

Background: PPARγ serves as a master transcriptional regulator of glucose and lipid metabolism, but it also plays an important role in carcinogenesis.
Results: Up-regulation of KLF4 upon PPARγ activation is mediated through the PPRE in the KLF4 promoter.
Conclusion: KLF4 partly mediates PPARγ-induced cell proliferation inhibition.
Significance: This study provides further insights into the PPARγ signal transduction pathway as well as a novel cancer therapeutic strategy.
Keywords: Cell Cycle, Krüppel-like Factor (KLF), Peroxisome Proliferator-activated Receptor (PPAR), Signal Transduction, Transcriptional Promoter, PPRE, TGZ
Abstract
Peroxisome proliferator-activated receptor γ (PPARγ), a subgroup of ligand-activated nuclear receptors, plays critical roles in cell cycle regulation, differentiation, apoptosis, and invasion. PPARγ is involved in tumorigenesis and is a potent target for cancer therapy. PPARγ transactivation of KLF4 has been demonstrated in various studies; however, how PPARγ regulates KLF4 expression is not clear. In this study, we reveal that PPARγ regulates the expression of KLF4 by binding directly to the PPAR response element (PPRE) within the KLF4 promoter. The PPRE resides at −1657 to −1669 bp upstream of the KLF4 ATG codon, which is essential for the transactivation of troglitazone-induced KLF4 expression. Furthermore, we found that stable silencing of KLF4 obviously suppressed the G1/S arrest and anti-proliferation effects induced by PPARγ ligands. Taken together, our data indicate that up-regulation of KLF4 upon PPARγ activation is mediated through the PPRE in the KLF4 promoter, thus providing further insights into the PPARγ signal transduction pathway as well as a novel cancer therapeutic strategy.
Introduction
The peroxisome proliferator-activated receptors (PPARs)3 are a subgroup of the ligand-activated nuclear receptor superfamily, and three isoforms have been identified: PPARα, PPARβ, and PPARγ (1). Like other members of this superfamily, PPARs mediate transcriptional regulation by binding to their central DNA domain, which recognizes response elements in the promoters of specific target genes (2). The most widely studied form among the three known forms of PPARs is PPARγ (3), which is expressed in a wide variety of cell types, including adipocytes, macrophages, and others (4, 5). It has been found that PPARγ expression is not limited to cells of adipocytic lineage but is also in various other tissues, particularly malignant tissues such as human prostate cancer, gastric cancer, liposarcoma, etc. (6–8). PPAR subtypes form functional heterodimers with members of the retinoid X receptor (RXR) family of nuclear receptors. The PPAR/RXR heterodimer regulates expression of target genes by binding to the PPAR response elements (PPREs) (9). 15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), a metabolite of prostaglandin D2, is a potential endogenous ligand for PPARγ, and thiazolidinediones such as troglitazone (TGZ), rosiglitazone (RGZ), ciglitazone, and pioglitazone (PGZ) are specific exogenous ligands for PPARγ (10). PPARγ is generally accepted as a master transcriptional regulator of glucose and lipid metabolism and also behaves as a tumor suppressor. Recent studies showed that PPARγ activation inhibits cell proliferation (11, 12), promotes differentiation (13), induces apoptosis (14), and inhibits angiogenesis (15), leading researchers to postulate that PPARγ may play an important role in tumorigenesis (14). However, the molecular mechanisms that mediate anticancer effects of dual PPARγ agonists in either a PPARγ-dependent or PPARγ-independent manner are not yet clear, and further studies for the identification of a unique PPARγ target gene are required.
The Krüppel-like family of zinc finger transcription factors regulates numerous biological processes, including proliferation, differentiation, apoptosis, development, and inflammation. It is reported to have a dual function as a tumor suppressor or an oncogene depending on the types of tissues in which it is expressed. Increased expression of KLF4 was reported in the ductal carcinoma of breast cancer and oral squamous cell carcinomas (16, 17), indicating that activation of KLF4 is one of the frequent steps toward carcinogenesis. In contrast, KLF4 is down-regulated in many human cancers, including esophageal, colorectal, bladder, and lung cancer and lymphoma (18–22). In line with a tumor suppressor function of KLF4, it has been found that KLF4 is down-regulated by promoter hypermethylation and loss of heterozygosity in colorectal cancer, and its overexpression reduces tumorigenesis in colon cancer cells in vivo (19, 23). These studies indicate that KLF4 negatively regulates cell cycle progression, but the mechanisms of by which KLF4 affects tumorigenesis in colorectal cancer have yet to be elucidated. It has been shown that some PPARγ agonists up-regulate the expression of KLF4 in colon cancer cells (24, 25). However, the mechanisms by which how PPARγ agonists active the expression of KLF4 are still unknown. Bearing in mind growth inhibition of KLF4, it might be interesting to increase KLF4 expression in these cancer cells by activating PPARγ.
In this study, we demonstrated that KLF4 is a direct transcriptional target of PPARγ. Computer-aided transcription factor-binding site analysis identified one consensus PPRE at −1657 to −1669 bp upstream of the KLF4 ATG codon. Further analysis by ChIP, EMSA, and luciferase reporter assay revealed that PPARγ can specifically bind to the PPRE in the promoter region of KLF4, which is required for PPARγ to transactivate KLF4. Furthermore, we found that stable silencing of KLF4 obviously suppressed the G1/S arrest and anti-proliferation effects induced by PPARγ ligands, providing further insights into the PPARγ signal transduction pathway as well as a novel cancer therapeutic strategy.
EXPERIMENTAL PROCEDURES
Reagents, Cells Transfection, and RNA Interference
TGZ, RGZ, ciglitazone, PGZ, 15d-PGJ2, GW9662, WY14643, and GW0742 were purchased from Cayman Chemical Co. (Ann Arbor, MI). All synthetic ligands were dissolved in dimethyl sulfoxide (DMSO). The final DMSO concentration in the culture medium in all experiments was kept constant at 0.1%. Unless stated otherwise, cells were exposed to these ligands for 24 h. HEK293, HCT116, HT29, LoVo, and HCT15 cells were maintained in DMEM and RPMI 1640 medium supplemented with 10% fetal bovine serum and antibiotics. siRNAs (Shanghai GenePharma Co., Ltd., Shanghai, China) targeting PPARγ (GCCCTTCACTACTGTTGAC) and a control siRNA (AATTCTCCGAACGTGTCACGT) were used at 50 nmol/liter. Stable silencing of KLF4 was achieved using the shRNA-based vector with the target sequence GGACGGCTGTGGATGGAAA. Transfections were performed using Lipofectamine 2000 (Invitrogen) for plasmids according to the manufacturer's recommendations.
Plasmid Construction
DNA fragments of the KLF4 and PPARγ cDNA coding regions were amplified by PCR and subcloned into the pCDE-HA vector as described previously (26). For generation of the KLF4 promoter-reporters, the KLF4 −2051/+252 (designated as P1) and −1597/+252 (designated as P2) sequences were PCR-amplified and subcloned into the pGL3-Basic vector.
Western Blotting
Cell lysates were size-fractionated by SDS-PAGE and transferred onto polyvinylidene difluoride membrane. The following antibodies were used to detect specific proteins: anti-KLF4, anti-p27, anti-p21, anti-PPARγ, and anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-HA (Tiangen Biotech Co. Ltd., Beijing, China).
RNA Isolation and PCR Analysis
Total RNA was isolated by using TRIzol reagent (Invitrogen), and conventional RT-PCR and RT-quantitative PCR (RT-qPCR) were done using a One-Step RT-PCR kit (Qiagen) and SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA), respectively. The primers used are listed in Table 1.
TABLE 1.
RT-qPCR primers
| RT-qPCR primer | Sequences |
|---|---|
| KLF4 mRNA | |
| Forward | CGAACCCACACAGGTGAGAA |
| Reverse | TACGGTAGTGCCTGGTCAGTTC |
| KLF4 pre-mRNA | |
| Forward | AAAGGGGCGATTCT |
| Reverse | CCCCGTGTGTTTACGGTAGT |
| p27 | |
| Forward | GGGGCTCGTCTTTTCGGGGTGTTT |
| Reverse | GAGCGGGAGGGCGGAGAGGAG |
| p21 | |
| Forward | GGGGAAGGGACACACAAGAAGA |
| Reverse | AATGAACTGGGGAGGGATGG |
| GAPDH | |
| Forward | GTCGGAGTCAACGGATTTGG |
| Reverse | AAAAGCAGCCCTGGTGACC |
Reporter Assay
Luciferase assay was performed using the Dual-Luciferase reporter assay system (Promega) as described (26). HCT116 cells were transfected with pGL3-Basic (control) or KLF4 gene promoter constructs; 24 h after transfection, cells were left untreated or treated with 10 μm TGZ for 18 h. Luciferase activity was then determined and normalized to an internal cytomegalovirus-Renilla luciferase control. Each experiment was done in triplicate and repeated at least three times.
EMSA
EMSA was performed using a gel shift assay system (Pierce) according to the manufacturer's protocol. Biotin-labeled and unlabeled wild-type oligonucleotide probes contained the binding motif of PPARγ (5′-ACGAGTTGTCCTTTGACCTTTACTGG-3′), and unlabeled mutant probes contained a substitution of 2 bp (underlined, 5′-ACGAGTTGTCCTTTGATATTTACTGG-3′ (designated M1) and 5′-ATAAGTTGTCCTTTGACCTTTACTGG-3′ (designated M2)). Briefly, 3 fmol of labeled oligonucleotides was incubated with nuclear extracts (5 μg), which were obtained from HEK293 cells transfected with pcDNA3.1-PPARγ and pCDE-HA-RXRα using a nuclear extract kit (Pierce) according to the manufacturer's instructions. Competitor oligonucleotides were added at a 100-fold molar excess of the labeled probe except where indicated otherwise. In experiments involving antibodies, 2 μg of the anti-PPARγ or anti-HA polyclonal antibody was added to the reaction except where indicated otherwise. DNA-protein complexes were resolved on 6% nondenaturing polyacrylamide gel and transferred to a nylon membrane, followed by detection of the biotin-labeled probe by the chemiluminescent nucleic acid detection module (Pierce) according to the manufacturer's instructions. The membranes were exposed to x-ray films to visualize the signal.
Chromatin Immunoprecipitation
HCT116 cells were treated with DMSO or TGZ (10 μm) for 24 h and subjected to ChIP with a ChIP assay Kit (Upstate Cell Signaling Solutions, Charlottesville, VA). Briefly, formaldehyde was used to cross-link proteins with DNA, and cells were lysed in SDS lysis buffer. The cell lysate was sonicated to shear the DNA. Chromatin samples were precleared with protein G-agarose for 2 h at 4 °C and immunoprecipitated overnight at 4 °C with normal mouse IgG or anti-PPARγ antibody bound to protein G-agarose. Formaldehyde cross-linking was reversed by incubation at 65 °C, followed by proteinase K digestion and DNA purification. qPCR was then performed as described above using specific primers.4
BrdU Incorporation
Proliferation was measured using a BrdU cell proliferation ELISA kit (Roche Applied Science) according to the instructions of the manufacturer with minor modifications. Briefly, after labeling the cells with BrdU for 1 h, coverslips bearing treated cells were washed with PBS, fixed with 3% paraformaldehyde in PBS, permeabilized with 0.25% Triton X-100 in PBS for 10 min, and washed with PBS. Coverslips were then treated with 4 n HCl at room temperature for 30 min to denature the DNA, neutralized with 0.1 m sodium borate at room temperature for 5 min, and washed with PBS. A BrdU antibody was added to the coverslips at 37 °C for 1 h. Following washing with PBS, DAPI was added to the coverslips at room temperature for 3 min, after which they were washed with PBS, mounted, and visualized under a fluorescence microscope.
Cell Cycle Analysis
Cells were rinsed in Dulbecco's phosphate-buffered saline (Mediatech, Herndon, VA), trypsinized, resuspended in McCoy's medium containing 10% fetal bovine serum and 1% penicillin/streptomycin, collected by centrifugation, washed with Dulbecco's phosphate-buffered saline, again collected by centrifugation, resuspended in 70% ethanol, and fixed overnight at 4 °C. Cells were pelleted again by centrifugation and resuspended in staining solution containing 50 μg/ml propidium iodide, 50 μg/ml RNase A, 0.1% Triton X-100, and 0.1 mm EDTA for 30 min. Flow cytometry was performed on a FACSCalibur cytometer (BD Biosciences).
MTS Cell Proliferation Assay
Cells in DMEM containing 10% fatty acid-free BSA were seeded in 96-well plates at a density of 2 × 103 cells/well. The cells were then incubated in the absence or presence of 10 μm TGZ, which was renewed daily. CellTiter 96 AQueous (MTS) One Solution reagent (Promega) was added to each well, and absorbance was recorded at 570 nm using a BioTek ELx800 absorbance microplate reader.
RESULTS
PPARγ Agonists Up-regulate KLF4 mRNA and Protein Expression in HCT116 Cells
To examine whether PPARγ influences KLF4 activity, we first analyzed the expression of KLF4 in HCT116 cells treated with TGZ. RT-PCR, RT-qPCR, and Western blot results clearly showed that TGZ induced the expression of KLF4. The expression of KLF4 was detectably increased after TGZ treatment (Fig. 1, A and B). This increase was confirmed at the protein level by immunoblotting experiments (Fig. 1C). We subsequently examined the expression of KLF4 upon treatment with different doses of TGZ ranging from 2 to 20 μm, and a dose-dependent stimulation of KLF4 expression by TGZ was observed (Fig. 1D). Additionally, other PPARγ agonists (RGZ, PGZ, and 15d-PGJ2) were tested, and increased KLF4 expression was found as well. In contrast, WY14643 (PPARα ligand) and GW0742 (PPARβ ligand) did not affect KLF4 expression (Fig. 1E). These results indicate that PPARγ agonists can specifically induce KLF4 expression, which is consistent with previous reports (25).
FIGURE 1.

PPARγ agonists up-regulate levels of KLF4 mRNA and protein expression in HCT116 cells. A and B, HCT116 cells were grown in the absence of serum for 18 h and then incubated with 10 μm TGZ for the indicated times. Total RNA was prepared, and the expression of KLF4 was determined by RT-PCR (A) or RT-qPCR (B). Data represent the mean ± S.D. of triplicate experiments. C, HCT116 cells were grown in the absence of serum for 18 h and then incubated with 10 μm TGZ for the indicated times. The expression of KLF4 was determined by Western blotting. β-Actin served as an internal control. D, HCT116 cells were grown in the absence of serum for 18 h and then incubated with increasing concentrations (0–20 μm) of TGZ for 8 h. The expression of KLF4 was determined by RT-qPCR. Data represent the mean ± S.D. of triplicate experiments. E, cells were serum-starved for 18 h and treated with control vehicle (0.2% DMSO; Ctr), RGZ (10 μm), 15d-PGJ2 (10 μm), PGZ (10 μm), WY14643 (10 μm), and GW0742 (10 μm) for 12 h. The expression of KLF4 was determined by RT-qPCR. Data represent the mean ± S.D. of triplicate experiments.
Induction of KLF4 by PPARγ Agonists Is PPARγ-dependent
Previous studies showed that PPARγ agonists can alter gene expression in a PPARγ-dependent and PPARγ-independent manner. To clarify this, we tested KLF4 expression in wild-type PPARγ cell lines HT29 and LoVo and mutant PPARγ(K422Q) cell line HCT15 after TGZ treatment. RT-qPCR analysis and Western blotting revealed that PPARγ activation led to an increase in KLF4 expression in HT29 and LoVo cells, which was consistent with the change in HCT116 cells, but not in HCT15 cells (Fig. 2A). Additionally, this induction was blocked by cotreatment with the PPARγ antagonist GW9662 (Fig. 2B), suggesting that this induction is PPARγ-dependent. Furthermore, we performed experiments using pCDE-HA-PPARγ or pCDE-HA-PPARγ siRNA to study KLF4 expression in HCT116 cells. As shown in Fig. 2C, PPARγ agonist-induced KLF4 expression was up-regulated upon overexpression of PPARγ and down-regulated in the absence of PPARγ, suggesting partial regulation of KLF4 through PPARγ activation. These results indicate that the expression of KLF4 is regulated, at least in part, in a PPARγ-dependent manner.
FIGURE 2.

Induction of KLF4 mRNA by PPARγ agonists is PPARγ-dependent. A, HT29 and LoVo (wild-type PPARγ) and HCT15 (mutant PPARγ) cells were grown in the absence of serum for 18 h (control (Ctr)) and then incubated with 10 μm TGZ for 24 h. The expression of KLF4 was determined by RT-qPCR and Western blot analysis. Data represent the mean ± S.D. of triplicate experiments. B, HCT116 cells were treated with TGZ or GW9662 or preincubated with 20 μm GW9662 for 30 min and then treated with 10 μm TGZ for 24 h. The levels of KLF4 expression were determined by RT-qPCR and Western blot analysis. C, HCT116 cells were transiently transfected with either pCDE-HA-PPARγ or PPARγ siRNA (siPPARγ) (50 nm) as described under “Experimental Procedures.” After 24 h of transfection, the cells were treated with vehicle (DMSO; −) or TGZ (10 μm; +) for an additional 24 h. PPARγ and KLF4 protein expression was analyzed by Western blotting. β-Actin was used as a loading control. A representative image of three independent experiments is shown.
KLF4 Is Directly Induced by PPARγ Agonists
To determine a direct link between TGZ and KLF4 expression, we performed experiments using cycloheximide to block de novo protein translation to study the expression of KLF4 in HCT116 cells. As shown in Fig. 3A, TGZ-induced KLF4 expression was not influenced by cycloheximide treatment, suggesting that KLF4 is directly induced by TGZ and that this regulation may not require de novo protein synthesis. To explore if the increase in KLF4 mRNA levels triggered by TGZ treatment was linked to post-transcriptional regulation, we measured the half-life of KLF4 mRNA by incubating cells with actinomycin D to block de novo gene transcription. RT-qPCR analysis revealed that the mRNA stability of KLF4 was not influenced by TGZ treatment (Fig. 3B). To further determine whether up-regulation of KLF4 induced by TGZ was transcriptional regulation, we measured KLF4 pre-mRNA and mRNA levels with or without TGZ treatment by RT-qPCR. As shown in Fig. 3C, both KLF4 pre-mRNA and mRNA levels increased after TGZ treatment, suggesting that up-regulation of KLF4 induced by TGZ is the result of transcriptional rather than post-transcriptional events.
FIGURE 3.
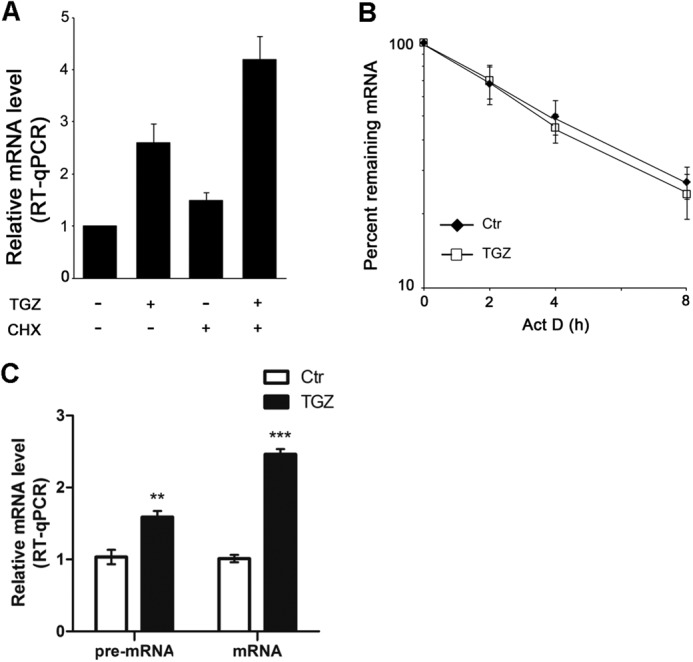
KLF4 is directly induced by PPARγ agonists. A, HCT116 cells were treated with 10 μm TGZ and/or 10 mg/ml cycloheximide (CHX) for 24 h. The levels of KLF4 mRNA were analyzed by RT-qPCR. Data represent the mean ± S.D. of triplicate experiments. B, HCT116 cells were untreated (control (Ctr)) or treated with 10 μm TGZ for 1 h, followed by actinomycin D (Act D; 5 mg/ml) treatment for the indicated times. KLF4 mRNA levels were measured by RT-qPCR, normalized to 18 S rRNA levels, and plotted on a logarithmic scale. Data represent the mean ± S.D. of triplicate experiments. C, HCT116 cells were treated with or without 10 μm TGZ for 12 h. KLF4 pre-mRNA and mRNA levels were measured by RT-qPCR. Data represent the mean ± S.D. of triplicate experiments. **, p < 0.01; ***, p < 0.001.
Up-regulation of KLF4 upon PPARγ Activation Is Mediated through the PPRE in the KLF4 Promoter
PPARγ regulates gene transcription via binding to PPREs located in the promoter regions of target genes. To further determine the direct regulation of KLF4 by PPARγ, we searched for putative PPARγ-binding sites in the human KLF4 promoter. Remarkably, we identified one potential PPARγ-binding site located at −1657 to −1669 bp upstream of the KLF4 ATG codon. Moreover, the putative PPRE site is highly conserved among different species (Fig. 4A), suggesting that KLF4 mRNA might be a direct target of PPARγ. To determine whether the putative PPRE is involved in transactivation, we next measured the KLF4 promoter activity using KLF4 gene promoter-reporter construct P1 (with the putative PPRE) and depletion mutant reporter construct P2 (lacking the putative PPRE) (Fig. 4B). As shown in Fig. 4C (left panel), a 2-fold increase in P1 was seen with TGZ treatment. In contrast, no significant activation of P2 was observed upon treatment with TGZ. Additionally, we performed cotransfection experiments using KLF4 promoter-reporter construct P1 and pCDE-HA-PPARγ or PPARγ siRNA; our data revealed that overexpression of PPARγ significantly activated P1 and that knockdown of PPARγ reduced the promoter activity (Fig. 4C, right panel). These findings demonstrate unequivocally that PPARγ transactivates KLF4 by binding the putative PPRE. To further examine whether PPARγ can bind the putative PPRE of KLF4, EMSA was performed using biotinylated probes and pcDNA3.1-PPARγ and pCDE-HA-RXR vectors expressing PPARγ and HA-tagged RXR protein. The results show that PPARγ bound a single-strand probe containing the putative PPRE of KLF4 (Fig. 4D, lane 2) and that excess specific competitor KLF4 PPRE (with the putative PPRE) inhibited formation of the probe-PPARγ/RXR complex (lane 3), whereas mutant competitor M1 (with the mutant PPRE) failed to inhibit the biotinylated probe binding to PPARγ (lane 4). In contrast, competitor M2, which contained a mutation near the PPRE, inhibited the biotinylated probe binding to PPARγ (Fig. 4D, lane 5). Furthermore, when anti-PPARγ or anti-HA antibody was applied to the probe/protein mixture, the binding was further shifted (Fig. 4D, lanes 6 and 7). These results demonstrate that the putative PPRE of KLF4 represents a bona fide site for the PPARγ-binding site. PPARγ may regulate KLF4 gene transcription in vivo by its binding activity for chromatin. We performed a quantitative ChIP assay using samples with or without TGZ treatment and antibody to PPARγ. The results show that PPARγ specifically bound the promoter region encompassing the PPRE of the KLF4 gene especially after TGZ treatment. In contrast, normal IgG did not precipitate detectable DNA, and the control sequence near the PPRE lost the immunoprecipitation signal (Fig. 4E). This provides additional evidence to support the active role of PPARγ in KLF4 gene transcription in vivo.
FIGURE 4.

Up-regulation of KLF4 upon PPARγ activation is mediated through the PPRE in the KLF4 promoter. A, the PPRE is located at −1657 to −1669 bp upstream of the KLF4 ATG codon and is highly conserved in different species. B, schematic representation of the KLF4 promoter-luciferase (Luc) reporter constructs. C, left panel, HCT116 cells were transfected with pGL3-Basic (control) or KLF4 promoter constructs. At 24 h after transfection, cells were left untreated (control (Ctr)) or treated with 10 μm TGZ for 18 h. Luciferase activity was determined and normalized to an internal cytomegalovirus-Renilla luciferase control. Data are shown as the relative luciferase value (mean ± S.D. of triplicate experiments) compared with untreated cells, which were set to 1. Right panel, HT29 cells were transfected with either pCDE-HA-PPARγ or PPARγ siRNA (siPPARγ; 50 nm) as indicated together with pGL3-Basic (control) or KLF4 promoter constructs for 48 h. Luciferase activity was determined and normalized to an internal cytomegalovirus-Renilla luciferase control. Data represent the mean ± S.D. of triplicate experiment *, p < 0.05; ***, p < 0.001. D, EMSAs were performed using 3 fmol of labeled oligonucleotides and 5 μg of nuclear extracts (NE) obtained from HEK293 cells transfected with pcDNA3.1-PPARγ and pCDE-HA-RXRα. Competitor oligonucleotides were added at a 100-fold molar excess of the labeled probe. In experiments involving antibodies, 2 μg of anti-PPARγ (Ab1) or anti-HA (Ab2) polyclonal antibody was added to the reaction. The positions of free probe (F), shifted bands (S), and supershifted bands (SS) are indicted. M1 and M2 are the mutant oligonucleotides used as competitor DNA. E, HCT116 cells were treated with 10 μm TGZ for 8 h, and ChIP assay were carried out with antibody against PPARγ or control IgG. The percent input of coprecipitating DNAs was calculated by RT-qPCR. Data represent the mean ± S.D. of triplicate experiments.
Stable Silencing of KLF4 Attenuates the Effect of PPARγ Agonists on the Modulation of KLF4 Target Genes
To determine whether the PPARγ-induced KLF4 increase would modify the expression levels of known KLF4 target genes, we measured the expression levels of p21Waf1, p27Kip1, and cyclins D1 and E2. HCT116 cells stably silenced with KLF4 or control shRNA were used to delineate the role of KLF4 as a mediator of these TGZ-modulatable genes. As shown in Fig. 5A, shRNA markedly inhibited KLF4 mRNA expression following TGZ treatment. The response of the KLF4 target genes to TGZ was determined by real-time RT-qPCR and Western blotting in KLF4 or control shRNA-silenced cells. TGZ treatment resulted in an up-regulation of p21Waf1 and p27Kip1 but not cyclin D1 or E2 in the control shRNA-silenced cells (Fig. 5, B–D, and supplemental Fig. S1). Ablation of KLF4 expression significantly muted the effect of TGZ on the expression of these genes.
FIGURE 5.

Stable silencing of KLF4 attenuates the effect of PPARγ agonists on the modulation of KLF4 target genes. A–C, RNA was extracted from HCT116 cells stably silenced with KLF4 (sh-KLF4) or control (sh-Ctr) shRNA and treated for 24 h with DMSO (control (Ctr)) or TGZ (10 μm). KLF4, p21Waf1, and p27Kip1 expression was measured by real-time qPCR and normalized to GAPDH. Results are the mean ± S.D. of three different experiments. *, p < 0.05; ***, p < 0.001. D, HCT116 cells stably silenced with control or KLF4 shRNA were grown in the absence of serum for 18 h and then incubated with DMSO (−) or TGZ (10 μm; +) for 48 h. The expression of KLF4, p21, and p27 was determined by Western blotting. β-Actin served as an internal control.
Ablation of KLF4 Expression Reduces PPARγ Agonist-mediated G1/S Arrest and Anti-proliferation Effects in HCT116 Cells
In an effort to evaluate the biological significance of KLF4 up-regulation by TGZ, we assessed the response of the stable silencing of KLF4 in HCT116 cells to TGZ-mediated cell proliferation inhibition. The growth curves for control and KLF4 shRNA-silenced cells treated with or without TGZ were compared. Ablation of KLF4 expression reduced PPARγ agonist-mediated cell proliferation arrest (Fig. 6A) but had very subtle effects on long-term cell growth arrest both in vitro and in vivo (supplemental Figs. S2 and S3). To clarify the mechanisms underlying cell proliferation inhibition, BrdU incorporation and cell cycle analysis were conducted at 48 h post-TGZ treatment. TGZ treatment inhibited DNA synthesis by >50% in the control shRNA-silenced cells as opposed to 20% in the KLF4 shRNA-silenced cells (Fig. 6B and supplemental Fig. S4). The difference is statistically significant (p < 0.05). Additionally, as shown in Fig. 6C, TGZ-induced G1/S arrest was markedly muted as a result of KLF4 ablation. Taken together with the KLF4 silencing data, our results show the important role of KLF4 up-regulation in mediating the effect of TGZ on cell proliferation inhibition.
FIGURE 6.

Ablation of KLF4 expression reduces PPARγ agonist-mediated G1/S arrest and anti-proliferation effects in HCT116 cells. A, HCT116 cells stably silenced with control (sh-ctr) or KLF4 (sh-KLF4) shRNA were incubated with DMSO (control) or TGZ (10 μm) for the indicated times, and cell growth inhibition was detected using MTS assay. Values were the mean ± S.D. of absorbance at 570 nm for three independent experiments. B, HCT116 cells stably silenced with control or KLF4 shRNA were left untreated (control (Ctr)) or treated with 10 μm TGZ for 48 h and then analyzed by BrdU incorporation using a BrdU cell proliferation ELISA kit. Similar results were observed in a triplicate analysis. *, p < 0.05. C, cell cycle analysis was conducted at 48 h post-TGZ treatment in control and KLF4 shRNA-silenced HCT116 cells. The percentage of the G1 phase cells was determined and is shown in a graph. Similar results were observed in a triplicate analysis. *, p < 0.05.
DISCUSSION
PPARγ is known as a master transcriptional regulator of glucose and lipid metabolism, but it also plays an important role in carcinogenesis. This receptor has the ability to bind a variety of small lipophilic compounds. In turn, these ligands direct cofactor recruitment to PPARγ, regulating the transcription of genes in a variety of complex metabolic pathways. Indeed, PPARγ ligands such as thiazolidinediones (RGZ, TGZ, and PGZ), which are commonly used in the clinical setting as anti-diabetic medications, are potent and selective activators of PPARγ. The role of thiazolidinediones in growth of cancer cells has been elucidated in some studies. In a phase II study of the use of TGZ in the treatment of patients with advanced breast cancer, no objective tumor response was observed (27). However, the study was incomplete. On the other hand, it is important to note that neither hormone status of the tumors nor the amount of PPARγ protein was assessed before patients were included in the study. In contrast, some studies have suggested that PPARγ ligands inhibit growth of malignant human cells; cause cell cycle arrest and apoptosis in a broad spectrum of tumor cell lines; and can be used as chemopreventive agents for colon, breast, and prostate carcinogenesis (28). Another study showed that up-regulation of p27 and p21 plays a role in the regulation of TGZ-induced G1 arrest in colon cancer cell lines (29). However, up to now, no functional PPREs were definitively identified in the p27Kip1 and p21Waf1 promoter regions, which means that the regulation of p27 and p21 by PPARγ is indirect, involving other factors.
Previous studies showed that some PPARγ agonists induce KLF4 expression in a receptor-dependent manner in colorectal cancer cells (24, 25). In line with this, we have shown that TGZ specifically increased KLF4 mRNA and protein levels and that this regulation is direct because the PPARγ-induced expression of KLF4 was not influenced by co-incubation with cycloheximide, suggesting that this regulation does not require de novo protein synthesis. Additionally, our data show that the mRNA stability of KLF4 was not influenced after TGZ treatment by incubating cells with actinomycin D. Furthermore, both KLF4 pre-mRNA and mRNA were induced by TGZ treatment, indicating that the up-regulation of KLF4 induced by TGZ is at the transcriptional level rather than the post-transcriptional level. In contrast, Chen and Tseng (30) concluded that 15d-PGJ2 up-regulates KLF4 expression independently of PPARγ through activation of the MAPK signaling pathway in HT29 colon cancer cells. The differences we have confirmed in ligand-receptor activity may provide an explanation for differences of action in different colorectal cancer cells. Thus, we have shown that TGZ-induced KLF4 expression was abolished in the presence of GW9662, a PPARγ-specific antagonist, suggesting the PPARγ-dependent manner of TGZ-induced KLF4 expression. Remarkably, we identified one functional PPARγ-binding site with computer-assisted transcription factor-binding site analysis. This site is located at −1657 to −1669 bp upstream of the KLF4 ATG codon and is highly conserved among different species. The specificity of the KLF4-binding sites was also confirmed by EMSA and ChIP assay, as shown in Fig. 4 (D and E, respectively). Additionally, our data indicate that PPARγ binds to its PPRE as a heterodimer with RXR. Upon binding a ligand, the conformation of PPARγ is altered and stabilized such that a binding cleft is created, and recruitment of transcriptional coactivators occurs. It has been reported that activation of the PPARγ/RXR heterodimer can generate a synergistic response in inhibiting cell growth in colon cancer cell lines (31). Therefore, further work will be necessary to determine whether activation of the PPARγ/RXR heterodimer generates a synergistic response in upon KLF4 expression. Furthermore, luciferase assay demonstrated unequivocally that PPARγ transactivates KLF4 by binding the putative PPRE (Fig. 4C).
Notably, the biological function of KLF4 depends on the genetic context, and many studies have shown that the expression of KLF4 is associated with both inhibition and induction of cell proliferation, affecting tumorigenesis both positively and negatively (23, 32, 33). However, recent studies showed that KLF4 is down-regulated during tumorigenesis of the gastrointestinal epithelium and is frequently lost in other human cancer types. Consistent with a tumor suppressor function of KLF4, its overexpression reduces tumorigenesis in colonic cancer cells. Although KLF4 may play a protumorigenic role in other cancer types (16), our data provide evidence that KLF4 has a role as a tumor suppressor in colorectal cancer. In this study, we have presented three lines of evidence to support the role of KLF4 in mediating the effect of TGZ on inhibition of colorectal cancer cell proliferation. First, TGZ treatment led to a direct induction of KLF4 expression. Second, stable silencing of KLF4 by shRNA significantly diminished the responsiveness to TGZ with respect to the expression of KLF4 target genes p21Waf1 and p27Kip1. Third, ablation of KLF4 expression reduced TGZ-mediated cell proliferation arrest in HCT116 cells. Our preliminary findings indicate that knockdown of PPARγ resulted in down-regulation of KLF4 and increased cell growth (supplemental Fig. S5). Furthermore, we found that TGZ-induced anti-proliferation effects were markedly muted as a result of KLF4 ablation (Fig. 6, A–C). However, it is important to know that KLF4 is one of several transcription factors whose expression is known to be modulated by TGZ treatment. Thus, it is not surprising to find that silencing KLF4 alone may not completely block the effect of TGZ on cell proliferation arrest. Further work will be necessary to identify additional genes involved in this network.
In conclusion, our data clearly demonstrate for the first time that PPARγ regulates the expression of KLF4 by binding directly to the PPRE within the KLF4 promoter, leading to activation of a tumor suppressor network in colorectal cancer. Furthermore, our results provide a novel cancer therapeutic strategy and will help define the mechanisms by which PPARγ and KLF4 are involved in colorectal cancer cell biology.
Supplementary Material
This work was supported by National Basic Research Program of China Grant 2011CB504205 and National Natural Science Foundation of China Grants 81130043 and 81021061.

This article contains supplemental Figs. S1–S5.
Primer sequences are available upon request.
- PPAR
- peroxisome proliferator-activated receptor
- RXR
- retinoid X receptor
- PPRE
- PPAR response element
- 15d-PGJ2
- 15-deoxy-Δ12,14-prostaglandin J2
- TGZ
- troglitazone
- RGZ
- rosiglitazone
- PGZ
- pioglitazone
- DMSO
- dimethyl sulfoxide
- qPCR
- quantitative PCR
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt.
REFERENCES
- 1. Vamecq J., Latruffe N. (1999) Medical significance of peroxisome proliferator-activated receptors. Lancet 354, 141–148 [DOI] [PubMed] [Google Scholar]
- 2. Kersten S., Desvergne B., Wahli W. (2000) Roles of PPARs in health and disease. Nature 405, 421–424 [DOI] [PubMed] [Google Scholar]
- 3. Lehrke M., Lazar M. A. (2005) The many faces of PPARγ. Cell 123, 993–999 [DOI] [PubMed] [Google Scholar]
- 4. Tontonoz P., Hu E., Spiegelman B. M. (1994) Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 79, 1147–1156 [DOI] [PubMed] [Google Scholar]
- 5. Chinetti G., Griglio S., Antonucci M., Torra I. P., Delerive P., Majd Z., Fruchart J. C., Chapman J., Najib J., Staels B. (1998) Activation of proliferator-activated receptors α and γ induces apoptosis of human monocyte-derived macrophages. J. Biol. Chem. 273, 25573–25580 [DOI] [PubMed] [Google Scholar]
- 6. Kubota T., Koshizuka K., Williamson E. A., Asou H., Said J. W., Holden S., Miyoshi I., Koeffler H. P. (1998) Ligand for peroxisome proliferator-activated receptor γ (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Res. 58, 3344–3352 [PubMed] [Google Scholar]
- 7. Takahashi N., Okumura T., Motomura W., Fujimoto Y., Kawabata I., Kohgo Y. (1999) Activation of PPARγ inhibits cell growth and induces apoptosis in human gastric cancer cells. FEBS Lett. 455, 135–139 [DOI] [PubMed] [Google Scholar]
- 8. Tontonoz P., Singer S., Forman B. M., Sarraf P., Fletcher J. A., Fletcher C. D., Brun R. P., Mueller E., Altiok S., Oppenheim H., Evans R. M., Spiegelman B. M. (1997) Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor γ and the retinoid X receptor. Proc. Natl. Acad. Sci. U.S.A. 94, 237–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kliewer S. A., Umesono K., Noonan D. J., Heyman R. A., Evans R. M. (1992) Convergence of 9-cis-retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 358, 771–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Forman B. M., Tontonoz P., Chen J., Brun R. P., Spiegelman B. M., Evans R. M. (1995) 15-Deoxy-Δ12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell 83, 803–812 [DOI] [PubMed] [Google Scholar]
- 11. Toyota M., Miyazaki Y., Kitamura S., Nagasawa Y., Kiyohara T., Shinomura Y., Matsuzawa Y. (2002) Peroxisome proliferator-activated receptor γ reduces the growth rate of pancreatic cancer cells through the reduction of cyclin D1. Life Sci 70, 1565–1575 [DOI] [PubMed] [Google Scholar]
- 12. Itami A., Watanabe G., Shimada Y., Hashimoto Y., Kawamura J., Kato M., Hosotani R., Imamura M. (2001) Ligands for peroxisome proliferator-activated receptor γ inhibit growth of pancreatic cancers both in vitro and in vivo. Int. J. Cancer 94, 370–376 [DOI] [PubMed] [Google Scholar]
- 13. Barak Y., Nelson M. C., Ong E. S., Jones Y. Z., Ruiz-Lozano P., Chien K. R., Koder A., Evans R. M. (1999) PPARγ is required for placental, cardiac, and adipose tissue development. Mol. Cell 4, 585–595 [DOI] [PubMed] [Google Scholar]
- 14. Ohta K., Endo T., Haraguchi K., Hershman J. M., Onaya T. (2001) Ligands for peroxisome proliferator-activated receptor γ inhibit growth and induce apoptosis of human papillary thyroid carcinoma cells. J. Clin. Endocrinol. Metab. 86, 2170–2177 [DOI] [PubMed] [Google Scholar]
- 15. Panigrahy D., Singer S., Shen L. Q., Butterfield C. E., Freedman D. A., Chen E. J., Moses M. A., Kilroy S., Duensing S., Fletcher C., Fletcher J. A., Hlatky L., Hahnfeldt P., Folkman J., Kaipainen A. (2002) PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J. Clin. Invest. 110, 923–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Foster K. W., Frost A. R., McKie-Bell P., Lin C. Y., Engler J. A., Grizzle W. E., Ruppert J. M. (2000) Increase of GKLF messenger RNA and protein expression during progression of breast cancer. Cancer Res. 60, 6488–6495 [PubMed] [Google Scholar]
- 17. Foster K. W., Ren S., Louro I. D., Lobo-Ruppert S. M., McKie-Bell P., Grizzle W., Hayes M. R., Broker T. R., Chow L. T., Ruppert J. M. (1999) Oncogene expression cloning by retroviral transduction of adenovirus E1A-immortalized rat kidney RK3E cells: transformation of a host with epithelial features by c-MYC and the zinc finger protein GKLF. Cell Growth Differ. 10, 423–434 [PubMed] [Google Scholar]
- 18. Wang N., Liu Z. H., Ding F., Wang X. Q., Zhou C. N., Wu M. (2002) Down-regulation of gut-enriched Krüppel-like factor expression in esophageal cancer. World J. Gastroenterol. 8, 966–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wei D., Kanai M., Huang S., Xie K. (2006) Emerging role of KLF4 in human gastrointestinal cancer. Carcinogenesis 27, 23–31 [DOI] [PubMed] [Google Scholar]
- 20. Ohnishi S., Ohnami S., Laub F., Aoki K., Suzuki K., Kanai Y., Haga K., Asaka M., Ramirez F., Yoshida T. (2003) Down-regulation and growth inhibitory effect of epithelial-type Krüppel-like transcription factor KLF4, but not KLF5, in bladder cancer. Biochem. Biophys. Res. Commun. 308, 251–256 [DOI] [PubMed] [Google Scholar]
- 21. Hu W., Hofstetter W. L., Li H., Zhou Y., He Y., Pataer A., Wang L., Xie K., Swisher S. G., Fang B. (2009) Putative tumor-suppressive function of Krüppel-like factor 4 in primary lung carcinoma. Clin. Cancer Res. 15, 5688–5695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guan H., Xie L., Leithäuser F., Flossbach L., Möller P., Wirth T., Ushmorov A. (2010) KLF4 is a tumor suppressor in B-cell non-Hodgkin lymphoma and in classic Hodgkin lymphoma. Blood 116, 1469–1478 [DOI] [PubMed] [Google Scholar]
- 23. Dang D. T., Chen X., Feng J., Torbenson M., Dang L. H., Yang V. W. (2003) Overexpression of Krüppel-like factor 4 in the human colon cancer cell line RKO leads to reduced tumorigenicity. Oncogene 22, 3424–3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rageul J., Mottier S., Jarry A., Shah Y., Théoleyre S., Masson D., Gonzalez F. J., Laboisse C. L., Denis M. G. (2009) KLF4-dependent, PPARγ-induced expression of GPA33 in colon cancer cell lines. Int. J. Cancer 125, 2802–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cekanova M., Lee S. H., McEntee M. F., Baek S. J. (2010) MCC-555-induced NAG-1 expression is mediated in part by KLF4. Eur. J. Pharmacol. 637, 30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou Q., Hong Y., Zhan Q., Shen Y., Liu Z. (2009) Role for Krüppel-like factor 4 in determining the outcome of p53 response to DNA damage. Cancer Res. 69, 8284–8292 [DOI] [PubMed] [Google Scholar]
- 27. Burstein H. J., Demetri G. D., Mueller E., Sarraf P., Spiegelman B. M., Winer E. P. (2003) Use of the peroxisome proliferator-activated receptor (PPAR) γ ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res. Treat. 79, 391–397 [DOI] [PubMed] [Google Scholar]
- 28. Koeffler H. P. (2003) Peroxisome proliferator-activated receptor γ and cancers. Clin. Cancer Res. 9, 1–9 [PubMed] [Google Scholar]
- 29. Ming M., Yu J. P., Meng X. Z., Zhou Y. H., Yu H. G., Luo H. S. (2006) Effect of ligand troglitazone on peroxisome proliferator-activated receptor γ expression and cellular growth in human colon cancer cells. World J. Gastroenterol. 12, 7263–7270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen Z. Y., Tseng C. C. (2005) 15-deoxy-Δ12,14-prostaglandin J2 up-regulates Krüppel-like factor 4 expression independently of peroxisome proliferator-activated receptor γ by activating the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signal transduction pathway in HT-29 colon cancer cells. Mol. Pharmacol. 68, 1203–1213 [DOI] [PubMed] [Google Scholar]
- 31. Papi A., Rocchi P., Ferreri A. M., Orlandi M. (2010) RXRγ and PPARγ ligands in combination to inhibit proliferation and invasiveness in colon cancer cells. Cancer Lett. 297, 65–74 [DOI] [PubMed] [Google Scholar]
- 32. Rowland B. D., Bernards R., Peeper D. S. (2005) The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat. Cell Biol. 7, 1074–1082 [DOI] [PubMed] [Google Scholar]
- 33. Rowland B. D., Peeper D. S. (2006) KLF4, p21 and context-dependent opposing forces in cancer. Nat. Rev. Cancer 6, 11–23 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.