, 14028–14041 [DOI] [PubMed] [Google Scholar]
- 18. Kuwata H., Fujimoto C., Yoda E., Shimbara S., Nakatani Y., Hara S., Murakami M., Kudo I. (2007) A novel role of group VIB calcium-independent phospholipase A2 (iPLA2γ) in the inducible expression of group IIA secretory PLA2 in rat fibroblastic cells. J. Biol. Chem. 282, 20124–20132 [DOI] [PubMed] [Google Scholar]
- 19. Balsinde J., Balboa M. A., Dennis E. A. (1997) Antisense inhibition of group VI Ca2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D1 macrophages. J. Biol. Chem. 272, 29317–29321 [DOI] [PubMed] [Google Scholar]
- 20. Pérez R., Melero R., Balboa M. A., Balsinde J. (2004) Role of group VIA calcium-independent phospholipase A2 in arachidonic acid release, phospholipid fatty acid incorporation, and apoptosis in U937 cells responding to hydrogen peroxide. J. Biol. Chem. 279, 40385–40391 [DOI] [PubMed] [Google Scholar]
- 21. Ramanadham S., Hsu F. F., Bohrer A., Ma Z., Turk J. (1999) Studies of the role of group VI phospholipase A2 in fatty acid incorporation, phospholipid remodeling, lysophosphatidylcholine generation, and secretagogue-induced arachidonic acid release in pancreatic islets and insulinoma cells. J. Biol. Chem. 274, 13915–13927 [DOI] [PubMed] [Google Scholar]
- 22. Bao S., Bohrer A., Ramanadham S., Jin W., Zhang S., Turk J. (2006) Effects of stable suppression of Group VIA phospholipase A2 expression on phospholipid content and composition, insulin secretion, and proliferation of INS-1 insulinoma cells. J. Biol. Chem. 281, 187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saavedra G., Zhang W., Peterson B., Cummings B. S. (2006) Differential roles for cytosolic and microsomal Ca2+-independent phospholipase A2 in cell growth and maintenance of phospholipids. J. Pharmacol. Exp. Ther. 318, 1211–1219 [DOI] [PubMed] [Google Scholar]
- 24. Zhang X. H., Zhao C., Seleznev K., Song K., Manfredi J. J., Ma Z. A. (2006) Disruption of G1-phase phospholipid turnover by inhibition of Ca2+-independent phospholipase A2 induces a p53-dependent cell-cycle arrest in G1 phase. J. Cell Sci. 119, 1005–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cummings B. S., McHowat J., Schnellmann R. G. (2002) Role of an endoplasmic reticulum Ca2+-independent phospholipase A2 in oxidant-induced renal cell death. Am. J. Physiol. Renal Physiol. 283, F492–F498 [DOI] [PubMed] [Google Scholar]
- 26. Kinsey G. R., Blum J. L., Covington M. D., Cummings B. S., McHowat J., Schnellmann R. G. (2008) Decreased iPLA2γ expression induces lipid peroxidation and cell death and sensitizes cells to oxidant-induced apoptosis. J. Lipid Res. 49, 1477–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun B., Zhang X., Talathi S., Cummings B. S. (2008) Inhibition of Ca2+-independent phospholipase A2 decreases prostate cancer cell growth by p53-dependent and independent mechanisms. J. Pharmacol. Exp. Ther. 326, 59–68 [DOI] [PubMed] [Google Scholar]
- 28. Roshak A. K., Capper E. A., Stevenson C., Eichman C., Marshall L. A. (2000) Human calcium-independent phospholipase A2 mediates lymphocyte proliferation. J. Biol. Chem. 275, 35692–35698 [DOI] [PubMed] [Google Scholar]
- 29. Tran K., Wang Y., DeLong C. J., Cui Z., Yao Z. (2000) The assembly of very low density lipoproteins in rat hepatoma McA-RH7777 cells is inhibited by phospholipase A2 antagonists. J. Biol. Chem. 275, 25023–25030 [DOI] [PubMed] [Google Scholar]
- 30. Atsumi G., Murakami M., Kojima K., Hadano A., Tajima M., Kudo I. (2000) Distinct roles of two intracellular phospholipase A2s in fatty acid release in the cell death pathway. Proteolytic fragment of type IVA cytosolic phospholipase A2α inhibits stimulus-induced arachidonate release, whereas that of type VI Ca2+-independent phospholipase A2 augments spontaneous fatty acid release. J. Biol. Chem. 275, 18248–18258 [DOI] [PubMed] [Google Scholar]
- 31. McHowat J., Kell P. J., O'Neill H. B., Creer M. H. (2001) Endothelial cell PAF synthesis following thrombin stimulation utilizes Ca2+-independent phospholipase A2. Biochemistry 40, 14921–14931 [DOI] [PubMed] [Google Scholar]
- 32. Mancuso D. J., Jenkins C. M., Sims H. F., Cohen J. M., Yang J., Gross R. W. (2004) Complex transcriptional and translational regulation of iPLAγ resulting in multiple gene products containing dual competing sites for mitochondrial or peroxisomal localization. Eur. J. Biochem. 271, 4709–4724 [DOI] [PubMed] [Google Scholar]
- 33. Yang J., Han X., Gross R. W. (2003) Identification of hepatic peroxisomal phospholipase A2 and characterization of arachidonic acid-containing choline glycerophospholipids in hepatic peroxisomes. FEBS Lett. 546, 247–250 [DOI] [PubMed] [Google Scholar]
- 34. Tanaka H., Minakami R., Kanaya H., Sumimoto H. (2004) Catalytic residues of group VIB calcium-independent phospholipase A2 (iPLA2γ). Biochem. Biophys. Res. Commun. 320, 1284–1290 [DOI] [PubMed] [Google Scholar]
- 35. Mansour S. J., Matten W. T., Hermann A. S., Candia J. M., Rong S., Fukasawa K., Vande Woude G. F., Ahn N. G. (1994) Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265, 966–970 [DOI] [PubMed] [Google Scholar]
- 36. Winzen R., Kracht M., Ritter B., Wilhelm A., Chen C. Y., Shyu A. B., Müller M., Gaestel M., Resch K., Holtmann H. (1999) The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 18, 4969–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Waskiewicz A. J., Flynn A., Proud C. G., Cooper J. A. (1997) Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 16, 1909–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Coers W., Reivinen J., Miettinen A., Huitema S., Vos J. T., Salant D. J., Weening J. J. (1996) Characterization of a rat glomerular visceral epithelial cell line. Exp. Nephrol. 4, 184–192 [PubMed] [Google Scholar]
- 39. Cybulsky A. V., Papillon J., McTavish A. J. (1998) Complement activates phospholipases and protein kinases in glomerular epithelial cells. Kidney Int. 54, 360–372 [DOI] [PubMed] [Google Scholar]
- 40. Cybulsky A. V., Takano T., Papillon J., Khadir A., Liu J., Peng H. (2002) Complement C5b-9 membrane attack complex increases expression of endoplasmic reticulum stress proteins in glomerular epithelial cells. J. Biol. Chem. 277, 41342–41351 [DOI] [PubMed] [Google Scholar]
- 41. Takano T., Cybulsky A. V. (2000) Complement C5b-9-mediated arachidonic acid metabolism in glomerular epithelial cells. Role of cyclooxygenase-1 and -2. Am. J. Pathol. 156, 2091–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pattabiraman P. P., Lih F. B., Tomer K. B., Rao P. V. (2012) The role of calcium-independent phospholipase A2γ in modulation of aqueous humor drainage and Ca2+ sensitization of trabecular meshwork contraction. Am. J. Physiol. Cell Physiol. 302, C979–C991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kinsey G. R., McHowat J., Beckett C. S., Schnellmann R. G. (2007) Identification of calcium-independent phospholipase A2γ in mitochondria and its role in mitochondrial oxidative stress. Am. J. Physiol. Renal Physiol. 292, F853–F860 [DOI] [PubMed] [Google Scholar]
- 44. Cargnello M., Roux P. P. (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Börsch-Haubold A. G., Pasquet S., Watson S. P. (1998) Direct inhibition of cyclooxygenase-1 and -2 by the kinase inhibitors SB 203580 and PD 98059. SB 203580 also inhibits thromboxane synthase. J. Biol. Chem. 273, 28766–28772 [DOI] [PubMed] [Google Scholar]
- 46. Takahashi S., Keto Y., Fujita T., Uchiyama T., Yamamoto A. (2001) FR167653, a p38 mitogen-activated protein kinase inhibitor, prevents Helicobacter pylori-induced gastritis in mongolian gerbils. J. Pharmacol. Exp. Ther. 296, 48–56 [PubMed] [Google Scholar]
- 47. Kyriakis J. M., Avruch J. (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81, 807–869 [DOI] [PubMed] [Google Scholar]
- 48. Winstead M. V., Balsinde J., Dennis E. A. (2000) Calcium-independent phospholipase A2. Structure and function. Biochim. Biophys. Acta 1488, 28–39 [DOI] [PubMed] [Google Scholar]
- 49. Obenauer J. C., Cantley L. C., Yaffe M. B. (2003) Scansite 2.0. Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Deng N., Zhang J., Zong C., Wang Y., Lu H., Yang P., Wang W., Young G. W., Korge P., Lotz C., Doran P., Liem D. A., Apweiler R., Weiss J. N., Duan H., Ping P. (2011) Phosphoproteome analysis reveals regulatory sites in major pathways of cardiac mitochondria. Mol. Cell. Proteomics 10, M110000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cybulsky A. V., Takano T., Papillon J., Bijian K., Guillemette J. (2005) Activation of the extracellular signal-regulated kinase by complement C5b-9. Am. J. Physiol. Renal Physiol. 289, F593–F603 [DOI] [PubMed] [Google Scholar]
- 52. Panesar M., Papillon J., McTavish A. J., Cybulsky A. V. (1997) Activation of phospholipase A2 by complement C5b-9 in glomerular epithelial cells. J. Immunol. 159, 3584–3594 [PubMed] [Google Scholar]
- 53. Yellaturu C. R., Rao G. N. (2003) A requirement for calcium-independent phospholipase A2 in thrombin-induced arachidonic acid release and growth in vascular smooth muscle cells. J. Biol. Chem. 278, 43831–43837 [DOI] [PubMed] [Google Scholar]
- 54. Beckett C. S., Pennington K., McHowat J. (2006) Activation of MAPKs in thrombin-stimulated ventricular myocytes is dependent on Ca2+-independent PLA2. Am. J. Physiol. Cell Physiol. 290, C1350–C1354 [DOI] [PubMed] [Google Scholar]
- 55. Aoto M., Shinzawa K., Suzuki Y., Ohkubo N., Mitsuda N., Tsujimoto Y. (2009) Essential role of p38 MAPK in caspase-independent, iPLA2-dependent cell death under hypoxia/low glucose conditions. FEBS Lett. 583, 1611–1618 [DOI] [PubMed] [Google Scholar]
- 56. Wolf M. J., Wang J., Turk J., Gross R. W. (1997) Depletion of intracellular calcium stores activates smooth muscle cell calcium-independent phospholipase A2. A novel mechanism underlying arachidonic acid mobilization. J. Biol. Chem. 272, 1522–1526 [DOI] [PubMed] [Google Scholar]
- 57. Moon S. H., Jenkins C. M., Liu X., Guan S., Mancuso D. J., Gross R. W. (2012) Activation of mitochondrial calcium-independent phospholipase A2γ (iPLA2γ) by divalent cations mediating arachidonate release and production of downstream eicosanoids. J. Biol. Chem. 287, 14880–14895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hefner Y., Borsch-Haubold A. G., Murakami M., Wilde J. I., Pasquet S., Schieltz D., Ghomashchi F., Yates J. R., 3rd, Armstrong C. G., Paterson A., Cohen P., Fukunaga R., Hunter T., Kudo I., Watson S. P., Gelb M. H. (2000) Serine 727 phosphorylation and activation of cytosolic phospholipase A2 by MNK1-related protein kinases. J. Biol. Chem. 275, 37542–37551 [DOI] [PubMed] [Google Scholar]
- 59. Takano T., Cybulsky A. V., Cupples W. A., Ajikobi D. O., Papillon J., Aoudjit L. (2003) Inhibition of cyclooxygenases reduces complement-induced glomerular epithelial cell injury and proteinuria in passive Heymann nephritis. J. Pharmacol. Exp. Ther. 305, 240–249 [DOI] [PubMed] [Google Scholar]
- 60. Aoudjit L., Potapov A., Takano T. (2006) Prostaglandin E2 promotes cell survival of glomerular epithelial cells via the EP4 receptor. Am. J. Physiol. Renal Physiol. 290, F1534–F1542 [DOI] [PubMed] [Google Scholar]
- 61. Liu J., Takano T., Papillon J., Khadir A., Cybulsky A. V. (2001) Cytosolic phospholipase A2α associates with plasma membrane, endoplasmic reticulum, and nuclear membrane in glomerular epithelial cells. Biochem. J. 353, 79–90 [PMC free article] [PubMed] [Google Scholar]