

Background: Yap1 regulates cardiac development, yet the function of Yap1 in the adult heart remains unknown.
Results: Yap1 promotes cardiomyocyte survival, hypertrophy, and proliferation and protects against chronic myocardial infarction (MI).
Conclusion: Yap1 is critical for basal heart homeostasis, and Yap1 deficiency exacerbates myocardial injury.
Significance: Increasing cardiomyocyte survival and proliferation may afford protection in vivo against MI injury.
Keywords: Apoptosis, Cardiac Hypertrophy, Cardiac Muscle, Heart Failure, Myocardial Infarction, Yap1
Abstract
Yap1 is an important regulator of cardiomyocyte proliferation and embryonic heart development, yet the function of endogenous Yap1 in the adult heart remains unknown. We studied the role of Yap1 in maintaining basal cardiac function and in modulating injury after chronic myocardial infarction (MI). Cardiomyocyte-specific homozygous inactivation of Yap1 in the postnatal heart (YapF/FCre) elicited increased myocyte apoptosis and fibrosis, dilated cardiomyopathy, and premature death. Heterozygous deletion (Yap+/FCre) did not cause an overt cardiac phenotype compared with YapF/F control mice at base line. In response to stress (MI), nuclear Yap1 was found selectively in the border zone and not in the remote area of the heart. After chronic MI (28 days), Yap+/FCre mice had significantly increased myocyte apoptosis and fibrosis, with attenuated compensatory cardiomyocyte hypertrophy, and further impaired function versus Yap+/F control mice. Studies in isolated cardiomyocytes demonstrated that Yap1 expression is sufficient to promote increased cell size and hypertrophic gene expression and protected cardiomyocytes against H2O2-induced cell death, whereas Yap1 depletion attenuated phenylephrine-induced hypertrophy and augmented apoptosis. Finally, we observed a significant decrease in cardiomyocyte proliferation in Yap+/FCre hearts compared with Yap+/F controls after MI and demonstrated that Yap1 is sufficient to promote cardiomyocyte proliferation in isolated cardiomyocytes. Our findings suggest that Yap1 is critical for basal heart homeostasis and that Yap1 deficiency exacerbates injury in response to chronic MI.
Introduction
Acute myocardial infarction (MI)4 and ischemic heart disease contribute significantly to the development of heart failure and are leading healthcare concerns worldwide (1). Despite recent progress, interventions aimed at treating patients with MI still result in high rates of chronic heart failure. Loss of cardiomyocytes and the subsequent remodeling of the myocardium are indicative of chronic MI and contribute to the decline in heart function (2–4). Therefore, it is imperative to elucidate fundamental molecular mechanisms underlying cardiomyocyte growth and survival in response to stress.
The Hippo signaling pathway, originally discovered in Drosophila, regulates organ size by modulating cell proliferation and survival (5–7). This kinase cascade is highly conserved from flies to humans and culminates with the phosphorylation and inactivation of the transcriptional coactivator Yes-associated protein isoform 1 (Yap1). Increased activation of Yap1 has been detected in certain tumors, and overexpression of Yap1 in the mouse liver causes robust overgrowth and hepatocellular carcinoma (8, 9). However, mice with targeted deletion of Yap1 in the liver have increased hepatocyte apoptosis and impaired function, suggesting an important physiological role of Yap1 in maintaining organ homeostasis (10). Recent work has investigated the role of Yap1 in heart development. Independent groups observed diminished cardiomyocyte proliferation, hypoplastic hearts, and embryonic lethality in mice lacking Yap1 during development, suggesting that Yap1 is critical to achieve normal cardiomyocyte numbers and normal heart size (11, 12). However, the function of endogenous Yap1 in the postnatal heart has not been investigated.
The aim of this study was to determine the physiological role of Yap1 in maintaining cardiac homeostasis as well as its function in responding to chronic myocardial ischemia in the adult mouse. To achieve this aim we utilized bigenic mice harboring a floxed Yap1 allele and inactivated gene expression selectively in cardiomyocytes in the postnatal heart by breeding with α-MHC Cre transgenic animals.
EXPERIMENTAL PROCEDURES
Animals
Generation of transgenic mice harboring a floxed Yap1 allele (C57BL/6 background) and α-MHC Cre recombinase transgenic mice (C57BL/6 background) has been previously reported (10, 13). All experiments involving animals were approved by the New Jersey Medical School Institutional Animal Care and Use Committee.
MI Surgery
Pathogen-free mice were housed in a temperature-controlled environment with 12 h of light/dark cycles where they received food and water ad libitum. Mice were anesthetized by intraperitoneal injection of pentobarbital sodium (60 mg/kg). A rodent ventilator (model 683; Harvard Apparatus Inc.) was used with 65% oxygen during the surgical procedure. The animals were kept warm using heat lamps and heating pads. Rectal temperature was monitored and maintained between 36.5 and 37.5 °C. The chest was opened by a horizontal incision through the muscle between the ribs (third intercostal space). Ischemia was achieved by permanent ligation of the anterior descending branch of the left coronary artery (LAD) using an 8-0 nylon suture with a silicon tubing (1 mm OD) placed on top of the LAD 2 mm below the border between left atrium and left ventricle (LV). Regional ischemia was confirmed by ECG change (S-wave/T-wave segment elevation). After 2 days or 4 weeks of ligation, the heart was perfused with saline. The size of infarct at 4 weeks was determined as previously described (14). Briefly, the LV was cut into four transverse slices (1-mm thick), subjected to Masson's Trichrome staining, and mounted. The length of the circumference in the infarct portion and the normal area was obtained from each of the 4 LV slices. Infarct size at 2 days was determined using 2,3,5-triphenyltetrazolium chloride as described (15).
Echocardiography
Echocardiography was performed after mice were anesthetized with 12 μl/g body weight of 2.5% Avertin as described previously (16).
Primary Cardiomyocyte Isolation and Culture
Ventricular cardiomyocytes were prepared from 1-day-old Crl: (WI) BR Wistar rats (Charles River Laboratories). A cardiomyocyte-rich fraction was obtained by centrifugation through a discontinuous Percoll gradient. Cells were cultured in a complete medium (cardiomyocyte) containing Dulbecco's modified Eagle's medium/F-12 supplemented with 5% horse serum, 4 μg/ml transferrin, 0.7 ng/ml sodium selenite (Invitrogen), 2 g/liter bovine serum albumin (fraction V), 3 mmol/liter pyruvic acid, 15 mmol/liter HEPES, 100 μmol/liter ascorbic acid, 100 μg/ml ampicillin, 5 μg/ml linoleic, acid and 100 μmol/liter 5-bromo-2′-deoxyuridine (Sigma). The inhibitors LY294002, PD98059, and Akt inhibitor V (triciribine) were obtained from EMD, Calbiochem. Phenylephrine was purchased from Sigma.
Generation of Adenovirus
Recombinant adenovirus vectors were constructed, propagated, and titered as previously described (17). Briefly, pBHGloxΔE1,3Cre (Microbix), including the ΔE1 adenoviral genome, was co-transfected with the pDC shuttle vector containing the gene of interest into HEK293 cells using Lipofectamine 2000 (Invitrogen). Through homologous recombination, the test genes were integrated into the E1-deleted adenoviral genome. The viruses were propagated in HEK293 cells. We made replication-defective human adenovirus type 5 (devoid of E1) harboring Yap1. Adenovirus harboring β-galactosidase (LacZ) was used as a control.
Generation of Yap1 Short Hairpin RNA (shRNA) Adenovirus
pSilencer 1.0-U6 expression vector was purchased from Ambion. The U6 RNA polymerase III promoter and the polylinker region were subcloned into the adenoviral shuttle vector pDC316 (Microbix). The hairpin-forming oligo, corresponding to bases 553–572 of the rat yap1 cDNA, and its antisense with ApaI and HindIII overhangs were synthesized, annealed, and subcloned distal to the U6 promoter.
Histological Analysis
Heart specimens were fixed with formalin, embedded in paraffin, and sectioned at 6-μm thickness. Interstitial fibrosis was evaluated by Masson's Trichrome staining (16). The myocyte cross-sectional area was measured from images captured from wheat germ agglutinin-stained sections (14). The outlines of 100–200 myocytes were traced in each section using NIH ImageJ.
Evaluation of Apoptosis in Tissue Sections and Isolated Cardiomyocytes
DNA fragmentation was detected in situ using TUNEL as described (17). Briefly, deparaffinized sections were incubated with proteinase K, and DNA fragments were labeled with fluorescein-conjugated dUTP using TdT (Roche Applied Science). Nuclear density was determined by manual counting of DAPI-stained nuclei in six fields for each animal using the 40× objective, and the number of TUNEL-positive nuclei was counted by examining the entire section using the same power objective. DNA fragmentation was detected in cultured cells using TUNEL as described previously (17). Nuclear density was determined by counting DAPI-stained nuclei in 20 different fields for each sample.
Evaluation of Necrosis
Cardiomyocytes were transduced with LacZ or Yap1 adenovirus and treated with H2O2 (100 μm) or vehicle for 2 h. Propidium iodide (500 nm) was added to the culture medium for 10 min before DAPI counterstain and fluorescent imaging.
Evaluation of Cardiomyocyte Hypertrophy
After treatment, cardiomyocytes were fixed in 3.7% paraformaldehyde and permeabilized in 0.1% Triton X-100. The cardiomyocyte surface area was measured using troponin T-stained cardiomyocytes, and NIH Image J. Troponin T was detected with Alexa Fluor 594-conjugated goat anti-mouse IgG (Invitrogen). Activity of the ANF promoter was evaluated using ANF-luciferase reporter plasmid (ANF-Luc) as described (16).
Quantitative RT-PCR
RNA was isolated, cDNA was generated, and quantitative RT-PCR performed as described previously (16). Primers used were: Akt1, sense (5′-ACTCATTCCAGACCCACGAC-3′) and antisense (5′-CCGGTACACCACGTTCTTCT-3′); Akt2, sense (5′-GAGCATCGGTTCTTCCTCAG-3′) and antisense (5′-AGAACTGGGGGAAGTGTGTG-3′); Nppa, sense (5′-ATACAGTGCGGTGTCCAACA-3′) and antisense (5′-CGAGAGCACCTCCATCTCTC-3); Nppb, sense (5′-GGAAATGGCTCAGAGACAGC-3′) and antisense (5′-CGATCCGGTCTATCTTCTGC-3′); Myh7, sense (5′-TGATGACTCCGAGGAGCTTT-3′) and antisense (5′-TGACACAGACCCTTGAGCAG-3′); Gapdh, sense (5′-AGACAGCCGCATCTTCTTGT-3′) and antisense (5′-CTTGCCGTGGGTAGAGTCAT-3′). Data were normalized to Gapdh.
Immunoblot Analysis
Ventricular tissue and cardiomyocytes were lysed in buffer containing 50 mm Tris·HCl (pH 7.5), 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholic acid, 1 mm EDTA, 0.1 mm Na3VO4, 1 mm NaF, 50 μm phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, and 5 μg/ml leupeptin. For immunoblotting, antibodies against Akt, phospho-AktSer-473, glycogen synthase kinase 3 (GSK-3β), phospho-GSK-3βSer-9, Yap1, phospho-YapSer-127, ERK, phospho-ERKThr-202/Tyr-204, cleaved caspase-3, FLAG, and GAPDH were purchased from Cell Signaling Technology. Phospho-Lats2Ser-872 antibody was generated previously as described (18). Lats2 antibody was purchased from Bethyl Laboratories. TEAD1/TEF antibody was purchased from BD Biosciences. Tubulin antibody was purchased from Sigma.
Immunofluorescence
Heart sections or cardiomyocytes were stained with anti-Ki-67 rabbit polyclonal antibody (Vector Laboratories), anti-phospho(Ser-10)-histone H3 rabbit polyclonal antibody (Cell Signaling), anti-Yap1 rabbit polyclonal antibody (Cell Signaling), anti-troponin-T mouse monoclonal antibody (Neomarkers), Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen), Alexa Fluor 594-conjugated goat anti-mouse IgG (Invitrogen), and Vectashield mounting medium with DAPI (Vector Laboratories). Analyses were performed using fluorescence microscopy (Zeiss).
Measurement of Cardiomyocyte Number in the Heart
Total cardiomyocyte numbers were estimated using a published method (19). Briefly, the number of nuclei per unit area of myocardium (N(n)A) was determined in 20 fields (40×). The average nuclear length (Dn) was determined in longitudinally oriented myocytes. The number of myocyte nuclei per unit volume of myocardium (N(n)V) was calculated using the equation N(n)V = N(n)A/Dn. The estimated total number of myocyte nuclei in the LV was calculated as the product of the number of myocyte nuclei per unit volume, N(n)V and the total ventricular volume, which was derived from the LV weight using the specific gravity of muscle tissue, 1.06 g/ml.
Statistics
All values are expressed as the mean ± S.E. Statistical analyses were performed by either t test or analysis of variance and the Tukey post-test procedure, with p < 0.05 considered significant.
RESULTS
Postnatal Deletion of Yap1 in Cardiomyocytes Causes Dilated Cardiomyopathy and Premature Death
To determine the effect of postnatal deletion of Yap1 in the heart, we inactivated a conditional Yap1 allele (YapF/F) (10) specifically in cardiomyocytes using α-MHC Cre recombinase transgenic mice (13). Depletion of Yap1 protein levels was confirmed by immunoblot analysis using ventricular tissue from adult Yap+/FCre, YapF/FCre, and control YapF/F mice (Fig. 1A). We also determined the specificity of Yap1 disruption and found that Yap1 protein expression was unaffected in other tissues (Fig. 1B). Baseline gravimetric analysis was performed on 8 week-old male mice. We observed a significant increase in both heart weight and LV weight normalized to tibia length in the YapF/FCre mice compared with controls, whereas no difference between Yap+/FCre and control mice was evident (Fig. 1, C and D). The lung weight-to-tibia length ratio was also significantly increased in YapF/FCre mice, suggesting pulmonary congestion (Fig. 1E). To determine differences in cardiac geometry and function, echocardiography was performed. No significant differences in heart shape or function were present between YapF/F and Yap+/FCre mice; however, YapF/FCre mice demonstrated clear septal and posterior wall thinning as well as chamber dilation compared with control mice (Table 1). Furthermore, myocardial function, as determined by left ventricular ejection fraction (LVEF) and fractional shortening (FS), was strikingly reduced in YapF/FCre mice (Fig. 1F, Table 1). Ultimately, the YapF/FCre mice died prematurely due to dilated cardiomyopathy and heart failure, with a median lifespan between 10 and 11 weeks (Fig. 1, G and H). No difference in survival was observed between YapF/F and Yap+/FCre mice.
FIGURE 1.

Base-line cardiac characterization of Yap1-deficient mice. A, shown is immunoblot analysis of Yap1 expression in ventricular tissue of YapF/F, Yap+/FCre, and YapF/FCre adult (8-week-old) mice. B, shown is immunoblot analysis of Yap1 expression in the heart (he), lung (lg), liver (lv), and skeletal muscle (sk) from 8-week-old YapF/F control and YapF/FCre mice. C—E, heart weight (HW), left ventricular weight (LVW), and lung weight were measured and normalized to tibia length (TL) in adult mice. F, echocardiography was performed, and LVEF was determined. Values are the means ± S.E. *, p < 0.05. N.S. = not significant. G, representative M-mode tracings are shown. H, shown is a Kaplan-Meier survival curve demonstrating the premature death of YapF/FCre mice. Log-rank (Mantel-Cox) test for survival, YapF/FCre versus YapF/F, p < 0.0001.
TABLE 1.
Base-line echocardiographic analysis of 8-week-old Yap1-deficient mice
Data are presented as the mean ± S.E. DSEP WT, diastolic septal wall thickness; LVEDD, left ventricle end diastolic dimension; DPWT, diastolic posterior wall thickness; SSEP WT, systolic septal wall thickness; LVESD, left ventricle end systolic dimension; SPWT, systolic posterior wall thickness; HR, heart rate; BW, body weight.
| Parameter | YapF/F | Yap+/FCre | YapF/FCre |
|---|---|---|---|
| n | 9 | 10 | 5 |
| DSEP WT (mm) | 0.64 ± 0.05 | 0.55 ± 0.06 | 0.55 ± 0.09 |
| LVEDD (mm) | 3.52 ± 0.11 | 3.56 ± 0.11 | 4.85 ± 0.42a |
| DPWT (mm) | 0.79 ± 0.05 | 0.65 ± 0.04 | 0.59 ± 0.08a |
| SSEP WT (mm) | 1.14 ± 0.10 | 0.88 ± 0.08a | 0.66 ± 0.07a |
| LVESD (mm) | 2.30 ± 0.10 | 2.48 ± 0.14 | 4.14 ± 0.45a |
| SPWT (mm) | 1.10 ± 0.06 | 0.84 ± 0.04a | 0.60 ± 0.11a |
| LVEF (%) | 71.6 ± 2.8 | 65.8 ± 2.9 | 26.7 ± 7.3a |
| %FS | 34.5 ± 1.9 | 30.6 ± 2.0 | 10.3 ± 3.1a |
| HR | 451 ± 27 | 478 ± 27 | 419 ± 22 |
| BW (g) | 24.2 ± 1.1 | 22.9 ± 0.9 | 21.9 ± 1.4 |
a p < 0.05 versus YapF/F control mice.
Yap1 Deletion Promotes Myocardial Fibrosis, Cardiomyocyte Apoptosis, and Impaired Heart Function
Histological analysis revealed a robust increase in myocardial fibrosis in 8-week-old YapF/FCre mice, whereas no significant difference between Yap+/FCre and control mice was observed at base line (Fig. 2, A–C). Cardiomyocyte apoptosis was also determined by TUNEL. We found no difference in TUNEL-positive cardiomyocytes present between YapF/F and Yap+/FCre heart sections. On the other hand, there was a significant increase in the number of apoptotic cardiomyocytes present in YapF/FCre hearts at base line (Fig. 2, D and E). To determine whether the increase in YapF/FCre LV weight was a result of increased growth of individual myocytes, we stained heart sections with wheat germ agglutinin and analyzed the cross-sectional area (CSA) for each group. We found no significant difference between Yap+/FCre and YapF/F control hearts; however, relative myocyte CSA was significantly increased in YapF/FCre mice (Fig. 2, F and G). Yap1 is an important promoter of cardiomyocyte proliferation in the developing embryonic heart (11, 12, 20). Therefore, we also investigated the effect of postnatal Yap1 deletion on cardiomyocyte proliferation. We determined the number of cardiomyocytes present in hearts from YapF/F, Yap+/FCre, and YapF/FCre mice and found no significant difference between the three groups (Fig. 2H). Taken together, our base-line characterization indicates that adult mice with heterozygous Yap1 deletion in cardiomyocytes showed no significant difference in heart size or function compared with YapF/F control mice, but that homozygous deletion of cardiomyocyte Yap1 caused significant increases in cardiomyocyte apoptosis and interstitial fibrosis, leading to ventricular remodeling associated with severely impaired cardiac function and premature death.
FIGURE 2.

Histological analysis of Yap1-deficient mice. A–G, ventricles from adult mice were fixed, sectioned, and stained. A, shown is hematoxylin and eosin staining demonstrating disorganized cardiomyocytes and increased cell infiltration in YapF/FCre hearts. B and C, Masson's Trichrome staining revealed a significant increase in interstitial fibrosis in YapF/FCre hearts. D and E, TUNEL shows a significant increase in apoptotic cardiomyocytes in YapF/FCre hearts. Arrows indicate TUNEL-positive nuclei. F and G, wheat germ agglutinin (WGA) staining reveals increased myocyte CSA in YapF/FCre hearts. H, shown is the estimated number of cardiomyocytes present in the hearts of Yap1 mutant and control mice. Values are the means ± S.E. *, p < 0.05. N.S. = not significant. Scale bars, 100 μm.
Yap1 Promotes Akt Signaling to Prevent Cardiomyocyte Apoptosis
Yap1 is a transcriptional coactivator; therefore, we reasoned that gene expression is likely altered in hearts deficient for Yap1. To test this hypothesis, we performed a PCR-based and apoptosis-focused gene expression array (SA Biosciences; Ref. 21) using ventricular homogenates from 8-week-old YapF/F and YapF/FCre mice. We observed a striking down-regulation of Akt1 expression in the YapF/FCre heart. To confirm this, we performed immunoblots using YapF/F, Yap+/FCre, and YapF/FCre hearts and found a dose-dependent decrease in both total and phosphorylated Akt (Fig. 3A). To determine the effect of Yap1 expression in vitro, we engineered adenovirus to overexpress Yap1 (2–3-fold) in isolated neonatal rat cardiomyocytes. We then transduced cardiomyocytes with Yap1 or LacZ control and performed quantitative real-time-PCR to detect mRNA expression of Akt1 and Akt2. We found that Yap1 expression significantly increased Akt1 mRNA levels (Fig. 3B). Consistent with these results, Yap1 expression also caused an increase in total Akt protein levels and Akt activation, the latter being sensitive to PI3K inhibition (Fig. 3, C and D). On the other hand, no change in ERK activation or expression was observed in Yap1 mutant hearts or in cardiomyocytes transduced with Yap1 adenovirus (Fig. 3, A, C, and D). Akt is an established protective molecule in many cell types including cardiomyocytes (22). Because Yap1 deletion caused a robust increase in cardiomyocyte apoptosis, we sought to determine if increased Yap1 expression confers protection against apoptosis and if this response is Akt-dependent. We found that transduction of cardiomyocytes with Yap1 adenovirus, but not LacZ, inhibited H2O2-induced apoptosis, as determined by TUNEL and cleaved caspase-3, and that this protection was abolished by treatment with the PI3K inhibitor LY294002 (Fig. 3, E and F). Furthermore, Yap1 expression also attenuated necrotic cell death, as determined by propidium iodide uptake, in response to H2O2 compared with LacZ control (Fig. 3G). Taken together these data indicate that Yap1 has a cell autonomous protective capacity in cardiomyocytes that is mediated by Akt.
FIGURE 3.

Yap1 promotes cardiomyocyte survival through up-regulation of Akt. A, representative immunoblots detect p-Akt(S473), total Akt, p-ERK(T202/Y204), and total ERK from Yap+/F, Yap+/FCre, and YapF/FCre hearts. B, neonatal rat cardiomyocytes were transduced with LacZ control or Yap1 adenovirus, and quantitative real-time-PCR was performed to determine mRNA expression of Akt1 and Akt2. Results are normalized to Gapdh expression. *, p < 0.05. N.S. = not significant. C, cardiomyocytes were transduced with LacZ or Yap1 adenovirus and treated with LY294002 (10 μm) or vehicle. Representative immunoblots are shown. D, quantification of results was obtained from C. *, p < 0.05 versus LacZ. #, p < 0.05 versus Yap1. E, cardiomyocytes were transduced with LacZ or Yap1 adenovirus and treated with LY294002 (10 μm) or vehicle. Cells were then treated with vehicle or H2O2 (100 μmol/liter), and apoptosis was evaluated by TUNEL. Values are the means ± S.E. *, p < 0.05 versus LacZ + vehicle. #, p < 0.05 versus LacZ + H2O2 + vehicle. §, p < 0.05 versus Yap1 + H2O2 + vehicle. F, cardiomyocyte apoptosis was also determined by detection of cleaved caspase-3 (17kD). A representative immunoblot is shown. G, cardiomyocyte viability was determined by propidium iodide (PI) uptake after H2O2 (100 μmol/liter) treatment. Values are the means ± S.E. *, p < 0.05 versus LacZ + vehicle. #, p < 0.05 versus LacZ + H2O2.
Yap1 Induces Akt-independent Cardiomyocyte Hypertrophy
To determine the effect of Yap1 on cardiomyocyte growth, we transduced cardiomyocytes with Yap1 or LacZ adenovirus and determined cardiomyocyte cell surface area. We found that Yap1 expression significantly increased cell size compared with LacZ. We then treated cells with LY294002 or PD98059 to test the role of PI3K/Akt and MEK1/ERK respectively, in this response. We found no significant difference between vehicle and LY294002- or PD98059-treated cardiomyocytes transduced with Yap1, indicating that neither Akt nor ERK activity mediates Yap1-induced hypertrophy (Fig. 4, A and B). Next we measured fetal gene expression associated with hypertrophy. We found that Yap1 expression caused significant increases in Nppa (ANF), Nppb (brain natriuretic protein) and Myh7 (β-MHC) compared with control (Fig. 4C). To test the role of Akt and ERK signaling in fetal gene expression, we performed luciferase reporter assays using the ANF-luc reporter gene in cardiomyocytes. Transduction with Yap1 induced a 4–5-fold increase in ANF luciferase activity compared with LacZ control (Fig. 4D). Interestingly, treatment with LY294002, the Akt inhibitor triciribine (Akt V), or PD98059 did not prevent Yap1-induced ANF-luciferase expression (Fig. 4D), indicating that Akt and ERK do not mediate fetal gene expression elicited by Yap1. Taken together these data indicate that Yap1 is sufficient to activate a hypertrophic response independent of Akt and ERK activity.
FIGURE 4.

Yap1-induced cardiomyocyte hypertrophy is Akt-independent. A, cardiomyocytes were transduced with LacZ or Yap1 adenovirus and treated with LY294002 (10 μm), PD98059 (10 μm), or vehicle and stained to detect troponin T. Scale bars, 10 μm. B, quantification of cardiomyocyte surface area is shown. Values are the means ± S.E. *, p < 0.05. N.S. = not significant. C, cardiomyocytes were transduced with LacZ or Yap1 adenovirus, and quantitative real-time-PCR was performed to detect ANF (Nppa), brain natriuretic protein (Nppb), and β-MHC (Myh7) gene expression. Results were normalized to Gapdh. *, p < 0.05. D, cardiomyocytes were transfected with LacZ or Yap1 in combination with ANF-luc reporter plasmid. Cells were treated with LY294002 (10 μm), Akt inhibitor V (triciribine; 10 μm), PD98059 (10 μm), or vehicle control, and luciferase activity was determined. Values are the means ± S.E. *, p < 0.05 versus LacZ + vehicle. N.S. = not significant.
Yap1 Depletion Inhibits Hypertrophy and Augments Apoptosis in Cardiomyocytes
To test the effect of Yap1 depletion, we generated an adenoviral shRNA construct targeting Yap1 (shYap1) that effectively decreased protein expression in cardiomyocytes (Fig. 5A). We then tested the effect of Yap1 knockdown on phenylephrine-induced hypertrophy. We found that shYap1, but not a scrambled control shRNA adenovirus (shCtrl), significantly attenuated the increases in cell size and ANF-luciferase reporter activity induced by phenylephrine (Fig. 5, B–D). We also found that shYap1 caused a significant increase in H2O2-induced apoptosis compared with shCtrl, additional evidence of the protective function of Yap1 in cardiomyocytes (Fig. 5E).
FIGURE 5.

Depletion of Yap1 attenuates cardiomyocyte hypertrophy and promotes apoptosis. A, shown is a representative immunoblot demonstrating endogenous Yap1 knockdown using adenoviral shRNA. Cardiomyocytes were infected with scrambled control (shCtrl) or Yap1-targeted (shYap1) virus, and cells were harvested 72 h later. B, cardiomyocytes were transduced with shCtrl or shYap1, treated with phenylephrine (PE; 100 μm) or vehicle, and stained to detect troponin T. Scale bars, 10 μm. C, quantification of the cardiomyocyte surface area is shown. Values are the means ± S.E. *, p < 0.05 versus shCtrl + vehicle. #, p < 0.05 versus shCtrl + phenylephrine. D, cardiomyocytes were transfected with shCtrl or shYap1 in combination with ANF-luc reporter plasmid. Cells were treated with phenylephrine (100 μm) or vehicle control, and luciferase activity was determined. Values are the means ± S.E. *, p < 0.05 versus shCtrl + vehicle. #, p < 0.05 versus shCtrl + phenylephrine. E, cardiomyocytes were transduced with shCtrl or shYap1 adenovirus and treated with vehicle or H2O2 (100 μmol/liter), and apoptosis was evaluated by TUNEL. Values are the means ± S.E. *, p < 0.05 versus shCtrl + vehicle. #, p < 0.05 versus shCtrl + H2O2.
MI Increases the Nuclear Localization of Yap1
These data demonstrate the pro-survival and pro-hypertrophic effects of Yap1 on cardiomyocytes. Therefore, we set out to determine the consequence of Yap1 deletion on myocardial injury in vivo. Yap1 is negatively regulated through Ser-127 phosphorylation and cytosolic retention (5). To determine whether Yap1 nuclear localization/activation is increased in response to stress, we subjected wild-type (WT) mice to MI by applying permanent ligation of the left descending coronary artery. Whole ventricle analysis showed increased phosphorylation of Lats2 and Yap1 in MI hearts compared with sham operated controls, consistent with our previous finding that the core components of the mammalian Hippo signaling pathway, namely Mst1/Lats2, are activated during MI and can broadly target Yap1 for phosphorylation and nuclear exclusion (Fig. 6, A and B) (14). However, a more precise evaluation of Yap1 revealed that at 4 days post-occlusion, Yap1 was localized predominantly in the nuclei of cardiomyocytes in the border zone, but not in the remote region of the heart, as evaluated by double staining with anti-troponin T and anti-Yap1 antibodies (Fig. 6, C and D). This was in contrast to sham-operated hearts, which had nearly exclusive cytosolic Yap1. These data indicate that a subpopulation of Yap1 is selectively activated at the site of injury during MI.
FIGURE 6.

Yap1 deficiency exacerbates injury after chronic MI. A, shown is a representative immunoblot demonstrating increased Yap1 phosphorylation at Ser-127 and Lats2 at Ser-872 in hearts of 28-day post-MI mice compared with sham-operated controls. B, shown is quantitation of immunoblot results. *, p < 0.05. C and D, nuclear Yap1 is increased in cardiomyocytes present in the border zone but not in the ischemic or remote zones of MI hearts. C, mice were subjected to permanent coronary ligation. Yap1 staining of the ischemic, border, and remote zones after 4 days is shown. Note that Yap1 accumulates in the nucleus in the border zone (B, arrow), but it is excluded from the nucleus in the ischemic (I, arrowhead) and remote zones comparable to control (sham, thin arrow). Green, anti-Yap1 staining; red, anti-troponin T staining; blue, DAPI. Scale bars, 100 μm. D, shown is quantification of nuclear Yap1-positive cardiomyocytes in the border zone. E, heart weight (HW) normalized to tibia length (TL) was determined after 28 days MI. F, heart sections were stained with wheat germ agglutinin, and CSA was determined in the remote region after MI. G, shown are representative images of Masson's Trichrome-stained Yap+/F and Yap+/FCre hearts after 28 days MI. Scale bars, 1 mm. H, shown is quantification of infarct size as a percentage of total endocardial circumference. I, quantification of fibrosis is shown. J, heart sections were stained by TUNEL to detect cardiomyocytes undergoing apoptosis. K, a representative immunoblot demonstrates increased Akt phosphorylation in hearts from Yap+/F but not Yap+/FCre mice subjected to 2-day MI compared with sham operated controls. Values are the means ± S.E. *, p < 0.05. N.S. = not significant.
Yap1 Deficiency Modulates Cardiac Remodeling in Response to MI
Using this same model, we subjected Yap+/F and Yap+/FCre mice to chronic MI for a 4-week period. YapF/FCre mice were not used due to their severe phenotype at baseline. First, we confirmed that the size of the initial myocardial infarction (at day 2), as evaluated by 2,3,5-triphenyltetrazolium chloride staining, was similar between Yap+/F and Yap+/FCre mice (data not shown). Postmortem measurements revealed that heart weight/tibia length did not differ between Yap+/F and Yap+/FCre mice after a 28-day MI (Fig. 6E). However, there was a modest but significant decrease in CSA of cardiomyocytes present in the remote region of Yap+/FCre hearts compared with Yap+/F, indicating a blunted hypertrophic response (Fig. 6F). Heart sections were stained with Masson's Trichrome to analyze the size of MI. We found that infarct size, as determined by planimetry of the infarct normalized to the endocardial circumference, was larger in Yap+/FCre compared with Yap+/F mice, suggesting that impaired healing or expansion of MI occurred in Yap+/FCre mice (Fig. G and H). Myocardial fibrosis and cardiomyocyte apoptosis, as determined by Masson's Trichrome and TUNEL, respectively, were also significantly increased in the border and remote regions of Yap+/FCre versus Yap+/F hearts after MI (Fig. 6, I and J). Interestingly, we found that Akt is activated after a 2-day MI in Yap+/F hearts; however, this response is dramatically attenuated in Yap+/FCre hearts, indicating impaired Akt signaling in response to injury (Fig. 6K). We also assessed cardiac remodeling and function by performing serial echocardiography at 1, 2, and 4 weeks post-MI. We observed significant decreases in LVEF and FS between Yap+/F and Yap+/FCre mice at each time point after MI (Fig. 7, A and B). As expected Yap+/F hearts became dilated in response to MI; however, Yap+/FCre ventricular chambers were further enlarged at each time point (Fig. 7, C and D). We also determined wall thickness and found that Yap+/FCre hearts had significantly thinner posterior walls compared with Yap+/F control mice at 1, 2, and 4 weeks post-MI (Fig. 7, E and F, Table 2). Taken together, these findings demonstrate that Yap1 is activated at the site of myocardial injury and that Yap+/FCre mice have an impaired ability to compensate in response to chronic myocardial stress.
FIGURE 7.

Cardiac function is further impaired in Yap-deficient hearts after MI. A–F, serial echocardiography was performed at 0, 7, 14, and 28 days post-MI. LVEF (A), fractional shortening (% FS; B), left ventricular end diastolic dimension (LVEDD; C), left ventricular end systolic dimension (LVESD; D), systolic posterior wall thickness (SPWT; E), and diastolic posterior wall thickness (DPWT; F) were determined in Yap+/F and Yap+/FCre mice. Values are the means ± S.E. *, p < 0.05 versus Yap+/F control at the same time point.
TABLE 2.
Echocardiographic analysis of control and Yap1-deficient mice after 28-d MI.
Data are presented as mean ± S.E. DSEP WT, diastolic septal wall thickness; LVEDD, left ventricle end diastolic dimension; DPWT, diastolic posterior wall thickness; SSEP WT, systolic septal wall thickness; LVESD, left ventricle end systolic dimension; SPWT, systolic posterior wall thickness; HR, heart rate; BW, body weight.
| Parameter | Yap+/F | Yap+/FCre |
|---|---|---|
| n | 10 | 13 |
| DSEP WT (mm) | 0.77 ± 0.05 | 0.59 ± 0.02a |
| LVEDD (mm) | 4.15 ± 0.15 | 5.05 ± 0.15a |
| DPWT (mm) | 0.82 ± 0.05 | 0.60 ± 0.04a |
| SSEP WT (mm) | 1.25 ± 0.06 | 0.84 ± 0.04a |
| LVESD (mm) | 2.87 ± 0.18 | 4.36 ± 0.17a |
| SPWT (mm) | 1.14 ± 0.06 | 0.74 ± 0.05a |
| LVEF (%) | 41.1 ± 3.9 | 17.9 ± 2.3a |
| %FS | 16.6 ± 1.9 | 6.5 ± 0.9a |
| HR | 535 ± 14 | 517 ± 16.8 |
| BW (g) | 25.9 ± 0.6 | 25.1 ± 0.6 |
a p < 0.05 versus Yap+/F control mice.
Cardiomyocyte Proliferation Is Attenuated in Yap1-deficient Mice
Yap1 promotes cell cycle progression and cell proliferation in non-cardiac cell types as well as cardiomyocytes in the embryonic heart (10–12). Although the adult heart has a limited capacity for regeneration, a small population of cardiomyocytes can cycle in response to injury and may confer a regenerative benefit to the heart (19). To test whether a deficiency in Yap1 affects the ability of adult cardiomyocytes to enter the cell cycle, we stained heart sections for the proliferative marker Ki-67. Quantitative analysis revealed a small population of Ki-67-positive cardiomyocytes in the border zone and remote regions of Yap+/F control hearts in response to MI. Interestingly, the number of Ki-67-positive myocytes observed in Yap+/FCre hearts was significantly reduced compared with Yap+/F controls (Fig. 8, A and B). To further test the effect of Yap1 deletion on cardiomyocyte proliferation, we stained for histone H3 phosphorylated at Ser-10 (p-HH3), a marker of M-phase, in hearts from both groups. p-HH3-positive cardiomyocytes were rare but detectable in post-MI hearts. We found significantly fewer p-HH3-positive myocytes in Yap+/FCre hearts compared with Yap+/F controls, further supporting the hypothesis that Yap1 is an important regulator of cardiomyocyte proliferation in response to chronic MI (Fig. 8, C and D). Finally, we used a gain-of-function approach to test the effect of increased Yap1 expression on the ability of cardiomyocytes to undergo DNA synthesis. We found a significant increase in Ki-67-positive staining in Yap1-transduced myocytes compared with LacZ control (Fig. 8, E and F). Similarly, we found that Yap1 also increased the percentage of p-HH3-positive cardiomyocytes compared with control treatment (Fig. 8, G and H). Importantly, we observed no effect of LY294002 treatment on Yap1-driven cardiomyocyte proliferation, indicating that Akt does not mediate this response. Taken together, these results demonstrate that loss of endogenous Yap1 impairs, whereas an increase in Yap1 promotes cardiomyocyte proliferation after injury.
FIGURE 8.
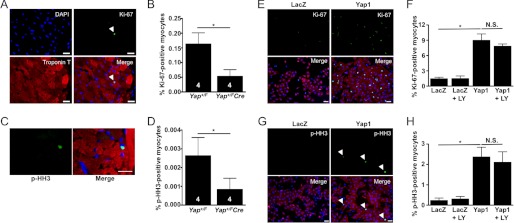
Cardiomyocyte proliferation is attenuated in Yap1-deficient hearts after MI. A, heart sections were stained with anti-Ki-67 (green), anti-troponin T (red), and DAPI (blue). The arrow indicates Ki-67-positive myocyte. B, shown is quantification of Ki-67-positive cardiomyocytes detected in Yap+/F and Yap+/FCre hearts. C and D, hearts were stained with anti-phospho(Ser-10)-histone H3 (green), anti-troponin T (red), and DAPI (blue). C, shown is a representative image of p-HH3-positive cardiomyocyte. D, quantification of p-HH3-positive cardiomyocytes is shown. Note that Ki-67 staining generally showed greater sensitivity than p-HH3 staining in our hands. Scale bars, 50 μm. E, neonatal rat cardiomyocytes were treated with Yap1 or LacZ adenovirus and vehicle or LY294002 (10 μm) then stained with anti-Ki-67 (green), anti-troponin T (red), and DAPI (blue). Scale bars, 30 μm. F, quantification of Ki-67-positive myocytes (%). G, cardiomyocytes were transduced with LacZ or Yap1 adenovirus and vehicle or LY294002 (10 μm) then stained with anti-p-HH3 (green), anti-troponin T (red), and DAPI (blue). Arrows indicate p-HH3-positive myocytes. Scale bars, 30 μm. H, quantification of p-HH3-positive cardiomyocytes (%) is shown. Values are the means ± S.E. *, p < 0.05. N.S. = not significant.
DISCUSSION
Here we investigated the function of endogenous Yap1 in the myocardium of adult mice both under basal conditions and in response to chronic MI by specifically disrupting postnatal Yap1 expression in cardiomyocytes. The major findings of this study are that 1) Yap1 is essential for homeostasis and normal function of the postnatal heart, 2) homozygous deletion of Yap1 in cardiomyocytes causes dilated cardiomyopathy and premature death, 3) Yap1 drives a pro-growth, pro-survival phenotype in a cell autonomous manner in cardiomyocytes, 4) heterozygous disruption of cardiomyocyte Yap1 is sufficient to accelerate myocyte loss, fibrosis, and cardiac dilation and further depress cardiac function following MI, and 5) cardiomyocyte compensatory hypertrophy and proliferation are impaired in post-MI mouse hearts deficient for Yap1.
Yap1 is a transcriptional coactivator and the most studied downstream effector of the mammalian Hippo signaling pathway. The core of this cascade involves the kinases Mst1 and Lats2 and, when activated, leads to increased phosphorylation of Yap1 at Ser-127, exclusion of Yap1 from the nucleus, and inhibition of Yap1-mediated gene expression. Whether or not upstream Hippo signaling intermediates can modulate Yap1 activation in the adult heart remains to be seen; however, disruption of Mst1/2, Salvador, and Lats1/2 modulates Yap1 phosphorylation in the mouse liver (23–25). Furthermore, our previous work has demonstrated that increased Mst1 expression in the mouse heart elicits increased cardiomyocyte apoptosis that is similar to what we observe in our current Yap1 loss-of-function model. Indeed, Mst1 transgenic mice developed dilated cardiomyopathy leading to premature death due to heart failure (17). We now show that homozygous disruption of myocardial Yap1 resulted in a similar dilated phenotype with accelerated mortality. Taken together, these observations raise the likelihood that Mst1 and Yap1 function in the same pathway and that Mst1 negatively regulates Yap1 in the adult heart.
Maintenance of cardiomyocyte number and the increased growth of surviving cardiomyocytes are two important factors that modulate preservation of myocardial function in response to stress. Our previous work has shown that activation of Mst1 or Lats2 in the heart promotes cardiomyocyte apoptosis while inhibiting myocyte hypertrophy after myocardial insult (16, 17). Similarly, deletion of Yap1 led to increased myocyte death and a blunted hypertrophic response at the cardiomyocyte level after chronic MI. This outcome is especially detrimental for the heart in that there are fewer remaining cardiomyocytes that cannot compensate through increased growth, leading to enhanced thinning of the myocardium, increased wall stress, and further impaired cardiac output. These findings highlight Yap1 signaling as a novel mechanism of myocardial protection and compensatory hypertrophy in response to stress.
Myocardial fibrosis and apoptosis are associated with, and contribute to, declining heart function (26, 27). Our data demonstrate significant increases in interstitial fibrosis and cardiomyocyte apoptosis in YapF/FCre hearts under basal conditions and in Yap+/FCre hearts in response to MI. Increased apoptosis contributes to overall myocyte loss, and it is likely that the increased fibrosis is primarily a result of the replacement of nonviable myocardium. We also predict that the increased fibrosis accounts for the maintenance of heart weight observed in Yap+/FCre mice despite impaired cardiomyocyte hypertrophy and reduced cardiomyocyte proliferation after MI.
Another key finding of this study is the attenuation of cardiomyocyte proliferation observed in Yap1 deficient hearts after chronic MI. Earlier reports have examined the function of proximal Hippo pathway members, Salvador, Mst1/2, and Lats1/2 as well as Yap1 directly in the embryonic mouse heart (11, 12, 20). These studies concluded that the mammalian Hippo signaling pathway and Yap1 in particular is critical in promoting cardiomyocyte proliferation during heart development. Disruption of Yap1 activation during gestation inhibited proliferation of cardiomyocytes, leading to hypoplastic hearts and embryonic lethality. Conversely, forced expression of activated Yap1 promoted myocyte proliferation both in vitro and in vivo (12). We have found that postnatal disruption of Yap1 did not affect total cardiomyocyte number, indicating that myocytes have most likely exited the cell cycle and stopped proliferating before Cre-mediated Yap1 deletion. However, we also demonstrate that after MI, Yap1 deficient mice had fewer cardiomyocytes positive for either Ki-67 or p-histone H3, markers of DNA synthesis and M-phase, respectively, indicating that Yap1 is important for cardiomyocyte proliferation in the injured adult heart. Additionally, increased Yap1 expression increased Ki-67 expression and histone H3 phosphorylation in isolated cardiomyocytes, suggesting that Yap1 is sufficient to promote cell autonomous proliferation. Although the absolute number of proliferating cardiomyocytes is far less in adult than in embryonic myocardium, this could nonetheless be an important endogenous mechanism of repair (19). In fact, we observed a significantly larger infarct size in Yap+/FCre hearts compared with Yap+/F controls. We propose that the increased myocyte loss due to increased apoptosis and the impaired cardiomyocyte proliferation in the Yap1-deficient mice contributes to the expansion of MI and resulting heart failure. Although our findings demonstrate that Yap1 is expressed in cardiomyocytes, its expression and function in stem cells (28) or cardiac progenitor cells is currently under investigation and may play an important role in contributing to regeneration in response to injury.
Importantly, we also provide evidence that Yap1 promotes cardiomyocyte hypertrophy in vitro. Furthermore, in Yap1-deficient hearts, cardiomyocyte CSA was significantly reduced compared with control hearts after MI. These knock-out data point to Yap1 as a critical endogenous regulator of cardiomyocyte growth in response to stress. The lack of compensatory hypertrophy may also contribute to the LV dysfunction observed in the post-MI heart as it should increase wall stress, which in turn increases oxygen consumption and myocardial cell death. Although we did observe larger cardiomyocytes in YapF/FCre compared with YapF/F hearts at base line, this difference was most likely due to the profound cardiac dysfunction present in the YapF/FCre mice, which acts as a stressor to drive secondary effects including increased myocyte growth. Activation of numerous signaling pathways can elicit cardiomyocyte hypertrophy at base line (normal growth), and we speculate that this effect is Yap1-independent. On the other hand, endogenous Yap1 plays an essential role in mediating hypertrophy in response to volume overload. A prior study tested the role of Yap1 in cardiomyocyte hypertrophy by injecting a Cre-expressing adenovirus into Yap1flox adult mutant mice. After aortic constriction, no difference between the size of Yap1-deleted myocytes (marked by GFP) and control myocytes was observed (12). This apparent discrepancy may be a result of the different method of Yap1 manipulation or the different stress used to stimulate hypertrophy. Based on our findings, we believe that compensatory cardiomyocyte hypertrophy after chronic MI is regulated at least in part through Yap1 and that a deficiency in Yap1 impairs heart endogenous growth response to stress, leading to worsened function.
Selective Yap1 deletion has also been reported in the mouse liver (10). These mice developed enlarged livers with cirrhosis and impaired function. Interestingly, Yap1 deficiency caused increases in hepatocyte apoptosis and fibrosis. Conversely, transgenic expression of Yap1 conferred resistance to apoptosis of hepatocytes, indicating a pro-survival function of Yap1 in the liver (8, 9) similar to what we observe in cardiomyocytes. Taken together with our current work, these findings suggest a conserved role of Yap1 to promote cell survival, maintain mammalian organ homeostasis, and preserve normal function.
We now demonstrate that Yap1 promotes Akt expression and activation and protects cardiomyocytes against apoptosis in an Akt-dependent manner. On the other hand, Yap1-induced hypertrophy occurs independent of Akt. It, therefore, remains fundamental to further elucidate the mechanism of Yap1-mediated compensatory hypertrophy and protection of cardiomyocytes in the adult myocardium. Yap1 functions as a coactivator for the TEAD/TEF family of transcription factors and regulates the expression of a growing list of gene targets that mediate its downstream effects (29). In addition to canonical Hippo signaling, prior work has also demonstrated cross-talk between Yap1 and the Wnt/β-catenin and IGF-1 signaling pathways to modulate embryonic cardiomyocyte proliferation (11, 20). Furthermore, microRNAs, whose function is homologous to Drosophila bantam (30), may also mediate the effect of Yap1 upon growth and death of cardiomyocytes. In fact, we have recently found that miR-206 may mediate Yap1-induced cardiomyocyte hypertrophy.5 Whether the lack of compensatory hypertrophy observed in Yap+/FCre mice after MI is mediated through down-regulation of miR-206 remains to be elucidated.
In summary, we demonstrate that, in response to stress, endogenous Yap1 is a critical regulator of cardiomyocyte growth and survival in the adult mouse myocardium. We have found that a deficiency in Yap1 exacerbates injury after chronic MI, suggesting that specific targeting of Yap1 activation in cardiomyocytes could serve to ameliorate the damage caused by myocardial ischemia. These findings highlight Yap1 as a possible therapeutic target of activation for patients suffering from MI.
Acknowledgment
We thank Daniela Zablocki for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL59139, HL67724, HL69020, HL91469, and AG27211 (United States Public Health Service). This work was also supported by the Fondation Leducq Transatlantic Network of Excellence (to J. S.).
Y. Yang, N. Nakano, J. Park, D. Sayed, S. Matsushima, S. Sciarretta, D. P. Del Re, Y. Park, B. Tian, M. Abdellatif, and J. Sadoshima, manuscript in preparation.
- MI
- myocardial infarction
- ANF
- atrial natriuretic factor
- CSA
- cross-sectional area
- FS
- fractional shortening
- LV
- left ventricle
- LVEF
- LV ejection fraction
- p-HH3
- phosphohistone H3
- TEAD/TEF
- TEA domain/transcriptional enhancer factor
- Yap1
- Yes-associated protein isoform 1.
REFERENCES
- 1. Velagaleti R. S., Pencina M. J., Murabito J. M., Wang T. J., Parikh N. I., D'Agostino R. B., Levy D., Kannel W. B., Vasan R. S. (2008) Long term trends in the incidence of heart failure after myocardial infarction. Circulation 118, 2057–2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Narula J., Haider N., Virmani R., DiSalvo T. G., Kolodgie F. D., Hajjar R. J., Schmidt U., Semigran M. J., Dec G. W., Khaw B. A. (1996) Apoptosis in myocytes in end-stage heart failure. N. Engl. J. Med. 335, 1182–1189 [DOI] [PubMed] [Google Scholar]
- 3. Olivetti G., Abbi R., Quaini F., Kajstura J., Cheng W., Nitahara J. A., Quaini E., Di Loreto C., Beltrami C. A., Krajewski S., Reed J. C., Anversa P. (1997) Apoptosis in the failing human heart. N. Engl. J. Med. 336, 1131–1141 [DOI] [PubMed] [Google Scholar]
- 4. Mani K., Kitsis R. N. (2003) Myocyte apoptosis. Programming ventricular remodeling. J. Am. Coll. Cardiol. 41, 761–764 [DOI] [PubMed] [Google Scholar]
- 5. Pan D. (2010) The Hippo signaling pathway in development and cancer. Dev. Cell 19, 491–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Badouel C., Garg A., McNeill H. (2009) Herding Hippos. Regulating growth in flies and man. Curr. Opin. Cell Biol. 21, 837–843 [DOI] [PubMed] [Google Scholar]
- 7. Halder G., Johnson R. L. (2011) Hippo signaling. Growth control and beyond. Development 138, 9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dong J., Feldmann G., Huang J., Wu S., Zhang N., Comerford S. A., Gayyed M. F., Anders R. A., Maitra A., Pan D. (2007) Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Camargo F. D., Gokhale S., Johnnidis J. B., Fu D., Bell G. W., Jaenisch R., Brummelkamp T. R. (2007) YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 17, 2054–2060 [DOI] [PubMed] [Google Scholar]
- 10. Zhang N., Bai H., David K. K., Dong J., Zheng Y., Cai J., Giovannini M., Liu P., Anders R. A., Pan D. (2010) The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev. Cell 19, 27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xin M., Kim Y., Sutherland L. B., Qi X., McAnally J., Schwartz R. J., Richardson J. A., Bassel-Duby R., Olson E. N. (2011) Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal. 4, ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. von Gise A., Lin Z., Schlegelmilch K., Honor L. B., Pan G. M., Buck J. N., Ma Q., Ishiwata T., Zhou B., Camargo F. D., Pu W. T. (2012) YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 109, 2394–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Agah R., Frenkel P. A., French B. A., Michael L. H., Overbeek P. A., Schneider M. D. (1997) Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Invest. 100, 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Odashima M., Usui S., Takagi H., Hong C., Liu J., Yokota M., Sadoshima J. (2007) Inhibition of endogenous Mst1 prevents apoptosis and cardiac dysfunction without affecting cardiac hypertrophy after myocardial infarction. Circ. Res. 100, 1344–1352 [DOI] [PubMed] [Google Scholar]
- 15. Sciarretta S., Zhai P., Shao D., Maejima Y., Robbins J., Volpe M., Condorelli G., Sadoshima J. (2012) Rheb is a critical regulator of autophagy during myocardial ischemia. Pathophysiological implications in obesity and metabolic syndrome. Circulation 125, 1134–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matsui Y., Nakano N., Shao D., Gao S., Luo W., Hong C., Zhai P., Holle E., Yu X., Yabuta N., Tao W., Wagner T., Nojima H., Sadoshima J. (2008) Lats2 is a negative regulator of myocyte size in the heart. Circ. Res. 103, 1309–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamamoto S., Yang G., Zablocki D., Liu J., Hong C., Kim S. J., Soler S., Odashima M., Thaisz J., Yehia G., Molina C. A., Yatani A., Vatner D. E., Vatner S. F., Sadoshima J. (2003) Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J. Clin. Invest. 111, 1463–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yabuta N., Okada N., Ito A., Hosomi T., Nishihara S., Sasayama Y., Fujimori A., Okuzaki D., Zhao H., Ikawa M., Okabe M., Nojima H. (2007) Lats2 is an essential mitotic regulator required for the coordination of cell division. J. Biol. Chem. 282, 19259–19271 [DOI] [PubMed] [Google Scholar]
- 19. Beltrami A. P., Urbanek K., Kajstura J., Yan S. M., Finato N., Bussani R., Nadal-Ginard B., Silvestri F., Leri A., Beltrami C. A., Anversa P. (2001) Evidence that human cardiac myocytes divide after myocardial infarction. N. Engl. J. Med. 344, 1750–1757 [DOI] [PubMed] [Google Scholar]
- 20. Heallen T., Zhang M., Wang J., Bonilla-Claudio M., Klysik E., Johnson R. L., Martin J. F. (2011) Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Del Re D. P., Matsuda T., Zhai P., Gao S., Clark G. J., Van Der Weyden L., Sadoshima J. (2010) Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J. Clin. Invest. 120, 3555–3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morisco C., Zebrowski D., Condorelli G., Tsichlis P., Vatner S. F., Sadoshima J. (2000) The Akt-glycogen synthase kinase 3β pathway regulates transcription of atrial natriuretic factor induced by β-adrenergic receptor stimulation in cardiac myocytes. J. Biol. Chem. 275, 14466–14475 [DOI] [PubMed] [Google Scholar]
- 23. Zhou D., Conrad C., Xia F., Park J. S., Payer B., Yin Y., Lauwers G. Y., Thasler W., Lee J. T., Avruch J., Bardeesy N. (2009) Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16, 425–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu L., Li Y., Kim S. M., Bossuyt W., Liu P., Qiu Q., Wang Y., Halder G., Finegold M. J., Lee J. S., Johnson R. L. (2010) Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. U.S.A. 107, 1437–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Song H., Mak K. K., Topol L., Yun K., Hu J., Garrett L., Chen Y., Park O., Chang J., Simpson R. M., Wang C. Y., Gao B., Jiang J., Yang Y. (2010) Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl. Acad. Sci. U.S.A. 107, 1431–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Conrad C. H., Brooks W. W., Hayes J. A., Sen S., Robinson K. G., Bing O. H. (1995) Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation 91, 161–170 [DOI] [PubMed] [Google Scholar]
- 27. Wencker D., Chandra M., Nguyen K., Miao W., Garantziotis S., Factor S. M., Shirani J., Armstrong R. C., Kitsis R. N. (2003) A mechanistic role for cardiac myocyte apoptosis in heart failure. J. Clin. Invest. 111, 1497–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao B., Tumaneng K., Guan K. L. (2011) The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 13, 877–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao B., Li L., Lei Q., Guan K. L. (2010) The Hippo-YAP pathway in organ size control and tumorigenesis. An updated version. Genes Dev. 24, 862–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thompson B. J., Cohen S. M. (2006) The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell 126, 767–774 [DOI] [PubMed] [Google Scholar]