

Background: Macrophages can adopt primed or tolerance state, depending upon the history of prior challenges.
Results: IRAK-1 and Tollip facilitate priming of macrophages by super low dose endotoxin.
Conclusion: Super low dose endotoxin primes macrophages by inactivating molecular suppressors, including RelB.
Significance: Revealing mechanisms of macrophage priming is crucial for understanding the pathogenesis of inflammatory diseases.
Keywords: Inflammation, Innate Immunity, Macrophages, Signal Transduction, Signaling, Innate Immunity, Macrophage, Memory, Molecular Mechanisms, Priming
Abstract
Host macrophages can be preprogrammed into opposing primed or tolerant states depending upon the nature and quantities of external stimulants. The paradigm of priming and tolerance has significant implications in the pathogenesis and resolution of both acute and chronic inflammatory diseases. However, the responsible mechanisms are not well understood. Here, we report that super low dose bacterial endotoxin lipopolysaccharide (LPS), as low as 5 pg/ml, primes the expression of proinflammatory mediators in macrophages upon a second high dose LPS challenge (100 ng/ml), although 5 pg/ml LPS itself does not trigger noticeable macrophage activation. Mice primed with super low dose LPS (0.5 μg/kg body weight) in vivo experience significantly elevated mortality following a second hit of high dose LPS as compared with saline-primed control mice. Mechanistically, we demonstrate that LPS primes macrophages by removing transcriptional suppressive RelB through interleukin receptor-associated kinase 1 and Tollip (Toll-interacting protein)-dependent mechanisms. This is in sharp contrast to the well documented RelB stabilization and induction by high dose LPS, potentially through the phosphoinositide 3-kinase (PI3K) pathway. Super low dose and high dose LPS cause opposing modulation of interleukin receptor-associated kinase 1 and PI3K pathways and lead to opposing regulation of RelB. The pathway switching induced by super low versus high dose LPS underscores the importance of competing intracellular circuitry during the establishment of macrophage priming and tolerance.
Introduction
The host innate immune system not only recognizes the structural nature of foreign molecular patterns but also discerns the history and concentration of foreign stimulants. This rudimentary innate immune memory is best reflected in the paradigm of endotoxin priming and tolerance (1). During disseminated endotoxin shock and acute sepsis, bacterial endotoxin (LPS) ranging from 1 to 300 ng/ml can induce a robust cytokine storm (2, 3). Intriguingly, cells pre-exposed to LPS are hyporesponsive to a second LPS challenge in terms of proinflammatory gene expression, a phenomenon known as endotoxin tolerance (4). Consequently, a rapid termination of inflammatory responses soon ensues, which serves to dampen collateral inflammatory damage.
In contrast, a subclinical super low dose of LPS (<100 pg/ml) causes a distinct effect by priming cells for more robust expression of proinflammatory mediators in response to a second LPS challenge (5–7). Recently, super low levels of LPS have been detected in circulation of humans and experimental animals with adverse health conditions, including obesity, chronic smoking, infection, and aging (8–12). Humans with these adverse conditions tend to have elevated mortality associated with septic shock (13, 14). Endotoxin priming and exacerbated mortality are also observed in experimental animals (15, 16).
The molecular mechanism responsible for the opposing effects of endotoxin priming and tolerance is not well understood. Endotoxin tolerance has drawn most of the research effort in the past. LPS (>1 ng/ml) is known to induce an initial wave of robust NFκB activation, followed by NFκB-induced negative regulators, including inhibitor of κ-B α (IκBα), MAPK phosphatase 1, interleukin receptor-associated kinase M (IRAK-M),2 and RelB (17–19). These negative regulators shut down signaling pathways and the expression of proinflammatory mediators at multiple levels. For example, IκBα suppresses p65/RelA by secluding it in the cytoplasm; MAPK phosphatase 1 suppresses the activities of MAP kinases (20); IRAK-M suppresses interleukin receptor-associated kinase 1 (IRAK-1) (21); and RelB blocks the transcription of proinflammatory mediators by assembling a suppressive complex on their promoters (19). In addition, LPS is known to activate the phosphoinositide 3-kinase (PI3K) pathway, which dampens the expression of proinflammatory mediators through inducing negative regulators, including MAPK phosphatase 1, IRAK-M, and suppressing IRAK-1 (22–26). As a consequence, LPS (>1 ng/ml) causes a robust yet transient expression of proinflammatory mediators, followed by a refractory tolerant state. However, the mechanism of priming effect is less studied and understood.
We first reported that low dose LPS (50–100 pg/ml) fails to activate either the robust NFκB pathway or the MAP kinase pathway (27). As a consequence, low dose LPS fails to induce robust expression of proinflammatory mediators. However, we documented that low dose LPS is capable of removing transcriptional suppressors from the promoters of inflammatory genes (27).
The objective of this study is to examine the molecular mechanism responsible for the paradigm of LPS priming and tolerance. By utilizing wild type (WT), IRAK-1, and Tollip (Toll-interacting protein)-deficient primary murine bone marrow-derived macrophages (BMDM), we identified an opposing effect of super low dose LPS versus high dose LPS on the regulation of PI3K and RelB. Although high dose LPS activates PI3K pathway and increases of RelB levels, super low dose LPS suppresses PI3K, and reduces the levels of RelB. The pathway switching triggered by super low dose versus high dose LPS underlies the paradigm of LPS priming and tolerance.
MATERIALS AND METHODS
Reagents
LPS (Escherichia coli 0111:B4) and lipoteichoic acid (LTA) were purchased from Sigma Aldrich. Anti-IκBα, ATF2, RelB, p65, IRAK-1, IRAK-M, GAPDH, β-actin antibodies were obtained from Santa Cruz Biotechnology. Anti-mouse IgG and anti-rabbit IgG horseradish peroxidase-linked (HRP) antibodies were purchased from Cell Signaling Technology. The proteasome inhibitor MG132 was from Calbiochem.
Mice and Cell Culture
Wild type (WT) C57BL/6 mice were purchased from the Charles River laboratory. IRAK-1-deficient mice from C57BL/6 background were kindly provided by Dr. James Thomas from the University of Texas Southwestern Medical School. Tollip deficient mice from C57BL/6 background were provided by Dr. Jürg Tschopp from the University of Lausanne at Switzerland. All mice were housed and bred at Derring Hall animal facility in compliance with the approved Animal Care and Use Committee protocols at Virginia Polytechnic Institute and State University. BMDM were isolated as we described previously (27). Wild type (WT) Raw264.7 and GP96 knocked down Raw264.7 cells defective in cell surface Toll-like receptor 4 (TLR4) were maintained as described previously (28).
Analysis of Protein and mRNA
Western blots and RNA extractions were performed as described previously (27). The relative levels of different transcripts were calculated using the ΔΔCt method, and results were normalized based on the expression of GAPDH. The relative level of mRNA in untreated WT cells was adjusted to 1 and served as the basal reference value. The following primer pairs were used: mouse Il-6, 5′-ATC CAG TTG CCT TCT TGG GAC TGA-3′ (forward) and 5′-TAA GCC TCC GAC TTG TGA AGT GGT-3′ (reverse); mouse Tnf-α, 5′-AGC CGA TGG GTT GTA CCT TGT CTA-3′ (forward) and 5′-TGA GAT AGC AAA TCG GCT GAC GGT-3′ (reverse); mouse Sr-α, 5′-AAG AAG AAC AAG CGC ACG TGG AAC-3′ (forward) and 5′-AGG AGG CCC TTG AAT GAA GGT GAT-3′ (reverse); mouse Kmo, 5′-CGT TTC CAA AGG TGT GCC CAT GAA-3′ (forward) and 5′-AAG TGC ACC TTC GCA TTG GCA TAG-3′ (reverse).
Western Blots and Analyses of Cytokines
Subsequent to indicated treatments and three washes with PBS, whole cell lysates were harvested from cells using 1× SDS lysis buffer containing protease inhibitor mixture (Sigma). Equal amounts of protein extracts were subjected to SDS-PAGE gel (12%), transferred to PVDF membranes, and visualized with specific Abs using the Amersham Biosciences ECL Plus chemiluminescent detection system (GE Healthcare). Meanwhile, plasma of mice or cell supernatants were collected after treatments by centrifugation and stored at −70 °C before examination. Analyses of cytokines, including interleukin-6 (IL-6) and TNFα, were carried out according to the manufacturer's instructions (Bio-Rad). Briefly, samples and standards were diluted and tested by ELISA, which involved several incubation and wash steps. Magnetic Beads coupled with antibodies, biotinylated detector antibodies, and phycoerythrin-conjugated streptavidin were added in samples subsequently and incubated at various times. The test plate was then read on a Bio-Plex system (Bio-Rad). Raw data were analyzed, and standard curves were generated by reference cytokine concentrations. All samples and standards were tested in duplicates.
Chromatin Immunoprecipitation Analysis
Chromatin immunoprecipitation (ChIP) analyses were performed as described previously using the Chip-IT Express kit (Active MotifTM) (27). The immunoprecipitated DNA fragments were analyzed by PCR using primers spanning the binding sites of RelB on the proximal promoter of mouse IL-6. The following are the primer sequences used to amplify the enriched chromatin samples: promoter of mouse IL-6, 5′-TCC CAT CAA GAC ATG CTC AAG TGC-3′ (forward) and 5′-AGC AGA ATG AGC TAC AGA CAT CCC-3′ (reverse).
Statistical Analysis
For all in vitro studies, statistical significances between groups were determined using the Student's t test and indicated by an asterisk in figures; p values < 0.05 were considered statistically significant. For the in vivo cytokine analyses, the Mann-Whitney U test was performed. The log-rank test was used to assess significant difference in the mortality rate, with p values < 0.05.
RESULTS
Super Low Dose LPS Primes, whereas High Dose LPS Tolerizes, the Induction of Proinflammatory Mediators in Macrophages
We first determined the expression of Il-6 and tumor necrosis factor α (Tnfα) in WT BMDM treated with varying dosages of LPS. As shown in Fig. 1A, high dose LPS (100 ng/ml) induced robust induction of both Il-6 and Tnfα. In contrast, low dose LPS (50 pg/ml) induced minimal yet statistically significant expression of Il-6 and Tnfα. This is consistent with our early report that ∼50 pg/ml LPS is the threshold concentration required to trigger mild and low grade expression of proinflammatory mediators. Indeed, we observed that a super low dose LPS (5 pg/ml) failed to induce noticeable expression of Il-6 and Tnfα (Fig. 1A).
FIGURE 1.

Super low dose LPS primes, whereas high dose LPS tolerizes, the expression of proinflammatory mediators in macrophages. A, WT BMDM were treated either a single dose of LPS (0, 5 pg/ml, 10 ng/ml, or 100 ng/ml) for 4 h or were treated with a second dose of 100 ng/ml LPS for 4 h after the first LPS treatment (0, 5 pg/ml, 10 ng/ml) for 4 h. The cells were washed twice with PBS before the second treatment. The relative transcript levels were standardized against GAPDH levels. B, culture supernatants from primed cells as indicated on the figure legend were used to measure the protein levels of IL-6 (n = 3); mean ± S.D.; *, p < 0.05.
We then tested whether super low dose and high dose LPS may cause differential priming and tolerance of macrophages in response to subsequent LPS challenge. WT macrophages were pretreated with 0 or 5 pg/ml or 10 ng/ml LPS for 4 h, followed by washes with PBS and fresh medium. Pretreated cells were restimulated with 100 ng/ml LPS for 4 h. We chose the 4-h time interval between the two consecutive LPS treatments based on well established observations regarding LPS priming and tolerance (5, 7, 29). Furthermore, we chose this shorter time interval rather than the alternative overnight interval to avoid potential complications of cell proliferation, death, and other paracrine effects with an extended LPS treatment. As shown in Fig. 1A, despites its failure to induce any expression of Il-6 and Tnfα, 5 pg/ml LPS potently primed the induction of Il-6 and Tnfα in macrophages challenged with a second dose LPS. In contrast, high dose LPS (10 ng/ml) significantly suppressed the induction of Il-6 and Tnfα upon secondary challenge. To further confirm that low dose LPS indicate prime the protein expression of proinflammatory mediators, we measured the protein levels of IL-6 in macrophages treated with a high dose LPS alone or primed with a low dose LPS prior to a high dose LPS treatment. As shown in Fig. 1B, low dose LPS significantly primed the production of IL-6 protein in WT macrophages.
Opposing Intracellular Effects of Super Low Dose and High Dose LPS
Our previously study revealed that low dose LPS (50–100pg/ml) fails to induce classical NFκB pathway and MAP kinases (27). Further confirming this finding, we observed that super low dose LPS fails to cause IκBα degradation or induction of activating transcription factor 2 (ATF2), a member of the AP-1 family of transcription factors (Fig. 2A). To further examine the molecular mechanisms responsible for the opposing effects of super low dose versus high dose LPS, we examined the proximal signaling components of the TLR4 pathway. As shown in Fig. 2B, super low dose LPS (5 pg/ml) did not trigger any noticeable IRAK-1 degradation (Fig. 2B). This is in contrast to the effect of high dose LPS (>100 ng/ml) that is known to cause IRAK-1 degradation (30).
FIGURE 2.

Opposing intracellular effects of super low dose and high dose LPS. Western blots of whole cell extracts from WT BMDM treated with super low dose LPS (5 pg/ml) or high dose LPS (10 ng/ml) for indicated time with antibodies against ATF2 and IκBα (A), IRAK-1 and IRAK-M (B), and phosphorylated Akt (C). The blots were probed with Abs specific for GAPDH or unphosphorylated Akt protein as loading controls. Figures were representative of three independent experiments. The resting levels of IRAK-1, IRAK-M, or p-Akt were set as 1, and its relative modulation after LPS treatment were compared and plotted (n = 3). *, p < 0.05 as compared with untreated controls.
High dose LPS is also known to induce IRAK-M, a negative regulator of IRAK-1 and the TLR4 pathway that contributes to LPS tolerance (30). In contrast, we observed that super low dose LPS failed to induce IRAK-M protein levels in macrophages (Fig. 2B). Instead, a slight yet significant reduction in IRAK-M occurs after 2 h of super low dose LPS treatment. Other negative regulatory pathways induced by high dose LPS include the PI3K pathway that contributes to the suppression of IRAK-1/TNF receptor-associated factor 6 (22), the induction of negative regulators, including IRAK-M (25, 31), and the induction of negative transcriptional suppressors of inflammatory genes such as RelB and cAMP response element-binding (32, 33). Thus, we tested the effects of super low dose LPS on the activation status of PI3K pathway. As shown in Fig. 2C, high dose LPS induced robust phosphorylation of Akt, an indication of PI3K activation. In contrast, super low dose LPS triggered a decrease in the basal levels of phosphorylated Akt.
Differential Regulation of RelB by Super Low Versus High Dose LPS
RelB has been identified as a crucial modulator of endotoxin tolerance by assembling a suppressive complex on the promoters of proinflammatory genes (19). Deletion of RelB leads to excessive proinflammatory responses both in vitro and in vivo (34–36). High dose LPS is well documented to induce RelB, through NFκB- as well as PI3K-mediated expression (33, 35). However, no report is available regarding the status of RelB in cells challenged with super low dose LPS. We observed that super low (5 pg/ml) or low dose (50 pg/ml) LPS led to a reduction of RelB protein in macrophages (Fig. 3A). This is in contrast to a robust induction of RelB by high dose LPS (Fig. 3A). It is worth mentioning that high dose LPS led to an initial transient decrease in RelB (at 0.5 h), followed by a significant increase at the 2-h time point. The initial decrease of RelB correlates with the transient induction of pro-inflammatory mediators by high dose LPS.
FIGURE 3.

Distinct regulation of RelB by super low and high dose LPS. (A) Western blot analysis of RelB expression in WT BMDM treated with super low dose LPS (5 pg/ml), low dose LPS (50 pg/ml), or high dose LPS (100 ng/ml) for indicated time. Relative levels of RelB were quantified using GAPDH as loading controls. *, p < 0.05 as compared with untreated controls. B, CHIP analysis to detect the binding of RelB to the Il-6 promoter in response to high and super low dose LPS. WT BMDM cells were treated with either super low or high dose LPS for 4 h or a sequential combination of LPS treatments for additional 2 h. The samples were immunoprecipitated using a RelB-specific antibody and analyzed by PCR using primers spanning the promoter region of mouse Il-6.
To further confirm the involvement of RelB in differential gene expression induced by super low and high dose LPS, we performed ChIP analysis on the promoter of the Il-6 gene. As shown in Fig. 3B, 4 h of priming with 5 pg/ml LPS removed RelB from the Il-6 promoter, and prevented RelB association with the Il-6 promoter following additional high dose LPS treatment.
IRAK-1 and Tollip Are Required for the Suppression of RelB
We then studied the upstream molecular mechanisms responsible for the opposing regulation of RelB by super low dose and high dose LPS. Based on previous studies that IRAK-1 and Tollip are preferentially responsible for mediating the proinflammatory responses of low dose LPS (27, 37, 38), we tested the regulation of RelB in WT, IRAK-1 knock-out, and Tollip knock-out BMDM. As shown in Fig. 4A, super low dose LPS preferentially reduced the levels of RelB in WT, but not in IRAK-1 or Tollip-deficient cells. It is interesting to note that the resting levels of RelB were significantly higher in untreated IRAK-1-deficient and Tollip-deficient cells. Given the fact that IRAK-M counteracts the function of IRAK-1, we also tested the levels of RelB in IRAK-M-deficient BMDM. We observed that RelB was lower in IRAK-M-deficient cells as compared with that in WT cells (Fig. 4B).
FIGURE 4.

IRAK-1 and Tollip are required for the suppression of RelB by super low dose LPS. Western blots analysis of RelB expression in whole cell lysates of WT, Tollip-deficient, and IRAK1-deficient BMDM (A); Western blots analysis of RelB expression in whole cell lysates of WT and IRAK-M-deficient BMDM (B); WT BMDM with or without proteasome inhibitor MG-132 (10 μm) pretreatment (C); GAPDH or actin were probed as equal loading controls. Western blots analysis of p-Tyr216 GSK3, p-Ser9 GSK3, and total GSK3 levels in whole cell lysates of WT, Tollip-deficient, and IRAK1-deficient BMDM (D).
In addition to its induction, RelB was recently reported to undergo proteasome-mediated degradation (39). To test whether proteasome-mediated degradation may account for LPS-induced decrease in RelB levels, we applied the proteasome inhibitor MG-132. As shown in Fig. 4C, MG-132 effectively stabilized RelB and blocked RelB level decrease initiated by super low dose LPS challenge.
The degradation of RelB was recently shown to be regulated by GSK3 (39). To further determine whether low dose LPS may differentially modulate GSK3 activation in WT, IRAK-1, and Tollip-deficient cells, we examined the phosphorylation status of GSK3. GSK3 activity can be differentially regulated by two distinct phosphorylation events at serine 9 and tyrosine 216. Ser9 phosphorylation suppresses, whereas Tyr216 phosphorylation activates GSK3 activity. We observed that 5 pg/ml LPS induced Tyr216 phosphorylation and suppressed Ser9 phosphorylation of GSK3 (Fig. 4D), an indication of GSK3 activation. The resting levels of p-Tyr216 GSK3 were lower, and the resting levels of p-Ser9 GSK3 were higher in IRAK-1 and Tollip-deficient cells. Furthermore, low dose LPS failed to induce Tyr216 in either IRAK-1 or Tollip-deficient macrophages (Fig. 4D).
IRAK-1 and Tollip Are Required for the Priming of Macrophages
Based on our findings, we tested whether IRAK-1 and Tollip may contribute to the priming effects of super low dose LPS in the induction of proinflammatory mediators. WT, IRAK-1-deficient, and Tollip-deficient BMDM were primed with 0 or 5 pg/ml LPS for 4 h. Following washes with PBS and fresh medium, they were treated with 100 ng/ml LPS for an additional 4 h. As shown in Fig. 5, super low dose LPS effectively primed the induction of Il-6, Tnfα, scavenger receptor A (Sr-a), and kynurenine 3-monooxygenase (Kmo) in WT cells. In sharp contrast, the priming effect of super low dose LPS was ablated in IRAK-1- or Tollip-deficient cells.
FIGURE 5.

IRAK-1 and Tollip are required for the priming of macrophages by super low dose LPS. WT, Tollip-deficient, and IRAK1-deficient BMDM were pretreated with or without a pretreatment of super low dose LPS (5 pg/ml) for 4 h, followed by a high dose LPS (100 ng/ml) treatment for 4 h. Quantitative RT-PCR analysis for fold change of indicated genes were carried out using total harvested RNA samples. The relative transcript levels were standardized against GAPDH levels. Data were representative of at least three independent experiments (mean ± S.D.; *, p < 0.05).
In Vivo Priming by Super Low Dose LPS
We then tested the in vivo effect of LPS priming. WT and IRAK1-deficient mice were injected with either PBS or an initial super low dose LPS (0.5 μg/kg body weight). Four hours later, they were injected with a high dose of LPS (25 mg/kg body weight). The mortality was closely observed every 4 h for 2 days. As shown in Fig. 6A, PBS-primed mice only experienced initial adverse signs, including lack of movement, eye discharge, and black, tar-like feces, but eventually survived. In sharp contrast, priming with super low dose LPS caused 70% mortality in WT mice. On the other hand, all IRAK-1-deficient mice survived the priming with either PBS or super low dose LPS, although some displayed the symptoms described above.
FIGURE 6.

In vivo priming of proinflammatory responses of super low dose LPS depends upon IRAK-1. A, WT and IRAK-1-deficient mice (n ≥ 8) were either injected i.v. with sterile PBS or 0.5 μg/kg LPS. 4 h later, they were injected intraperitoneally with 25 mg/kg LPS. Morbidity and mortality were closely monitored every 4 h interval for 2 days. *, p < 0.05. B, WT and IRAK-1-deficient mice (n = 5) were either injected i.v. with sterile PBS or 0.5 μg/kg LPS. 4 h later, they were injected i.p. with 25 mg/kg LPS. Tail vein blood were collected and used to measure plasma levels of TNFα and IL-6.
We also measured the plasma levels of TNFα and IL-6 in PBS and LPS-primed mice. As shown in Fig. 6B, priming by super low dose LPS alone caused only modest expression of IL-6 and TNFα. Expectedly, the high dose LPS dramatically increased the plasma levels of IL-6 and TNFα in WT mice primed by super low dose LPS, as compared with the PBS control group (a 6.2-fold increase for IL-6 and a 1.8-fold increase for TNFα). In contrast, the priming effect of super low dose LPS on the expression of IL-6 and TNFα was ablated in IRAK-1-deficient mice.
Cross-priming by Lipoteichoic Acid and oxLDL
To test whether priming can be induced by other TLR agonists in addition to LPS, we tested the effect of selected TLR2 agonist LTA and TLR4 agonist oxidized low density lipoprotein (oxLDL). As shown in Fig. 7, both low dose LTA (5 ng/ml) and oxLDL can significantly prime macrophages for a more robust expression of the Il-6 gene following a second dose LPS. In terms of the mechanism, we observed that LTA similarly reduces the levels of RelB, as well as the phosphorylation of Akt, and increase the GSK3 phosphorylation. The cross-priming by oxLDL may underlie the pathogenesis of atherosclerosis. A potential paradigm for the dynamic priming and tolerance is proposed in Fig. 8.
FIGURE 7.

Cross-priming of murine macrophages by lipoteichoic acid and oxLDL. A, WT BMDM were treated with medium control, 5 ng/ml LTA, 100 ng/ml LPS, or an initial LTA (5 ng/ml) for 4 h, followed by LPS (100 ng/ml) for 2 h. The levels of expressed Il-6 were measured by real-time RT-PCR. B, WT BMDM were treated with medium control, 10 μg/ml oxLDL, 100 ng/ml LPS for 4 h, or an initial oxLDL (10 μg/ml) for 4 h, followed by LPS (100 ng/ml) for 4 h. The levels of expressed Il-6 were measured by real-time RT-PCR. C, WT BMDM were treated as indicated in the legend to Fig. 7. The levels of phosphorylated Akt (p-AKT) and total Akt were detected by Western blot. *, p < 0.05.
FIGURE 8.
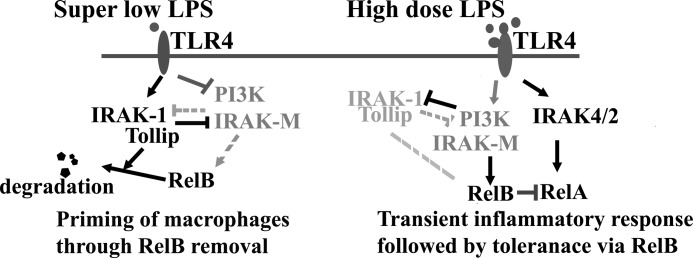
The pathway switching paradigm underlying LPS priming and tolerance. IRAK-1 and Tollip counteract the function of PI3K pathway and IRAK-M and set in motion a dynamic and competing intracellular circuit that controls the fate of RelB. Super low dose LPS skews the circuit toward the activation of IRAK-1, Tollip, and degradation of RelB. Although super low dose LPS fails to induce any noticeable expression of proinflammatory mediators due to the lack of robust NFκB pathway, the clearance of RelB on the promoters of inflammatory genes can prime their further induction when cells are further stimulated with a higher dose LPS. In contrast, high dose LPS switches to the robust activation of NFκB through IRAK-4/2 that leads to dramatic induction of proinflammatory mediators. High dose LPS also activates PI3K pathway and IRAK-M that suppresses IRAK-1 and induces RelB. This leads to subsequent tolerance.
DISCUSSION
This study reveals potential mechanisms responsible for the paradigm of LPS priming and tolerance (Fig. 7). Our data suggest that super low dose LPS primes the expression of proinflammatory mediators by removing the transcriptional suppressive RelB, thus enabling a more robust expression of proinflammatory mediators in cells challenged with an additional dose of LPS. On the other hand, high dose LPS initially reduces and later induces RelB. This may underlie the transient induction and eventual tolerization/suppression of proinflammatory mediators. IRAK-1 and Tollip contribute to RelB degradation induced by super low dose LPS. The differential modulation of RelB by super low and high dose LPS may be mediated through opposing regulation of upstream signaling molecules involving IRAK-1, PI3K. Super low dose LPS deactivates PI3K responsible for RelB induction. On the other hand, super low dose LPS leaves IRAK-1 intact and may enable IRAK-1 mediated degradation of RelB. In contrast, high dose LPS activates PI3K and induces RelB.
Our findings reveal anew the paradigm of opposing effects by varying dosages of stimulants. In addition to LPS, other highly important cellular mediators, including cytokines, hormones, and adipokines can manifest the similar paradigm. For example, high levels of interleukin-1β (IL-1β) (>1 ng/ml) are known to induce robust cytokine storm and cell death (40–42). In contrast, low levels of IL-1β (∼10 pg/ml) can induce cellular proliferation (42). Likewise, both insulin and leptin are known to trigger either cellular sensitivity or resistance/tolerance (43, 44). These phenomena underscore the complex adaptation of cellular systems that ensure proper physiological adjustments to changing environments, with the intended goal of bringing the cellular systems back to homeostatic states. However, unintended side effects of these adjustments often occur that lead to both chronic and acute human inflammatory diseases. For example, LPS tolerance during the late stage of septic shock is intended to reduce and minimize the harmful proinflammatory cytokine storm (45). Yet, this very hyporesponsiveness may subject the host to more severe secondary infection and death (4, 46). On the other hand, LPS priming may alert the host to the presence of bacterial infection and facilitate bacterial clearance (5). However, exacerbated responses in low dose LPS-primed individuals during severe bacterial infection and disseminated sepsis may contribute to higher mortality. Maintaining the proper balance between these adjustments is crucial for survival and long term health.
Our findings provide mechanistic insight regarding the dynamic maintenance of LPS priming and tolerance. Specifically, our data support the theory that competing intracellular players, including PI3K and IRAK-1, are differentially modulated by super low and high dose LPS, and that their opposing regulation may bear critical significance in LPS priming and tolerance. The competitive interactions between these molecules have been noticed previously (22, 26), although their opposing regulation by low and high dose LPS has not been studied previously. High dose LPS was extensively used in previous mechanistic studies and can degrade IRAK-1, activate the PI3K pathway, and induce IRAK-M (Fig. 6) (30, 47, 48). IRAK-M was shown to inhibit IRAK-1 function (21). PI3K pathway was shown to increase IRAK-M and down-regulate both the levels and activities of IRAK-1 (22, 23, 26, 31). Furthermore, PI3K also induces RelB (49). As a consequence, these studies explain the phenomenon of LPS tolerance. Our data indicate that the effect of super low dose LPS on these competing players flips as compared with high dose LPS. We found that super low dose LPS maintains IRAK-1 and deactivates the PI3K pathway. This may enable macrophage priming instead of tolerance. It is worth mentioning that an analogous paradigm has recently been defined during the differentiation of T helper 17 and T regulatory cells. Transforming growth factor β (TGFβ) was shown to be involved in the opposing differentiation of both T helper 17 and T regulatory cells (50, 51). The competing circuit between the transcription factors forkhead box P3 (FoxP3) and retinoic acid-related orphan receptor γ T can be differentially modulated by varying dosages of TGFβ, and as a consequence, the differentiation of CD4+ T cells can be skewed toward either T helper 17 or T regulatory predominance (52). Our current study adds another example of the dynamic and competing circuitry that controls the fate of immune cell function.
Although our findings provide a first step toward the complex regulation of LPS priming and tolerance, the proximal sensing of super low and high dose LPS remains a mystery. Although it is beyond the scope of this study, we hypothesize that differential dimerization of TLR4 with other co-receptors may play a role. It is known that TLR4 can heterodimerize with CD36, SR-A, or MAC-1/CD11b (53, 54). MAC-1/TLR4 may preferentially induce PI3K activation and suppression of inflammatory responses (55). In contrast, CD36/TLR4 was implicated in the propagation of chronic inflammatory processes (53). Future studies are clearly warranted to determine the usage of TLR4 co-receptors by super low dose LPS. Consequently, super low dose LPS and high dose LPS may cause pathway switching at multiple levels, ultimately leading to the opposing regulation of IRAKs and PI3K. Corroborating this, it has been reported that multiple upstream kinases including IRAK-4 and protein kinase C (PKC) can potentially activate IRAK-1 (56, 57). We postulate that low versus high dose LPS may differentially activate IRAK-1 upstream kinases that may either cause IRAK-1 degradation and subsequent tolerance or persistent activation and priming. Further detailed studies are warranted to test this hypothesis. In addition to IRAKs and PKC, TLR4 signaling is known to activate protein tyrosine kinases such as Btk, Hck, and Pyk2 (58–60). High dose LPS can activate PI3K through MyD88 tyrosine phosphorylation and recruitment of PI3K adaptor p85 (61). On the other hand, a recent report revealed that PI3K can be potently suppressed by PKC (62). It is reasonable to speculate that differential activation of protein tyrosine kinases, PKC, and IRAK-4 may be induced by super low dose versus high dose LPS. As a consequence, opposing regulation of PI3K and IRAK-1 may ensue, skewing their competitive balance toward a priming or tolerance phenotype.
Our data reveal the intriguing regulation of RelB by IRAK-1 and Tollip following super low dose LPS challenge. Although high dose LPS is known to induce RelB, we observed that super low dose LPS fails to induce RelB, potentially due to the lack of NFκB and PI3K pathway activation. Instead, super low dose LPS significantly decreased RelB levels, potentially through proteasome-mediated degradation. This unique regulation of RelB is through IRAK-1- and Tollip-mediated activation of GSK3. Based on our data, we postulate that RelB may be degraded through two-step modifications that involve phosphorylation and ubiquitination. LPS first induces GSK3 activation through IRAK-1 and Tollip. Activated GSK3 then contributes to RelB phosphorylation and subsequent ubiquitination and degradation. This is consistent with a recent report demonstrating that RelB can be ubiquitinated and degraded through a GSK3-dependent process (39). It was shown that GSK3 leads to RelB degradation through RelB phosphorylation (39). Although Tollip has been identified for more than a decade, its physiological function is poorly understood, perhaps due to the high dosage of TLR stimulants used in past studies. Biochemical analyses indicate that Tollip can bind with phospholipids as well as ubiquitin through its CUE domain (63, 64). Further biochemical studies are needed to clarify whether Tollip may also directly contribute to the ubiquitination process of RelB.
In addition to LPS priming, other agents can potentially cause cross-priming. One of the classical examples for cross-priming is mediated by interferon γ (IFNγ). IFNγ significantly increases the inflammatory response of macrophages to subsequent LPS challenge (65). Interestingly, the underlying mechanisms also involve suppression of PI3K pathway by IFNγ (66). We tested other TLR agonists, including LTA (a TLR2 agonist) and oxidized LDL (TLR4 agonist), and observed that low levels of LTA and oxidized LDL can prime macrophages for an elevated proinflammatory response to subsequent LPS challenge. Mechanistically, low levels of LTA suppress PI3K pathway and decrease basal levels of phosphorylated Akt. Our data indicate that TLR2 and TLR4 agonists may share similar mechanisms in priming the macrophage activation. Intriguingly, we observed that oxLDL failed to suppress PI3K and failed to reduce RelB levels. Thus, oxLDL may cause cross-priming of macrophage to subsequent LPS challenge through a distinct mechanism. Our data partially explain the detrimental proinflammatory effects of oxidized LDL during the pathogenesis of atherosclerosis.
This study reveals potential pathway switching mechanisms responsible for the paradigm of LPS priming and tolerance. These mechanisms may have general implications in diverse cellular processes that govern the fate of immune cell activation. Future studies are needed to further define these mechanisms, the critical thresholds for pathway switching, and intervention strategies to aid in the maintenance of a proper homeostatic balance. These studies may hold the keys for effective prevention and treatment of devastating inflammatory diseases.
Acknowledgment
We thank Alwiya Ahmed for the assistance of BMDM culture.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 HL115835 (to L. L.).
- IRAK-M
- interleukin receptor-associated kinase M
- BMDM
- bone marrow-derived macrophage(s)
- LTA
- lipoteichoic acid
- TLR4
- Toll-like receptor 4
- oxLDL
- oxidized low density lipoprotein
- GSK3
- glycogen synthase kinase 3.
REFERENCES
- 1. Morris M., Li L. (2012) Molecular mechanisms responsible for the reduced expression of cholesterol transporters from macrophages by low-dose endotoxin. Arch. Immunol. Ther. Exp. 60, 13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scheifele D. W., Olsen E. M., Pendray M. R. (1985) Endotoxinemia and thrombocytopenia during neonatal necrotizing enterocolitis. Am. J. Clin. Pathol. 83, 227–229 [DOI] [PubMed] [Google Scholar]
- 3. Dofferhoff A. S., Nijland J. H., de Vries-Hospers H. G., Mulder P. O., Weits J., Bom V. J. (1991) Effects of different types and combinations of antimicrobial agents on endotoxin release from gram-negative bacteria: an in-vitro and in-vivo study. Scand. J. Infect. Dis. 23, 745–754 [DOI] [PubMed] [Google Scholar]
- 4. West M. A., Heagy W. (2002) Endotoxin tolerance: a review. Crit. Care Med. 30, S64–73 [PubMed] [Google Scholar]
- 5. Zhang X., Morrison D. C. (1993) Lipopolysaccharide-induced selective priming effects on tumor necrosis factor α and nitric oxide production in mouse peritoneal macrophages. J. Exp. Med. 177, 511–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Henricson B. E., Manthey C. L., Perera P. Y., Hamilton T. A., Vogel S. N. (1993) Dissociation of lipopolysaccharide (LPS)-inducible gene expression in murine macrophages pretreated with smooth LPS versus monophosphoryl lipid A. Infect. Immun. 61, 2325–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hirohashi N., Morrison D. C. (1996) Low-dose lipopolysaccharide (LPS) pretreatment of mouse macrophages modulates LPS-dependent interleukin-6 production in vitro. Infect. Immun. 64, 1011–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cani P. D., Bibiloni R., Knauf C., Waget A., Neyrinck A. M., Delzenne N. M., Burcelin R. (2008) Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57, 1470–1481 [DOI] [PubMed] [Google Scholar]
- 9. Goto T., Edén S., Nordenstam G., Sundh V., Svanborg-Edén C., Mattsby-Baltzer I. (1994) Endotoxin levels in sera of elderly individuals. Clin. Diagn. Lab. Immunol. 1, 684–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ancuta P., Kamat A., Kunstman K. J., Kim E. Y., Autissier P., Wurcel A., Zaman T., Stone D., Mefford M., Morgello S., Singer E. J., Wolinsky S. M., Gabuzda D. (2008) Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One 3, e2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Szeto C. C., Kwan B. C., Chow K. M., Lai K. B., Chung K. Y., Leung C. B., Li P. K. (2008) Endotoxemia is related to systemic inflammation and atherosclerosis in peritoneal dialysis patients. Clin. J. Am. Soc. Nephrol. 3, 431–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rao R. (2009) Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 50, 638–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Gaudio A. R., Rinaldi S., Chelazzi C., Borracci T. (2009) Pathophysiology of sepsis in the elderly: clinical impact and therapeutic considerations. Curr. Drug Targets 10, 60–70 [DOI] [PubMed] [Google Scholar]
- 14. Zahr F., Genovese E., Mathier M., Shullo M., Lockard K., Zomak R., McNamara D., Toyoda Y., Kormos R. L., Teuteberg J. J. (2011) Obese patients and mechanical circulatory support: weight loss, adverse events, and outcomes. Ann. Thorac. Surg. 92, 1420–1426 [DOI] [PubMed] [Google Scholar]
- 15. Strandberg L., Verdrengh M., Enge M., Andersson N., Amu S., Onnheim K., Benrick A., Brisslert M., Bylund J., Bokarewa M., Nilsson S., Jansson J. O. (2009) Mice chronically fed high-fat diet have increased mortality and disturbed immune response in sepsis. PLoS One 4, e7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Turnbull I. R., Clark A. T., Stromberg P. E., Dixon D. J., Woolsey C. A., Davis C. G., Hotchkiss R. S., Buchman T. G., Coopersmith C. M. (2009) Effects of aging on the immunopathologic response to sepsis. Crit. Care Med. 37, 1018–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen P., Li J., Barnes J., Kokkonen G. C., Lee J. C., Liu Y. (2002) Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J. Immunol. 169, 6408–6416 [DOI] [PubMed] [Google Scholar]
- 18. Harada K., Isse K., Sato Y., Ozaki S., Nakanuma Y. (2006) Endotoxin tolerance in human intrahepatic biliary epithelial cells is induced by upregulation of IRAK-M. Liver Int. 26, 935–942 [DOI] [PubMed] [Google Scholar]
- 19. Chen X., El Gazzar M., Yoza B. K., McCall C. E. (2009) The NF-kappaB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 284, 27857–27865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Salojin K. V., Owusu I. B., Millerchip K. A., Potter M., Platt K. A., Oravecz T. (2006) Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J. Immunol. 176, 1899–1907 [DOI] [PubMed] [Google Scholar]
- 21. Kobayashi K., Hernandez L. D., Galán J. E., Janeway C. A., Jr., Medzhitov R., Flavell R. A. (2002) IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110, 191–202 [DOI] [PubMed] [Google Scholar]
- 22. Chaurasia B., Mauer J., Koch L., Goldau J., Kock A. S., Brüning J. C. (2010) Phosphoinositide-dependent kinase 1 provides negative feedback inhibition to Toll-like receptor-mediated NF-κB activation in macrophages. Mol. Cell Biol. 30, 4354–4366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zacharioudaki V., Androulidaki A., Arranz A., Vrentzos G., Margioris A. N., Tsatsanis C. (2009) Adiponectin promotes endotoxin tolerance in macrophages by inducing IRAK-M expression. J. Immunol. 182, 6444–6451 [DOI] [PubMed] [Google Scholar]
- 24. Medina E. A., Morris I. R., Berton M. T. (2010) Phosphatidylinositol 3-kinase activation attenuates the TLR2-mediated macrophage proinflammatory cytokine response to Francisella tularensis live vaccine strain. J. Immunol. 185, 7562–7572 [DOI] [PubMed] [Google Scholar]
- 25. Zhu Q. Y., Liu Q., Chen J. X., Lan K., Ge B. X. (2010) MicroRNA-101 targets MAPK phosphatase-1 to regulate the activation of MAPKs in macrophages. J. Immunol. 185, 7435–7442 [DOI] [PubMed] [Google Scholar]
- 26. Noubir S., Hmama Z., Reiner N. E. (2004) Dual receptors and distinct pathways mediate interleukin-1 receptor-associated kinase degradation in response to lipopolysaccharide. Involvement of CD14/TLR4, CR3, and phosphatidylinositol 3-kinase. J. Biol. Chem. 279, 25189–25195 [DOI] [PubMed] [Google Scholar]
- 27. Maitra U., Gan L., Chang S., Li L. (2011) Low-dose endotoxin induces inflammation by selectively removing nuclear receptors and activating CCAAT/enhancer-binding protein δ. J. Immunol. 186, 4467–4473 [DOI] [PubMed] [Google Scholar]
- 28. Liu B., Yang Y., Qiu Z., Staron M., Hong F., Li Y., Wu S., Li Y., Hao B., Bona R., Han D., Li Z. (2010) Folding of Toll-like receptors by the HSP90 paralogue gp96 requires a substrate-specific cochaperone. Nat. Commun. 1, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X., Morrison D. C. (1993) Lipopolysaccharide structure-function relationship in activation versus reprogramming of mouse peritoneal macrophages. J. Leukoc. Biol. 54, 444–450 [DOI] [PubMed] [Google Scholar]
- 30. Deng J. C., Cheng G., Newstead M. W., Zeng X., Kobayashi K., Flavell R. A., Standiford T. J. (2006) Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J. Clin. Invest. 116, 2532–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Androulidaki A., Iliopoulos D., Arranz A., Doxaki C., Schworer S., Zacharioudaki V., Margioris A. N., Tsichlis P. N., Tsatsanis C. (2009) The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity 31, 220–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin M., Rehani K., Jope R. S., Michalek S. M. (2005) Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 6, 777–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gustin J. A., Korgaonkar C. K., Pincheira R., Li Q., Donner D. B. (2006) Akt regulates basal and induced processing of NF-κB2 (p100) to p52. J. Biol. Chem. 281, 16473–16481 [DOI] [PubMed] [Google Scholar]
- 34. Xia Y., Pauza M. E., Feng L., Lo D. (1997) RelB regulation of chemokine expression modulates local inflammation. Am. J. Pathol. 151, 375–387 [PMC free article] [PubMed] [Google Scholar]
- 35. Yoza B. K., Hu J. Y., Cousart S. L., Forrest L. M., McCall C. E. (2006) Induction of RelB participates in endotoxin tolerance. J. Immunol. 177, 4080–4085 [DOI] [PubMed] [Google Scholar]
- 36. Weih F., Warr G., Yang H., Bravo R. (1997) Multifocal defects in immune responses in RelB-deficient mice. J. Immunol. 158, 5211–5218 [PubMed] [Google Scholar]
- 37. Swantek J. L., Tsen M. F., Cobb M. H., Thomas J. A. (2000) IL-1 receptor-associated kinase modulates host responsiveness to endotoxin. J. Immunol. 164, 4301–4306 [DOI] [PubMed] [Google Scholar]
- 38. Didierlaurent A., Brissoni B., Velin D., Aebi N., Tardivel A., Käslin E., Sirard J. C., Angelov G., Tschopp J., Burns K. (2006) Tollip regulates proinflammatory responses to interleukin-1 and lipopolysaccharide. Mol. Cell Biol. 26, 735–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Neumann M., Klar S., Wilisch-Neumann A., Hollenbach E., Kavuri S., Leverkus M., Kandolf R., Brunner-Weinzierl M. C., Klingel K. (2011) Glycogen synthase kinase-3β is a crucial mediator of signal-induced RelB degradation. Oncogene 30, 2485–2492 [DOI] [PubMed] [Google Scholar]
- 40. Dinarello C. A. (2011) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suzuki K., Murtuza B., Beauchamp J. R., Brand N. J., Barton P. J., Varela-Carver A., Fukushima S., Coppen S. R., Partridge T. A., Yacoub M. H. (2004) Role of interleukin-1β in acute inflammation and graft death after cell transplantation to the heart. Circulation 110, II219–224 [DOI] [PubMed] [Google Scholar]
- 42. Maedler K., Schumann D. M., Sauter N., Ellingsgaard H., Bosco D., Baertschiger R., Iwakura Y., Oberholzer J., Wollheim C. B., Gauthier B. R., Donath M. Y. (2006) Low concentration of interleukin-1β induces FLICE-inhibitory protein-mediated β-cell proliferation in human pancreatic islets. Diabetes 55, 2713–2722 [DOI] [PubMed] [Google Scholar]
- 43. Mantzoros C. S., Magkos F., Brinkoetter M., Sienkiewicz E., Dardeno T. A., Kim S. Y., Hamnvik O. P., Koniaris A. (2011) Leptin in human physiology and pathophysiology. Am. J. Physiol. Endocrinol. Metab. 301, E567–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Boura-Halfon S., Zick Y. (2009) Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 296, E581–591 [DOI] [PubMed] [Google Scholar]
- 45. Medvedev A. E., Lentschat A., Kuhns D. B., Blanco J. C., Salkowski C., Zhang S., Arditi M., Gallin J. I., Vogel S. N. (2003) Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections. J. Exp. Med. 198, 521–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Biswas S. K., Lopez-Collazo E. (2009) Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 30, 475–487 [DOI] [PubMed] [Google Scholar]
- 47. Li L., Cousart S., Hu J., McCall C. E. (2000) Characterization of interleukin-1 receptor-associated kinase in normal and endotoxin-tolerant cells. J. Biol. Chem. 275, 23340–23345 [DOI] [PubMed] [Google Scholar]
- 48. Ni M., MacFarlane A. W., 4th, Toft M., Lowell C. A., Campbell K. S., Hamerman J. A. (2012) B-cell adaptor for PI3K (BCAP) negatively regulates Toll-like receptor signaling through activation of PI3K. Proc. Natl. Acad. Sci. U.S.A. 109, 267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Terragni J., Graham J. R., Adams K. W., Schaffer M. E., Tullai J. W., Cooper G. M. (2008) Phosphatidylinositol 3-kinase signaling in proliferating cells maintains an anti-apoptotic transcriptional program mediated by inhibition of FOXO and non-canonical activation of NFκB transcription factors. BMC Cell Biol. 9, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lochner M., Peduto L., Cherrier M., Sawa S., Langa F., Varona R., Riethmacher D., Si-Tahar M., Di Santo J. P., Eberl G. (2008) In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORγ t+ T cells. J. Exp. Med. 205, 1381–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou L., Lopes J. E., Chong M. M., Ivanov I. I., Min R., Victora G. D., Shen Y., Du J., Rubtsov Y. P., Rudensky A. Y., Ziegler S. F., Littman D. R. (2008) TGF-β-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORγt function. Nature 453, 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hong T., Xing J., Li L., Tyson J. (2011) A mathematical model for the reciprocal differentiation of T helper 17 cells and induced regulatory T cells. PLoS Comput. Biol. 7, e1002122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stewart C. R., Stuart L. M., Wilkinson K., van Gils J. M., Deng J., Halle A., Rayner K. J., Boyer L., Zhong R., Frazier W. A., Lacy-Hulbert A., El Khoury J., Golenbock D. T., Moore K. J. (2010) CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vogel S. N., English K. E., Fertsch D., Fultz M. J. (1983) Differential modulation of macrophage membrane markers by interferon: analysis of Fc and C3b receptors, Mac-1 and Ia antigen expression. J. Interferon Res. 3, 153–160 [DOI] [PubMed] [Google Scholar]
- 55. Han C., Jin J., Xu S., Liu H., Li N., Cao X. (2010) Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat. Immunol. 11, 734–742 [DOI] [PubMed] [Google Scholar]
- 56. Li S., Strelow A., Fontana E. J., Wesche H. (2002) IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc. Natl. Acad. Sci. U.S.A. 99, 5567–5572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gan L., Li L. (2010) Interleukin-1 receptor-associated kinase-1 (IRAK-1) functionally associates with PKCϵ and VASP in the regulation of macrophage migration. Mol. Immunol. 47, 1278–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Medvedev A. E., Piao W., Shoenfelt J., Rhee S. H., Chen H., Basu S., Wahl L. M., Fenton M. J., Vogel S. N. (2007) Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance. J. Biol. Chem. 282, 16042–16053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ernst M., Inglese M., Scholz G. M., Harder K. W., Clay F. J., Bozinovski S., Waring P., Darwiche R., Kay T., Sly P., Collins R., Turner D., Hibbs M. L., Anderson G. P., Dunn A. R. (2002) Constitutive activation of the SRC family kinase Hck results in spontaneous pulmonary inflammation and an enhanced innate immune response. J. Exp. Med. 196, 589–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xi C. X., Xiong F., Zhou Z., Mei L., Xiong W. C. (2010) PYK2 interacts with MyD88 and regulates MyD88-mediated NF-κB activation in macrophages. J. Leukoc. Biol. 87, 415–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Laird M. H., Rhee S. H., Perkins D. J., Medvedev A. E., Piao W., Fenton M. J., Vogel S. N. (2009) TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J. Leukoc. Biol. 85, 966–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee J. Y., Chiu Y. H., Asara J., Cantley L. C. (2011) Inhibition of PI3K binding to activators by serine phosphorylation of PI3K regulatory subunit p85α Src homology-2 domains. Proc. Natl. Acad. Sci. U.S.A. 108, 14157–14162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li T., Hu J., Li L. (2004) Characterization of Tollip protein upon lipopolysaccharide challenge. Mol. Immunol. 41, 85–92 [DOI] [PubMed] [Google Scholar]
- 64. Burns K., Clatworthy J., Martin L., Martinon F., Plumpton C., Maschera B., Lewis A., Ray K., Tschopp J., Volpe F. (2000) Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat. Cell Biol. 2, 346–351 [DOI] [PubMed] [Google Scholar]
- 65. Bosisio D., Polentarutti N., Sironi M., Bernasconi S., Miyake K., Webb G. R., Martin M. U., Mantovani A., Muzio M. (2002) Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-γ: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood 99, 3427–3431 [DOI] [PubMed] [Google Scholar]
- 66. Hu X., Paik P. K., Chen J., Yarilina A., Kockeritz L., Lu T. T., Woodgett J. R., Ivashkiv L. B. (2006) IFN-γ suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity 24, 563–574 [DOI] [PubMed] [Google Scholar]