

Background: Little is known about the role of JNK mitochondrial signaling in cardiomyocyte cell death.
Results: Global and mitochondrial inhibition of JNK protects against I/R injury thus reducing infarct volume.
Conclusion: Blocking JNK mitochondrial translocation or JNK inhibition may be an effective treatment for I/R-induced cardiomyocyte death.
Significance: These findings suggest a new molecular target for JNK inhibition.
Keywords: Enzyme Inhibitors, Ischemia, c-Jun N-terminal Kinase (JNK), Mitochondria, Signal Transduction
Abstract
To build upon recent findings that mitochondrial JNK signaling is inhibited by selectively blocking the interaction between JNK and Sab, we utilized a cell-permeable peptide to demonstrate that ischemia/reperfusion (I/R) injury could be protected in vivo and that JNK mitochondrial signaling was the mechanism by which reactive oxygen species (ROS) generation, mitochondrial dysfunction, and cardiomyocyte cell death occur. We also demonstrated that 5 mg/kg SR-3306 (a selective JNK inhibitor) was able to protect against I/R injury, reducing infarct volume by 34% (p < 0.05) while also decreasing I/R-induced increases in the activity of creatine phosphokinase and creatine kinase-MB. TUNEL staining showed that the percent TUNEL positive nuclei in rat hearts increased 10-fold after I/R injury and that this was reduced 4-fold (p < 0.01) by SR-3306. These data suggest that blocking JNK mitochondrial translocation or JNK inhibition prevents ROS increases and mitochondrial dysfunction and may be an effective treatment for I/R-induced cardiomyocyte death.
Introduction
The role of mitochondrial JNK is emerging as a significant new area of study, and the resulting oxidative stress, mitochondrial dysfunction, and cell death caused by JNK translocation have been associated with numerous cell types, tissues, and disease states (1–6). Beginning in 2000, and following with a subsequent series of reports over the next 4 years, Davis, Flavell, and colleagues (7–10) utilized JNK-deficient mouse embryonic fibroblasts to establish that JNK is required for TNF-α-stimulated ROS production and cytochrome c-mediated cell death and that Bcl2 family members are essential components of this mitochondrial apoptotic machinery.
It has long been established that oxidative stress and ischemia/reperfusion injury cause cardiomyocyte death. Many studies have linked activation of the JNK mitochondrial pathway to cardiomyocyte cell death both in vitro and in vivo (1, 11–13). For example, Molkentin and colleagues (13) showed that jnk1−/− or jnk2−/−, or transgenic mice expressing dominant negative JNK1/2, have less injury and cellular apoptosis following I/R2 injury in vivo. Gottlieb and colleagues (12) demonstrated activation of JNK in rabbits subjected to coronary artery ligation followed by reperfusion in vivo as well as in vitro in isolated adult rabbit cardiomyocytes. In the latter case, virally expressed dominant negative JNK2 or JNK-interacting protein-1 (JIP) was shown to be protective against simulated I/R in these cells (12). Finally, Ferrandi et al. (11) showed that inhibition of JNK by AS601245, a JNK small molecule ATP competitive inhibitor, decreases cardiomyocyte apoptosis and infarct size in rats after I/R, suggesting a therapeutic benefit of JNK inhibition. Importantly, none of these reports studied the molecular interactions of JNK with the mitochondria, and none measured ROS generation, protein carbonylation, or mitochondrial function in vitro or in vivo.
Given our recent work in which we showed that mitochondrial JNK signaling initiates physiological changes in HeLa cells stressed with anisomycin resulting in amplification of ROS (6), and the ability to selectively block the subsequent mitochondrial dysfunction and cell death associated with translocation (14), we decided to see whether these effects could be generated in cardiomyocyte-like H9c2 cells and during I/R in vivo. In 2002 Wiltshire et al. (15) established the mitochondrial membrane protein Sab (SH3BP5) as the JNK-interacting binding partner for JNK mitochondrial association. Thus, we designed a retro-inverso peptide containing the HIV-Tat sequence along with 20 residues from the Sab KIM1 domain (Tat-SabKIM1), which could be used for in vivo purposes (14). The current study was designed to test whether blocking JNK translocation to the mitochondria would prevent oxidative stress-induced cardiomyocyte death in vitro or I/R-induced cardiomyocyte death in vivo. To do this we utilized rat H9c2 cardiomyocyte-like cells and the I/R model in rats and monitored ROS generation, mitochondrial membrane potential (MMP), and cell death in vitro, as well as protein carbonylation, lipid peroxidation, creatine phosphokinase (CPK) and creatine kinase (CK-MB) activity, apoptosis, and infarct volume in vivo. The major findings showed that oxidative and I/R-stress induced JNK translocation to the mitochondria and that blocking this translocation with Tat-SabKIM1 peptide reduced ROS generation, increased MMP, and reduced cell death in vitro while decreasing I/R-induced protein carbonylation, and lipid peroxidation levels and decreasing infarct volumes and apoptosis in vivo. Moreover, our results showed that blocking JNK translocation to the mitochondria was similarly as effective at reducing all of the in vitro endpoints as well as reducing infarct volumes as was inhibiting the catalytic activity of JNK. These results suggest that inhibition of JNK mitochondrial signaling may be a new mechanism and molecular target for treating I/R injury.
EXPERIMENTAL PROCEDURES
Materials
Tat-Scramble (LPSVFGDVGAPSRLPEVSLSPPRRRQRRKKRG-NH2) and Tat-SabKIM1 (GFESLSVPSPLDLSGPRVVAPPRRRQRRKKRG-NH2) peptides were purchased from NeoPeptide.
Cell Culture
H9c2 cells and primary human cardiomyocytes (from ATCC) were grown under normal cell culture conditions (37 °C and 5% CO2) in DMEM (Invitrogen) supplemented with 10% fetal bovine serum and penicillin/streptomycin. To assure that the cells were actively growing, only cells at ∼80% confluency and between passages 5 and 15 were used in our experiments. H9c2 cells and primary human cardiomyocytes were exposed to 500 nm SR-3306, 500 nm SR-3562, 0.01% DMSO vehicle control, 10 μm Tat-SabKIM1, and 10 μm Tat-scramble for 30 min prior to the addition of stress. To induce oxidative stress and mitochondrial dysfunction in H9c2 cells and primary human cardiomyocytes, we added 100 μm hydrogen peroxide (H2O2)/FeSO4 or 100 μm hydrogen peroxide (H2O2)/FeSO4 directly to the media of the cells. The cells were exposed to H2O2/FeSO4 for the times indicated in the experiments.
Mitochondria Enrichment
Mitochondria were isolated similarly to the method described by Palloti and Lenaz (16) and as we previously described (6, 14). Briefly, 3 × 108 H9c2 cells were grown to ∼75% confluency in 150-mm cell culture dishes. H9c2 cells were washed with PBS three times at room temperature. The cells were lifted from the surface in 0.05% trypsin-EDTA and washed twice with PBS. The cells were pelleted by centrifugation (1000 × g for 15 min) at room temperature. Cells were suspended in 6 times the pellet volume with homogenization buffer (150 mm MgCl2, 10 mm KCl, 10 mm Tris-HCl, pH 6.7). Cells were incubated in the solution on ice for 2 min. H9c2 cells were homogenized with a glass homogenizer using 10 up-down strokes, and cellular disruption was confirmed by microscopy. Homogenization buffer supplemented with 0.25 m sucrose was added at one-third the homogenate volume. The homegenate was mixed thoroughly with gently inversion. The homogenate was separated by centrifugation at 1,000 × g for 5 min at 4 °C to dissociate the nuclear fraction from the homogenate. That supernatant was centrifuged at 5,000 × g for 10 min at 4 °C. The resulting pellet was suspended in ice-cold sucrose-magnesium buffer (150 mm MgCl2, 250 mm sucrose, 10 mm Tris-HCl, pH 6.7), and it was disrupted with an ice-cold Dounce homogenizer (three strokes). The pellet homogenate was centrifuged at 5,000 × g for 10 min at 4 °C. The pellet was then suspended in lysis buffer for either Western blot analysis or immunoprecipitation. All buffers contained both phosphatase and protease inhibitors to protect signaling components.
Outer Mitochondrial Membrane and Mitoplast Preparation
The outer mitochondrial membrane preparation was obtained by methods described in Schnaitman et al. (17).
Nuclear Subcellular Fractionation
Mitochondrial Superoxide Generation
Mitochondrial superoxide production was monitored by utilizing the mitochondria-selective dye MitoSOX-Red (Invitrogen) (18).
Mitochondrial Membrane Potential
Mitochondrial membrane potential was monitored using the JC-1 dye kit (Cayman Chemical). Cells were seeded in a 96-well black-walled, clear-bottomed plate as described for MitoSOX staining above. The cells were then stained with 5 μm JC-1 for 20 min as indicated in the manufacturer's protocol. The cells were washed three times in Hanks' balanced salt solution and covered in prewarmed Hanks' balanced salt solution for fluorescence measurements. The green JC-1 species were detected by excitation at 460 nm and monitoring emission at 488 nm. JC-1 aggregate staining was normalized to JC-1 monomer staining.
Cell Viability
Cell viability of H9c2 cells and primary human cardiomyocytes was monitored by MTT assay (Cayman Chemical). Cells (H9c2 and primary human cardiomyocytes, 20,000 cells/well) were seeded in a 96-well plate (clear bottom) and treated as described below in the text. At the culmination of each treatment, the cells were treated with the MTT reagent. Absorbance was monitored in a SpectraMax e5 plate reader (Molecular Devices).
Biological Replicates and Statistics
A minimum of four biological replicates was considered for all cell-based studies. For mitochondrial enrichments, a minimum of three biological replicates was used. Biochemical assays, fluorometric detection of superoxide, and other cellular measures were done with a minimum of five experimental replicates. To determine statistical significance, Student's paired t test was employed. Statistical significance is indicated by an asterisk in figures in which the p value is less than 0.05.
Western Blotting
Western blot analysis was performed as described (6, 14, 19). The antibodies used for Western blot analysis were: phospho-JNK (Cell Signaling Technology, catalog No. 9255), JNK (Cell Signaling Technology, No. 9258), phospho-c-Jun (Cell Signaling Technology, No. 3270), c-Jun (Cell Signaling Technology, No. 2315), α-tubulin (Cell Signaling Technology, No. 2125), MKK4 (Cell Signaling Technology, No. 9152), Sab (Novus Biologicals, H00009467-M01), COX-IV (Cell Signaling Technology, No. 4850), enolase (Cell Signaling Technology, No. 3810), calnexin (Cell Signaling Technology, No. 2679), and histone H3 (Cell Signaling Technology, No. 4499).
Immunoprecipitation
Mitochondrial lysates were produced by adding Nonidet P-40 buffer supplemented with phosphatase and protease inhibitors to mitochondrially enriched pellets. After thorough mixing, immunoprecipitations were performed using the Pierce co-immunoprecipitation kit protocol. Briefly, protein-anti-Sab antibody (Novus Biologicals, H00009467-M01) was coupled to the coupling resin included in the kit. The antibody resin was next incubated with 85 μg of mitochondrial lysate overnight at 4 °C. The antibody resin and accompanying complexes were acquired by centrifugation followed by the removal of the protein complex by SDS-induced dissociation. Following another centrifugation, the immunoprecipitated components were identified by Western blot analysis.
Immunofluorescent Detection of Phospho-JNK and Sab
H9c2 cells were seeded at 2.5 × 105 cells/well in 6-well plates containing an 18 × 18-mm German glass coverslip in each well bottom. Cells were grown to 80% confluency and then treated with H2O2/FeSO4, Tat-scramble, and Tat-SabKIM1 as described earlier. Following 20 min of stress, cells were fixed in 2% paraformaldehyde for 30 min at room temperature. The coverslips were washed with PBS and stored at 4 °C until use. Coverslips were quenched in 100 mm glycine for 20 min. Cells were permeabilized in PBS supplemented with 0.1% Triton X-100, and then cells were blocked in PBS with 0.01% Triton X-100 and 5% BSA. Next, cells were incubated with primary antibodies recognizing active JNK (Phospho-JNK, 1:200) and Sab (1:150) for 2.5 h at room temperature. Cells were then incubated with goat anti-rabbit AlexaFluor 566 and donkey anti-mouse AlexaFluor 488 secondary antibodies for 1.5 h at room temperature. After each antibody incubation, the cells were washed in PBS. Coverslips were mounted using Vectasheild/DAPI on slides. Fluorescence microscopy was conducted using 100× magnification on an Olympus microscope. Exposure times for each channel were as follows: DAPI, 0.05 ms; FITC, 0.34 ms; and TRITC, 0.52 ms. The gain was sent at 4.3. Images were cropped in Microsoft Powerpoint without any prior alteration in a graphics suite.
Myocardial Ischemia/Reperfusion Injury
Adult Sprague-Dawley rats weighing 250–350 g were used. A left thoracotomy was made between the fourth and the fifth ribs and a 6–0 vicryl suture was passed under the left anterior descending coronary artery (LAD) just below the left atrium. To occlude the artery, a snare was formed by threading the suture ends through a hole made in a 0.4-mm piece of polyethylene tubing. The snare was tightened against the artery until blanching of the apex was evident and then secured tightly with a micro-clamp. The LAD was occluded for 30 min and then released. The chest cavity was closed and the animal was gradually weaned from the respirator. Sham-operated rats served as surgical controls and were subjected to the same surgical procedures as the experimental rats, with the exception that the LAD was not occluded in the sham-operated rats.
To measure phospho-c-Jun and phospho-JNK, animals were euthanized after 30 min and 1, 2, 6, and 24 h of reperfusion. Hearts were transcardially perfused with cold PBS, pH 7.4, and the myocardial tissue of the left ventricle was collected quickly and frozen in liquid nitrogen.
To measure the efficacy of a JNK inhibitor, SR-3306 (5 mg/kg) was administered as an intravenous bolus (1.5 mg/kg, ∼0.4 ml) 5 min before the end of ischemia followed by continuous intravenous infusion via a Harvard 22 syringe pump (18 μg/kg/min) for 18 h following reperfusion. The control group received DMSO/Tween 80/H20 (10/10/80), and the positive control group received 3-aminobenzamide (9 mg/ml). 18 h later, the rats were euthanized. The heart was removed and five to six biventricular sections of similar thickness were made perpendicular to the long axis of the heart and incubated in 1% triphenyl tetrazolium chloride (TTC) in PBS for 10 min at 37 °C. Samples were stored in 10% formalin.
SR-3306 Infarct Size Analysis
To measure the efficacy of a SR-3306, each heart section stained with TTC was photographed on both sides for each group (SR-3306, vehicle, 3-aminobenzamide; n = 8). The infarct size was delineated and calculated for both sides using Adobe Photoshop CS3 software. Infarct size was expressed as the ratio of the infarct area to the total area of the left ventricle. The cumulative area for all sections of each heart was used for comparisons. All results are expressed as means ± S.E. One-way analysis of variance followed by Tukey's post hoc test was used to compare differences in relative infarct size. p < 0.05 was considered to be statistically significant.
Tat-Sab Infarct Size Analysis
Direct injections (five 5-μl injections, 2 mg/kg) were delivered 5 min before reperfusion into the left ventricle wall in areas below the occlusion site of the LAD (four injections) and in the apex of the heart (one injection). Injections were made with a 30-gauge needle (45° bevel) and a 10-μl Hamilton syringe. Control groups subjected to the same I/R procedure included a saline injection (I/R + saline), Tat-scramble peptide injection (I/R + Tat-scramble), and a sham group without LAD occlusion. Following 24 h of reperfusion, the rats were euthanized, and hearts were perfused transcardially with cold PBS. The hearts were removed, and five to six biventricular sections of similar thickness, made perpendicular to the long axis of the heart, were incubated in 1% TTC in PBS for 10 min at 37 °C. Samples were stored in 10% formalin. To measure the protective effects of Tat-Sab peptide, each heart section stained with TTC was photographed on both sides for each group (sham, I/R + saline, I/R + Tat-scramble, and I/R + Tat-Sab; n = 4, 8, 7, and 8, respectively). The total area of the left ventricle, the total area of the apex, and infarct size were delineated and calculated for both sides using Adobe Photoshop CS3 software. The infarct area was calculated as the risk area, which became necrotic as distinguished by TTC staining. Infarct size was expressed as the ratio of infarct area to both the total area of the left ventricle and the total area of the apex. All results are expressed as means ± S.E. of the mean (S.E.). One-way analysis of variance followed by Tukey's post hoc test was used to compare differences in relative infarct size. p < 0.05 was considered statistically significant.
CPK, CK-MB, and TUNEL Staining Analysis
Blood samples were collected via the lateral tail vein in EDTA tubes for CPK and CK-MB analysis at 2 h post-reperfusion from rats receiving 5 mg/kg SR-3306 (n = 4) and vehicle (DMSO/Tween/H20; n = 3). Blood samples were also collected from naive rats (n = 4) that did not undergo any procedure. Quantification of apoptosis was measured using the Roche in situ cell death detection kit, fluorescein (catalog No. 11684795910). Assay procedures were followed for paraffin-embedded tissues sections. Following TUNEL and DAPI staining, 30 random sections within the apex and midsection of the left ventricle were imaged for each group (SR-3306, vehicle, and sham; n = 8, 8, and 5, respectively). The apoptotic index, defined as the number of TUNEL positive nuclei (green) divided by the total number of nuclei (blue), was determined by averaging the counts from 30 random images from each heart.
4-Hydroxynonenal (4-HNE), Protein Carbonyls, and Aconitase
Left ventricles were lysed on dry ice in M-PER (Thermo-Fisher Scientific) supplemented with protease and phosphatase inhibitors. Left ventricle fragments were crushed to powder using a cold mortar and pestle, and the powder was placed in M-PER and vortexed vigorously. Samples were stored on dry ice for 15 min. Samples were centrifuged at 14,000 × g for 15 min at 4 °C to remove tissue debris. The protein in the homogenate was quantitated using the Peirce BCA assay kit protocol. 4-HNE was measured using the Cell BioLabs 4-HNE ELISA kit. The protein carbonyls detection kit and aconitase assay kit were purchased from Cayman Chemicals. Left ventricle lysates were monitored for these oxidative stress markers according to the manufacturers' protocols.
RESULTS
Hydrogen Peroxide-induced Oxidative Stress Activates JNK Signaling in H9c2 Cells
To demonstrate that the JNK pathway was activated under H2O2/FeSO4 stress conditions in H9c2 cardiomyocyte-like cells, we analyzed the time course for JNK and c-Jun activation by Western analysis. Fig. 1A shows the immunoreactivity of the approximate 55- and 46-kDa isoforms of JNK. The levels of JNK immunoreactivity did not change with treatment of H2O2/FeSO4 over an 8-h time course as expected. In contrast, there was only a faint p-JNK signal at t = 0 h (for the 55-kDa isoform) with a time-dependent increase in p-JNK signal beginning at 1 h and showing a maximum at 6 and 8 h after H2O2/FeSO4 treatment (Fig. 1B). These results are consistent with a robust time-dependent activation of JNK in H9c2 cardiomyocyte-like cells after oxidative stress imposed by H2O2/FeSO4. Fig. 1C presents the time course for c-Jun immunoreactivity in H9c2 cells treated with H2O2/FeSO4 for up to 8 h. Like JNK, there was no change in the c-Jun immunoreactivity after treatment with H2O2. However, a time-dependent increase in p-c-Jun immunoreactivity was seen beginning at 1 h after H2O2 treatment, peaking between 4 and 6 h after treatment (Fig. 1D). These results also show activation of the JNK pathway, as monitored by the immediate downstream substrate c-Jun following oxidative stress in cardiomyocytes. The immunoreactivity of α-tubulin, used as a loading control, is also presented in Fig. 1E.
FIGURE 1.

H2O2 stress activates JNK signaling in rat H9c2 cardiomyocyte-like cells. JNK signaling was evaluated by Western blot following 1, 2, 4, 6, and 8 h of treatment with 25 μm H2O2. Proteins (30 μg) were resolved by SDS-PAGE, and antibodies for A, JNK; B, phospho-JNK; C, c-jun; and D, phospho-c-jun were used to detect proteins of interest. (E) α-Tubulin was used as a loading control.
Oxidative Stress Causes JNK and MKK4 to Localize to the Mitochondria
Mitochondria were isolated from H9c2 cells treated for 20 min with 100 μm H2O2/FeSO4, and Western analysis was used to determine whether JNK, MKK4, and Sab were localized to the mitochondria. Fig. 2 shows that when H9c2 cells were treated with PBS (control) there was no JNK localized to the mitochondria but when treated with 100 μm H2O2/FeSO4, the 55-kDa splice variant of JNK was found on the mitochondria (Fig. 2A). Similarly, a 42-kDa band corresponding to MKK4 (an upstream activator of JNK) was detected only in cells treated with 100 μm H2O2/FeSO4, suggesting that MKK4, like JNK, was brought to the mitochondria in H9c2 cells under oxidative stress conditions (Fig. 2B). The putative JNK mitochondrial scaffold protein Sab was also present on the mitochondria (Fig. 2C), and COX-IV (D) and SOD1 (E) were present in equal amounts in both samples suggesting equal loading and confirming JNK and MKK4 localization to the mitochondria. Finally, GAPDH, calnexin, and histone H3 immunoreactivity indicated no contamination from other subcellular fractions (Fig. 2, F–H). These data suggest that oxidative stress drives JNK and MKK4 to the mitochondria, which may have a significant role in cardiomyocyte function, survival, and death.
FIGURE 2.

Oxidative stress induced by H2O2/FeSO4 caused JNK and MKK4 translocation to the mitochondria in rat H9c2 cardiomyocyte-like cells. A–H, mitochondria isolations were prepared from rat H9c2 cardiomyocyte-like cells stressed with 100 μm H2O2/FeSO4 for 20 min. The mitochondria were then monitored for JNK, MKK4, Sab, COX-IV, SOD1, and GAPDH by Western analysis. COX-IV and SOD1 served as the mitochondria loading controls, and GAPDH, calnexin, and histone H3 served as controls for cytosolic, microsomal, and nuclear contamination, respectively. I, JNK interacts with Sab and MKK4 on the mitochondria in rat H9c2 cardiomyocyte-like cells. The mitochondrial complex was isolated from rat H9c2 cardiomyocyte-like cells stressed with 100 μm H2O2/FeSO4 for 20 min, immunoprecipitated with Sab antibody, and then immunoblotted for Sab, JNK, and MKK4. J, disruption of the JNK-Sab interaction by Tat-SabKIM1 peptide prevented JNK localization on the mitochondria. Mitochondria isolations were prepared from rat H9c2 cardiomyocyte-like cells stressed with 100 μm H2O2/FeSO4 for 20 min and probed for phospho-JNK and JNK in the presence and absence of 3 μm Tat-SabKIM1 or Tat-scramble peptide. COX-IV served as the mitochondrial loading control. K, cytosol/nuclear isolations from rat H9c2 cardiomyocyte-like cells stressed with 100 μm H2O2/FeSO4 for 20 min and probed for phospho-JNK, JNK, phospho-c-Jun, and c-Jun in the presence and absence of 3 μm Tat-Sab or Tat-scramble peptide. α-Tubulin was used as a loading control.
To establish that JNK interacts with Sab and MKK4 on the mitochondria under oxidative stress, we treated H9c2 cells with H2O2/FeSO4, immunoprecipitated the mitochondrial complex with Sab, and blotted for Sab, JNK, and MKK4. Fig. 2I presents Western analysis showing that Sab was located on the mitochondrial membrane (lane 1) when no oxidative stress was applied, as expected, whereas JNK and MKK4 were not found on the mitochondria (lane 1). However, upon treatment of the cells with H2O2/FeSO4, JNK and MKK4 were driven to the mitochondria and were associated with Sab (Fig. 2I, lane 2). These results suggest that oxidative stress caused JNK and MKK4 to be localized to the mitochondria and that this interaction may be mediated by Sab.
JNK Translocation to the Mitochondria Is Inhibited by the Tat-Sab Peptide during Oxidative Stress in H9c2 Cells
Fig. 2J presents Western blot analysis of phospho-JNK and JNK in mitochondrial enrichments from H9c2 cells. The data show JNK and p-JNK were translocated to the mitochondria upon stimulation of H9c2 cells with 100 μm H2O2/FeSO4 for 20 min (Fig. 2J, lane 2), and this translocation was completely blocked by preincubation of the cells with 3 μm Tat-Sab peptide (lane 3). The negative control Tat-Sab scramble peptide had no effect on JNK translocation. The cytosolic/nuclear preparations in Fig. 2K showed increased p-JNK and p-c-Jun after H2O2/FeSO4 treatment but no inhibition of c-Jun phosphorylation, suggesting that Tat-Sab prevents JNK translocation to the mitochondria but does not inhibit JNK activity. Finally, to firmly establish JNK translocation to the outer mitochondrial membrane, Fig. 3 shows the immunofluorescence from various treatment regimens. Notably, Sab (green) was found on the outer mitochondrial membrane in all groups; p-JNK (red) was absent from the outer mitochondrial membrane in the PBS-treated sample (Fig. 3b), moved to the outer mitochondrial membrane after H2O2/FeSO4 treatment (Fig. 3f), and was blocked from this translocation when Tat-SabKIM1 was present (Fig. 3n). These results confirm mitochondrial JNK translocation.
FIGURE 3.

Immunofluorescence of phospho-JNK and Sab in H9c2 cells. Immunofluorescence of H9c2 cells demonstrates JNK co-localization with mitochondrial Sab in the presence of H2O2/FeSO4. Unstressed cells treated with 1× PBS and cells treated with 100 μm H2O2/FeSO4 for 20 min in the presence and absence of Tat-scramble or Tat-SabKIM1 were incubated with antibodies specific for active JNK (Phospho-JNK) and Sab. Cells were then incubated with fluorescently conjugated secondary antibodies as described under “Experimental Procedures.” Cells were stained with DAPI following antibody incubations. Images were acquired using fluorescence microscopy at ×100 magnification.
JNK Inhibition and Inhibition of JNK Translocation to the Mitochondria Protect H9c2 Cells and Primary Human Cardiomyocytes from Mitochondrial Dysfunction and Cell Death Induced by Oxidative Stress
To test whether JNK mitochondrial localization affected mitochondrial function we measured the impact of Tat-Sab and small molecule JNK inhibitors SR-3306 (19) and SR-3562 (20) on ROS generation and mitochondrial membrane depolarization in H9c2 cells and primary human cardiomyocytes treated with H2O2/FeSO4. SR-3306 is a potent, highly selective JNK inhibitor, which demonstrates good pharmacokinetic properties in rats (19). Fig. 4A presents the normalized MitoSOX fluorescence in the absence of oxidative stress (PBS) compared with treatment with 100 μm H2O2/FeSO4, 100 μm H2O2/FeSO4 + 500 nm SR-3306, 100 μm H2O2/FeSO4 + 10 μm Tat-scramble peptide, or 100 μm H2O2/FeSO4 + 10 μm Tat-Sab peptide. The results show that 100 μm H2O2/FeSO4 increased ROS generation by ∼2-fold, and the addition of 500 nm SR-3306, 500 nm SR-3562, or 10 μm Tat-Sab reduced ROS generation by ∼2-fold compared with the 100 μm H2O2/FeSO4 treatment. The Tat-scramble peptide had no effect on reducing ROS. A similar effect was seen when MMP was measured by JC-1 detection (Fig. 4B). Finally, the effect of SR-3306 or Tat-Sab on cell viability in response to oxidative stress was measured by an MTT assay (Fig. 4C). H9c2 cells treated with 100 μm H2O2/FeSO4 were ∼40% viable, whereas the addition of 500 nm SR-3306 or 500 nm SR3562 to cells treated with 100 μm H2O2/FeSO4 increased viability to ∼90%, and the addition of 10 μm Tat-Sab peptide to cells treated with 100 μm H2O2/FeSO4 increased viability to ∼70% compared with 98% viability in untreated cells. Similar results were found for primary human cardiomyocytes (Fig. 4, D–F).
FIGURE 4.

Inhibition of JNK catalytic activity and mitochondrial translocation prevented H2O2-induced ROS generation and MMP dissipation and prevented cell death in rat H9c2 cells cardiomyocyte-like cells and primary human cardiomyocyte treated with 100 μm H2O2/FeSO4. Cells were stressed for 4 h with 100 μm H2O2/FeSO4 in the presence or absence of 500 nm SR-3306, 500 nm SR-3562, 10 μm Tat-SabKIM1, or 10 μm Tat-scramble. A and D, normalized MitoSOX fluorescence was detected with MitoSOX dye in 96-well plates. B and E, mitochondrial membrane potential was measured by JC-1 staining. C and F, percent viable cells was measured by MTT assay.
Activation of the JNK Pathway in Vivo during I/R
To demonstrate that the JNK pathway was indeed activated during ischemia/reperfusion, we measured the phospho-c-Jun and p-JNK Western blot signal after 30 min ischemia and at different time points of reperfusion after ischemia. Fig. 5 shows the Western blot analysis of p-c-Jun and p-JNK at different time points of reperfusion. The results show there was no p-c-Jun signal prior to I/R (Fig. 5, lane 1 (control)), and there was a robust increase in the p-c-Jun signal at 30 min after reperfusion (lane 2) persisting at 1 h (lane 3), 2 h (lane 4), and 6 h (lane 5) of reperfusion. Similarly, p-JNK levels showed a persistent increase beginning at 30 min and extending to 6 h. Tubulin is shown as a loading control. These results indicate that the JNK pathway was activated after I/R and the p-c-Jun signal endured during at least the first 6 h of the reperfusion component of the injury.
FIGURE 5.

Reperfusion time course for c-Jun phosphorylation and JNK phosphorylation after I/R injury. Phosphorylation of c-Jun and JNK was evaluated by Western blot following 30 min and 1, 2, and 6 h of reperfusion after 30 min ischemia. Rat hearts were perfused transcardially with cold PBS, pH 7.4, and myocardial tissue of the left ventricle was collected and frozen in liquid N2 prior to analysis. 30 μg of total protein was loaded on the gel and resolved by SDS-PAGE. Total c-Jun levels and α-tubulin were used as loading controls (C).
In Vivo Efficacy of SR-3306 in Rats after I/R
Fig. 6 presents the in vivo efficacy of SR-3306 in anesthetized rats after 30 min of ischemia and 24 h of reperfusion. Fig. 6A shows the total area of the left ventricle (LV) was the same for the three treatment groups (DMSO, control; 3-aminobenzamide, positive control; and SR-3306). The results show that the total area of the LV for each of the three groups was approximately the same, ranging from 749 ± 75 in the DMSO group to 700 ± 46 in the SR-3306-treated group. Fig. 6B shows the total LV percent infarct size. The DMSO vehicle-treated group showed 29.5 ± 2.7% infarct after I/R. Treatment with 40 mg/kg 3-aminobenzamide, a positive control for I/R protection, showed the decrease in LV percent infarct was 19.7 ± 1.5%, a statistically significant (p = 0.048) effect. Treatment with 5 mg/kg SR-3306 also showed a statistically significant (p < 0.05) decrease, where LV percent infarct was 20.6 ± 1.7% (a reduction of 30%, p < 0.0001). These results show that a modest dose of SR-3306 was protective in the I/R myocardial infarction (MI) rat model.
FIGURE 6.

The in vivo efficacy of SR-3306 in anesthetized rats after 30 min of ischemia and 24 h of reperfusion. SR-3306 was given first as a bolus dose 5 min prior to onset of reperfusion and then administered by constant infusion at a rate of 18 μg/kg/min during the reperfusion phase to yield a final total dose of 5 mg/kg. A, total area of LV volume for three treatment groups: vehicle (DMSO), 40 mg/kg 3-aminobenzamide (3AB) as a positive control, and 5 mg/kg SR-3306. B, infarct size as total LV percent infarct for three treatment groups as described in A. n = 8–9 rats/group.
SR-3306 Reduced Apoptosis and CPK and CK-MB Activity in Vivo
We were able to extend these findings to include quantitative analysis of apoptosis in vivo, as measured by TUNEL staining. Fig. 7, A–F, presents representative DAPI- and TUNEL-stained sections from two representative sections of the heart comparing sham-treated animals to a vehicle-treated I/R group of animals, and a 5 mg/kg SR-3306-treated I/R group of animals. The total number of nuclei (blue) and TUNEL positive cells (green) are shown for each representative section of the heart. The midsection and apex of the heart showed profound apoptosis (Fig. 7, C and D) for the I/R vehicle-treated hearts, whereas the SR-3306-treated I/R group showed essentially no apoptosis in the midsection with minimal apoptosis in the apex (Fig. 7, E and F). Fig. 7G presents the percent TUNEL positive nuclei determined by averaging the counts from 30 random images from each heart. The percent TUNEL positive nuclei was defined as the number of TUNEL positive nuclei divided by the total number of nuclei. The profound protection of SR-3306 was seen in the approximately 4-fold decrease (p < 0.01) in the percent TUNEL positive nuclei compared with the vehicle-treated group. These results extend the findings from the efficacy measures, further supporting the protective effect of JNK inhibition in I/R injury and the benefits of utilizing this compound for in vivo probing of the role of JNK in mitochondrial dysfunction. A third measure showing the efficacy of SR-3306 in the MI rat model is presented in Fig. 7, H and I. Fig. 7H presents the CPK activity given in units/liter for naive rats, rats exposed to I/R injury for 2 h, and rats treated with 5 mg/kg SR-3306. The results showed that naive rats had CPK levels of 22.7 units/liter compared with 136.4 units/liter for the vehicle-treated group. This 6-fold increase in CPK levels was reduced to 50.1 units/liter for the SR-3306-treated group, a 2.7-fold decrease compared with the infarcted group (p < 0.05). These results confirmed the protective effect of SR-3306 in this MI model. A similar trend was seen in the measure of CK-MB activity, where CK-MB levels (in units/liter) are reduced ∼2-fold by SR-3306 (Fig. 7I).
FIGURE 7.
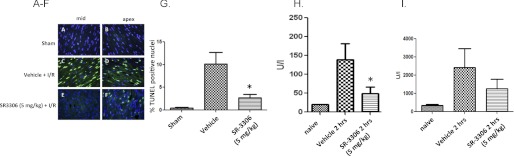
The selective JNK inhibitor SR-3306 decreases apoptosis and CPK and CK-MB levels in rat hearts during I/R injury in vivo. SR-3306 was given first as a bolus dose 5 min prior to onset of reperfusion and then administered by constant infusion at a rate of 18 μg/kg/min during the reperfusion phase to yield a final total dose of 5 mg/kg. A–F, DAPI- and TUNEL-stained sections from two representative sections of the heart comparing sham-treated animals with a vehicle-treated I/R group of animals and a 5 mg/kg SR-3306-treated I/R group of animals. Total number of nuclei (blue) and TUNEL positive cells (green) are shown for each representative section of the heart. G, percent TUNEL positive nuclei determined by averaging the counts from 30 random images from each heart. The percent TUNEL positive nuclei was defined as the number of TUNEL positive nuclei divided by the total number of nuclei. H, CPK activity given in units/liter for naive rats, rats exposed to I/R injury for 2 h, and rats treated with 5 mg/kg SR-3306. I, CK-MB activity given in units/liter for naive rats, rats exposed to I/R injury for 2 h, and rats treated with 5 mg/kg SR-3306.
Blocking JNK Translocation to the Mitochondria Protects against I/R Injury in Vivo
To demonstrate that the mechanism by which I/R injury occurred in vivo was through JNK translocation to the mitochondria, we designed a cell-permeable Sab peptide to block JNK interaction with Sab (14). Fig. 8 presents the protection afforded to both the whole heart and the apex of the left ventricle by 2 mg/kg Tat-Sab after I/R injury. Fig. 8, A and C, shows that the total area of the LV was the same for all groups in both the whole heart and the apex. Fig. 8B shows the whole heart percent infarct for the left ventricle, indicating 25.9 ± 3.9% infarct after I/R. Tat-scramble peptide did nothing to reduce the infarct volume, whereas treatment with 2 mg/kg Tat-Sab reduced the infarct volume to 16.1 ± 3.7% (a reduction of 38%, p < 0.0001). The protective effect of Tat-Sab is even more profound if only the apex is considered. Fig. 8D shows that the apex only percent infarct for the left ventricle was 45.1 ± 6.0% infarct after I/R. Again, Tat-scramble peptide did nothing to reduce the infarct volume, whereas treatment with 2 mg/kg Tat-Sab reduced the infarct volume to 19.9 ± 5.9% (a reduction of 56%, p < 0.0001).
FIGURE 8.

The in vivo efficacy of Tat-SabKIM1 in anesthetized rats after 30 min of ischemia and 24 h of reperfusion. Tat-SabKIM1 was given 5 min before reperfusion as a total of five direct injections into the left ventricle wall in areas below the occlusion site of the LAD (four injections) and in the apex of the heart (one injection). The total final dose of Tat-SabKIM1 was 2 mg/kg. A, the total area of the LV was measured by TTC staining for four treatment groups: sham, I/R + saline, I/R + 2 mg/kg Tat-scramble, and I/R + 2 mg/kg Tat-SabKIM1. B, total LV percent infarct for the four treatment groups described in A. C, the total area of the apex was measured by TTC staining for the four treatment groups described in A. D, total apex percent infarct for the four treatment groups described in A.
In Vivo Measures of Oxidative Stress in the Left Ventricle of Rat Hearts after I/R
To correlate the protected infarct volumes afforded by blocking JNK translocation to the mitochondria by Tat-Sab to measures of oxidative stress, we measured 4-HNE (a measure of lipid peroxidation), protein carbonyls, and aconitase activity in the presence and absence of Tat-Sab during I/R. 4-HNE levels were increased 8.8-fold (p < 0.05) during I/R compared with sham and were reduced by 2.5-fold (p < 0.05) when I/R occurred in the presence of 2 mg/kg Tat-Sab (Fig. 9A). Similarly, protein carbonylation was increased 9.4-fold (p < 0.05) during I/R compared with sham, but this change was reduced by 2.6-fold (p < 0.05) in the presence of 2 mg/kg Tat-Sab (Fig. 9B). Finally, aconitase activity, a measure of redox activity in the cell, was decreased by 13-fold (p < 0.05) during I/R compared with sham, and this was restored to ∼60% of the value of the sham group when I/R occurred in the presence of 2 mg/kg Tat-Sab (Fig. 9C).
FIGURE 9.

In vivo measures of oxidative stress in the left ventricle of rat hearts after 30 min of ischemia and 24 h of reperfusion. Tat-SabKIM1 was given 5 min before reperfusion as a total of five direct injections into the left ventricle wall in areas below the occlusion site of the LAD (four injections) and in the apex of the heart (one injection). The total final dose of Tat-SabKIM1 was 2 mg/kg. A, 4-HNE levels for four treatment groups: naive, sham, I/R, and I/R + 2 mg/kg Tat-SabKIM1. B, protein carbonyl levels for four treatment groups: naive, sham, I/R, and I/R + 2 mg/kg Tat-SabKIM1. C, aconitase activity for four treatment groups: naive, sham, I/R, and I/R + 2 mg/kg Tat-SabKIM1.
DISCUSSION
A growing body of evidence indicates that JNK-induced apoptosis is more dynamic than mere induction of AP-1-mediated transcription. Indeed, Aoki et al. (1) established that oxidative stress-induced apoptosis in cardiomyocytes does not require new protein synthesis and is more dependent on mitochondrial mechanisms. Our work in HeLa cells also showed that 80% of ROS generated after an initial oxidative stress was JNK-dependent, and this affects mitochondrial physiology and cell death (6). Hawana et al. (2) also observed a mitochondrially derived increase in ROS that was JNK-dependent in acetaminophen-induced liver injury where JNK was translocated to the mitochondria and mitochondrial dysfunction was observed. Given these findings, we set out to establish that blocking JNK translocation to the mitochondria could have a similar therapeutic benefit in cardiovascular I/R models as inhibition of JNK activity. To do this we developed a biochemical probe (Tat-SabKIM1) to interrogate mitochondrial function independently of transcriptional regulation.
Our results here support the following conclusions. First, the JNK pathway was activated in response to oxidative stress in rat cardiomyocyte-like H9c2 cells under the same time course as it was in rats exposed to 30 min of ischemia and varying time points of reperfusion injury (compare Figs. 1 and 5), suggesting that the H9c2 cell system is a reasonable model for in vivo I/R injury. Interestingly, the time course for JNK activation in H9c2 cells and in vivo in our study showed a maximal activation of JNK around 6 h, which was identical to the time course seen for JNK activation in whole cell fractions isolated from rat brains subjected to middle cerebral artery occlusion (21). The same was true for acetaminophen-induced liver injury, where JNK activation peaked at 6 h as measured by p-JNK Western; this correlated with peak serum alanine transaminase levels. These results suggest that JNK activation induced either by I/R injury or acetaminophen could be a universal response in various organs and that the time course for activation and cell death may be similar among these tissues.
Second, we can conclude from our data that JNK translocation to the mitochondria occurred rapidly in rat H9c2 cells and that the proportion of active JNK (i.e. p-JNK) increased over time, peaking near 6 h (Figs. 1 and 2). Again, our results correlate nicely with those found by Aoki et al. (1), who showed by immunofluorescence and confocal microscopy that endogenous MKK4 and JNK1 both translocate to the mitochondria very rapidly in adult rat primary cardiomyocytes exposed to oxidative stress. This time course is also consistent with what we found for anisomycin-induced stress in HeLa cells (6, 14). Thirdly, concurrent with the JNK activation, mitochondrial ROS generation and mitochondrial membrane dissipation occurred, leading to cell death (Fig. 4). It is here that our studies extend beyond what was found by Aoki et al. (1), as they did not have any measures for ROS generation or mitochondrial function. Thus, our study is the first report for these mitochondrial function measures in rat and human cardiomyocytes. Moreover, the near equal protection for mitochondrial function (Fig. 4, B and E) against oxidative stress by 500 nm SR-3306, 500 nm SR-3562, or 10 μm Tat-Sab in these cells suggests that inhibition of JNK mitochondrial signaling may be similarly effective as inhibition of JNK activity in protecting the cell against mitochondrial dysfunction and cell death initiated by oxidative stress.
It is important to note that whereas most studies in most cells and most tissues support our findings, there are some reports from the Webster group (22, 23) showing that JNK activation is protective against oxidative stress and reperfusion in primary neonatal rat cardiomyocytes; in those studies the Webster group either transfected primary neonatal rat cardiomyocytes with dominant negative (dn) JNK1 or infected these cells with adenoviral expressed dnJNK1. In each case, they saw an increase in apoptosis associated with dnJNK1 expression suggesting a protective effect for JNK1. This is opposite to what we see and what was seen by Gottlieb and colleagues (12), who saw protective effects from dnJNK2 that was adenovirally expressed in primary adult rabbit cardiomyocytes. The Gottlieb laboratory (12) also saw protective effects from adenoviral expression of JIP (a JNK inhibitor), suggesting that JNK inhibition, not activation, was protective in oxidative stress and I/R-stress models both in vitro and in vivo. Moreover, Gottleib and colleagues (12) found that TNFα-activated JNK activity in H9c2 cells was completed abolished by dnJNK2 and JIP-1, a result very much in line with our findings and again suggesting that JNK inhibition, not activation is protective. One potential reason for the discrepancy between our findings and those of Webster may be that like Aoki et al. (1) and the Gottlieb group (12), we utilized adult cardiomyocytes or tissues from adult animals, whereas the Webster findings all come from neonatal rat cardiomyocytes. This notion is supported by the fact that JNK1 is not expressed in human fetal heart (24), making the overexpression of dnJNK1 in the neonatal cells perhaps nonphysiologic. Along the same lines, a second potential reason for the discrepancy between our findings and those from Webster may be that our system utilized the endogenous expression levels of all JNKs in the cell, whereas the Webster work relied upon overexpression of dominant negative forms of JNK1, which may have altered the balance of JNK isoform functions within the cell that were not at physiological levels. A third reason may be that JNK isoform contributions to protection and cell death may vary. For example, Yu et al. (25) showed that jnk3 −/− mice had significantly reduced infarct volumes in the brain after 30 min of ischemia and 24 h of reperfusion in the middle cerebral artery occlusion model. JNK3 is expressed solely in the heart and brain and may have unique roles in those tissues (26). Similarly, Gottlieb and colleagues (12) showed protective effects in the dnJNK2 studies cited above, again suggesting that different JNK isoforms may have tissue-specific protective roles. In our studies we utilized physiologic levels of all three isoforms, which may best reflect native contributory responses to oxidative stress and I/R stress in vitro and in vivo.
It has already been established that inhibition of JNK by small molecule inhibitors reduces infarct size in rats after myocardial ischemia (11). Our goal was to compare the efficacy of SR-3306 in reducing infarct volume with that of Tat-Sab and correlating other biomarkers such as CPK and CK-MB and apoptotic index for SR-3306 with protection of oxidative stress in vivo by Tat-Sab, as well as to compare SR-3306 with AS601245. First, it should be noted that SR-3306 compared very favorably with AS601245 in that the reduction in infarct size with 5 mg/kg SR-3306 was equivalent to that of both 4.5 and 15 mg/kg AS601245. Because higher doses of SR-3306 were not tried, it is not known whether higher doses would produce a further lowering of infarct size than that obtained with higher doses (15 mg/kg) of AS601245. Similarly, both AS601245 and SR-3306 reduced the number of TUNEL positive cells by ∼4-fold, again suggesting equal efficacy for both compounds. The reductions in apoptosis and infarct size for SR-3306 also correlated with drops in CPK and CK-MB, suggesting that the reduction in infarct volume and apoptosis correlated well with these blood biomarkers of MI and that SR-3306 was indeed protective against the I/R injury.
In comparing the efficacy from small molecule JNK inhibitors such as SR-3306 and AS601245 with that of Tat-Sab, it was immediately evident that inhibiting JNK mitochondrial translocation provided a similar benefit for reducing infarct volume as did universal JNK inhibition (Figs. 6 and 8). A few caveats must be stated however. First, Tat-Sab was delivered directly into the rat myocardium, whereas SR-3306 was delivered by constant infusion. If the local dose of Tat-Sab was significantly higher than that of SR-3306, a direct comparison cannot be made. Conversely, rapid loss of Tat-Sab in vivo could theoretically produce lower concentrations than SR-3306. Nevertheless, it can be stated that similar protection was afforded by the two mechanisms of inhibition and that careful dose-response analysis would allow for quantitative comparison of efficacy for the two inhibition mechanisms. This result suggests that inhibition of JNK translocation to the mitochondria (and therefore JNK mitochondrial signaling) could possibly be at least partially responsible for the protective effects of JNK against I/R. This observation may have a profound effect on how therapeutic strategies may be considered and pursued. For example, if therapeutic benefit could be achieved by selectively inhibiting JNK mitochondrial signaling without affecting nuclear function as we have previously demonstrated (14), it can be conceived that any potential toxicities associated with inhibiting nuclear JNK signaling would be eliminated while still maintaining maximal therapeutic benefit. Given that the JNK-Sab interaction has significant affinity contributions from the 11-amino acid KIM domain, as evidenced by the 218 nm IC50 for Tat-SabKIM1 versus Sab protein (14), the notion of making Sab-competitive small molecule inhibitors or bidentate small molecule JNK inhibitors that span the ATP pocket as well as the substrate binding domain is feasible. Indeed, a few such compounds have been reported that are either competitive versus JIP (27) or that have some bidentate character (28–31). If these compounds can be improved in terms of potency, selectivity, and drug-like properties, our results now show that selective inhibition of JNK mitochondrial signaling might be achieved with therapeutic benefit. Finally, the changes in 4-HNE, protein carbonyl, and aconitase activity levels in the presence of Tat-Sab further illustrate that selective inhibition of mitochondrial JNK signaling impacts oxidative stress in vivo, suggesting key mechanistic roles for JNK in these processes.
In summary, this is the first demonstration on the molecular level that shows that blocking JNK mitochondrial signaling via inhibition of JNK interaction with Sab can be protective against in vivo I/R. Moreover, it is important to stress that the effect seen with inhibiting JNK mitochondrial signaling was similarly as efficacious as universal JNK inhibition. In addition, these results provide a mechanism by which JNK-driven, mitochondrially generated ROS causes mitochondrial dysfunction in cardiomyocytes; and blocking the molecular interaction between JNK and Sab mitigates these dysfunctions as manifested in significantly reduced infarct volumes, apoptosis, and oxidative stress in vitro and in vivo. These studies lay the foundation for designing mitochondria-specific JNK signaling inhibitors that may reduce any toxicities seen with JNK-dependent nuclear inhibition.
Acknowledgments
We are grateful to Dr. Carolyn Cray for running the CPK and CK-MB assays. We also thank Dr. Rudy Mora for help with the primary human cardiomyocytes and Melissa Tellinghuisen for help with the figures.
This work was supported, in whole or in part, by National Institutes of Health Grant NS057153 (to P. V. L.). This work was also supported by the Saul and Theresa Esman Foundation. Philip LoGrasso serves as a consultant for OPKO Health.
- I/R
- ischemia/reperfusion
- JIP
- JNK-interacting protein-1
- MMP
- mitochondrial membrane potential
- CPK
- creatine phosphokinase
- CK-MB
- creatine kinase-MB (muscle/brain type)
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- LAD
- left anterior descending coronary artery
- TTC
- triphenyl tetrazolium chloride
- 4-HNE
- 4-hydroxynonenal
- LV
- left ventricle
- dn
- dominant negative
- MI
- myocardial infarction.
REFERENCES
- 1. Aoki H., Kang P. M., Hampe J., Yoshimura K., Noma T., Matsuzaki M., Izumo S. (2002) Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. J. Biol. Chem. 277, 10244–10250 [DOI] [PubMed] [Google Scholar]
- 2. Hanawa N., Shinohara M., Saberi B., Gaarde W. A., Han D., Kaplowitz N. (2008) Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 283, 13565–13577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhao Y., Herdegen T. (2009) Cerebral ischemia provokes a profound exchange of activated JNK isoforms in brain mitochondria. Mol. Cell. Neurosci. 41, 186–195 [DOI] [PubMed] [Google Scholar]
- 4. Zhou Q., Lam P. Y., Han D., Cadenas E. (2008) c-Jun N-terminal kinase regulates mitochondrial bioenergetics by modulating pyruvate dehydrogenase activity in primary cortical neurons. J. Neurochem. 104, 325–335 [DOI] [PubMed] [Google Scholar]
- 5. Zhou Q., Lam P. Y., Han D., Cadenas E. (2009) Activation of c-Jun-N-terminal kinase and decline of mitochondrial pyruvate dehydrogenase activity during brain aging. FEBS Lett. 583, 1132–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chambers J. W., LoGrasso P. V. (2011) Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem. 286, 16052–16062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lei K., Davis R. J. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 2432–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lei K., Nimnual A., Zong W. X., Kennedy N. J., Flavell R. A., Thompson C. B., Bar-Sagi D., Davis R. J. (2002) The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol. Cell. Biol. 22, 4929–4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tournier C., Hess P., Yang D. D., Xu J., Turner T. K., Nimnual A., Bar-Sagi D., Jones S. N., Flavell R. A., Davis R. J. (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870–874 [DOI] [PubMed] [Google Scholar]
- 10. Ventura J. J., Coqswell P., Flavell R. A., Baldwin A. S., Jr., Davis R. J. (2004) JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 18, 2905–2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ferrandi C., Ballerio R., Gaillard P., Giachetti C., Carboni S., Vitte P. A., Gotteland J. P., Cirillo R. (2004) Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats. Br. J. Pharmacol. 142, 953–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. He H., Li H. L., Lin A., Gottlieb R. A. (1999) Activation of the JNK pathway is important for cardiomyocyte death in response to simulated ischemia. Cell Death Differ. 6, 987–991 [DOI] [PubMed] [Google Scholar]
- 13. Kaiser R. A., Liang Q., Bueno O., Huang Y., Lackey T., Klevitsky R., Hewett T. E., Molkentin J. D. (2005) Genetic inhibition or activation of JNK1/2 protects the myocardium from ischemia-reperfusion-induced cell death in vivo. J. Biol. Chem. 280, 32602–32608 [DOI] [PubMed] [Google Scholar]
- 14. Chambers J. W., Cherry L., Laughlin J. D., Figuera-Losada M., Lograsso P. V. (2011) Selective inhibition of mitochondrial JNK signaling achieved using peptide mimicry of the Sab kinase-interacting motif-1 (KIM1). ACS Chem. Biol. 6, 808–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wiltshire C., Matsushita M., Tsukada S., Gillespie D. A., May G. H. (2002) A new c-Jun N-terminal kinase (JNK)-interacting protein, Sab (SH3BP5), associates with mitochondria. Biochem. J. 367, 577–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pallotti F., Lenaz G. (2007) Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol. 80, 3–44 [DOI] [PubMed] [Google Scholar]
- 17. Schnaitman C., Erwin V. G., Greenawalt J. W. (1967) The submitochondrial localization of monoamine oxidase. An enzymatic marker for the outer membrane of rat liver mitochondria. J. Cell Biol. 32, 719–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mukhopadhyay P., Rajesh M., Haskó G., Hawkins B. J., Madesh M., Pacher P. (2007) Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat. Protoc. 2, 2295–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chambers J. W., Pachori A., Howard S., Ganno M., Hansen D., Jr., Kamenecka T., Song X., Duckett D., Chen W., Ling Y. Y., Cherry L., Cameron M. D., Lin L., Ruiz C. H., Lograsso P. (2011) Small molecule c-jun-N-terminal kinase (JNK) inhibitors protect dopaminergic neurons in a model of Parkinson's disease. ACS Chem. Neurosci. 2, 198–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kamenecka T., Jiang R., Song X., Duckett D., Chen W., Ling Y. Y., Habel J., Laughlin J. D., Chambers J., Figuera-Losada M., Cameron M. D., Lin L., Ruiz C. H., LoGrasso P. V. (2010) Synthesis, biological evaluation, X-ray structure, and pharmacokinetics of aminopyrimidine c-jun-N-terminal kinase (JNK) inhibitors. J. Med. Chem. 53, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Okuno S., Saito A., Hayashi T., Chan P. H. (2004) The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J. Neurosci. 24, 7879–7887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dougherty C. J., Kubasiak L. A., Frazier D. P., Li H., Xiong W. C., Bishopric N. H., Webster K. A. (2004) Mitochondrial signals initiate the activation of c-Jun N-terminal kinase (JNK) by hypoxia-reoxygenation. FASEB J. 18, 1060–1070 [DOI] [PubMed] [Google Scholar]
- 23. Dougherty C. J., Kubasiak L. A., Prentice H., Andreka P., Bishopric N. H., Webster K. A. (2002) Activation of c-Jun N-terminal kinase promotes survival of cardiac myocytes after oxidative stress. Biochem. J. 362, 561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dérijard B., Hibi M., Wu I. H., Barrett T., Su B., Deng T., Karin M., Davis R. J. (1994) JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76, 1025–1037 [DOI] [PubMed] [Google Scholar]
- 25. Yu J., Novgorodov S. A., Chudakova D., Zhu H., Bielawska A., Bielawska J., Obeid L. M., Kindy M. S., Gudz T. I. (2007) J. Biol. Chem. 282, 25940–25949 [DOI] [PubMed] [Google Scholar]
- 26. Mohit A. A., Martin J. H., Miller C. A. (1995) p493F12 kinase: a novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron 14, 67–78 [DOI] [PubMed] [Google Scholar]
- 27. Chen T., Kablaoui N., Little J., Timofeevski S., Tschantz W. R., Chen P., Feng J., Charlton M., Stanton R., Bauer P. (2009) Identification of small-molecule inhibitors of the JIP-JNK interaction. Biochem. J. 420, 283–294 [DOI] [PubMed] [Google Scholar]
- 28. De S. K., Chen V., Stebbins J. L., Chen L. H., Cellitti J. F., Machleidt T., Barile E., Riel-Mehan M., Dahl R., Yang L., Emdadi A., Murphy R., Pellecchia M. (2010) Synthesis and optimization of thiadiazole derivatives as a novel class of substrate competitive c-Jun N-terminal kinase inhibitors. Bioorg. Med. Chem. 18, 590–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De S. K., Chen L. H., Stebbins J. L., Machleidt T., Riel-Mehan M., Dahl R., Chen V., Yuan H., Barile E., Emdadi A., Murphy R., Pellecchia M. (2009) Discovery of 2-(5-nitrothiazol-2-ylthio)benzo[d]thiazoles as novel c-Jun N-terminal kinase inhibitors. Bioorg. Med. Chem. 17, 2712–2717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. De S. K., Stebbins J. L., Chen L. H., Riel-Mehan M., Machleidt T., Dahl R., Yuan H., Emdadi A., Barile E., Chen V., Murphy R., Pellecchia M. (2009) Design, synthesis, and structure-activity relationship of substrate competitive, selective, and in vivo active triazole and thiadiazole inhibitors of the c-Jun N-terminal kinase. J. Med. Chem. 52, 1943–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stebbins J. L., De S. K., Machleidt T., Becattini B., Vazquez J., Kuntzen C., Chen L. H., Cellitti J. F., Riel-Mehan M., Emdadi A., Solinas G., Karin M., Pellecchia M. (2008) Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc. Natl. Acad. Sci. U.S.A. 105, 16809–16813 [DOI] [PMC free article] [PubMed] [Google Scholar]