

Background: Oxidation of KCNB1 channels leads to oligomerization and apoptosis.
Results: KCNB1 oligomers aggregate in and disrupt glycolipid raft organization, promoting the activation of the Src/JNK pro-apoptotic pathway.
Conclusion: KCNB1 aggregates initiate an apoptotic cascade mediated by c-Src/JNK kinases.
Significance: Oxidized KCNB1 channels increase in aging mammalian brain. As such, this mechanism contributes to neuronal aging and neurodegeneration.
Keywords: Aging, Apoptosis, Membrane Biophysics, Membrane Proteins, Potassium Channels, Reactive Oxygen Species (ROS)
Abstract
Potassium (K+) channels are targets of reactive oxygen species in the aging nervous system. KCNB1 (formerly Kv2.1), a voltage-gated K+ channel abundantly expressed in the cortex and hippocampus, is oxidized in the brains of aging mice and of the triple transgenic 3xTg-AD mouse model of Alzheimer's disease. KCNB1 oxidation acts to enhance apoptosis in mammalian cell lines, whereas a KCNB1 variant resistant to oxidative modification, C73A-KCNB1, is cytoprotective. Here we investigated the molecular mechanisms through which oxidized KCNB1 channels promote apoptosis. Biochemical evidence showed that oxidized KCNB1 channels, which form oligomers held together by disulfide bridges involving Cys-73, accumulated in the plasma membrane as a result of defective endocytosis. In contrast, C73A-mutant channels, which do not oligomerize, were normally internalized. KCNB1 channels localize in lipid rafts, and their internalization was dynamin 2-dependent. Accordingly, cholesterol supplementation reduced apoptosis promoted by oxidation of KCNB1. In contrast, cholesterol depletion exacerbated apoptotic death in a KCNB1-independent fashion. Inhibition of raft-associating c-Src tyrosine kinase and downstream JNK kinase by pharmacological and molecular means suppressed the pro-apoptotic effect of KCNB1 oxidation. Together, these data suggest that the accumulation of KCNB1 oligomers in the membrane disrupts planar lipid raft integrity and causes apoptosis via activating the c-Src/JNK signaling pathway.
Introduction
Aging is associated with increased levels of reactive oxygen species (ROS)2 in the cell, a condition known as oxidative stress (1). ROS, which are synthesized during the metabolism of oxygen, play important roles in cell signaling. However, during aging or under conditions of cellular stress, such as in certain neurodegenerative diseases, they can diffuse in the cytoplasm and begin to oxidize a variety of essential components, including lipids, ribonucleic acids, and proteins (2). This may result in significant cellular damage (3). Potassium (K+) channels are targets of ROS in these conditions (4). The first experimental evidence for oxidation of K+ channels by ROS during aging came from the worm Caenorhabditis elegans (5). In this animal, oxidative modification of the voltage-gated K+ channel KVS-1 leads to progressive loss of neurosensory function (taste). KVS-1 is a homolog to mammalian KCNB1, which is abundantly expressed in the brain. Like KVS-1, KCNB1 is also a redox-susceptible channel (6). Oxidized KCNB1 channels are ∼10-fold more abundant in the brains of old, rather than young, mice. ROS effects on KCNB1 and KVS-1 are mediated by two conserved N-terminal cysteine residues, Cys-73 and Cys-113, respectively. The functional modifications resulting from their oxidation are different, though. KVS-1 is a rapidly activating-inactivating K+ channel (A-type). Oxidation of Cys-113 turns the channel in a non-inactivating channel. In contrast, KCNB1 can be described as a delayed rectifier (non-inactivating) channel, and its oxidation produces two major modifications. First, it induces the oligomerization of the channel through the formation of intersubunit disulfide bridges involving Cys-73 and a cysteine in the C terminus, Cys-710. Second, it decreases the open probability, giving rise to non-conducting channels. KCNB1 oxidation promotes apoptosis in cultured cells, an effect primarily caused by oligomer formation and not by changes in the magnitude of the current. Evidence further shows that oxidation of KCNB1 is associated with increased ROS. Thus, considering the central role played by KCNB1 in the cortex and hippocampus and the fact that this channel is oxidized in the aging mouse brain and in the brain of a mouse model of Alzheimer's disease (6), the elucidation of the mechanism through which KCNB1 oligomerization causes apoptosis deserves elucidation.
Here we show that oxidative conditions lead to the accumulation of KCNB1 channels in the plasma membrane by impairing their internalization. KCNB1 aggregates disrupt glycolipid raft organization, triggering a stress signal that results in the activation of an apoptotic cascade mediated by c-Src and JNK kinases.
EXPERIMENTAL PROCEDURES
Cell Cultures
Undifferentiated mouse neuroblastoma N2A cells were grown in DMEM supplemented with 10% fetal bovine serum and 1% sodium pyruvate. Cells were transfected at 90% confluence using Lipofectamine 2000 (Invitrogen).
Chemicals
Chemicals were prepared in stocks and freshly added prior the experiment. The chemicals used were as follows: cholesterol (Sigma), 3 mg/ml in DMEM, serum-free; methyl-β-cyclodextrin (MβCD) (Sigma), 20 mm in DMEM, serum-free; 2′,7′-dichlorofluorescein diacetate (Molecular Probes), 500 μm in 4.7% DMSO, 95.2% PBS; IETD-CHO (caspase-8 inhibitor I) (EMD Chemicals), 2.5 mm in DMSO; SP600125 (Sigma), 10 mm in DMSO; dTDP (Sigma), 100 mm in DMSO; l-glutathione, reduced (Sigma), 150 mm in PBS; and PP2 (Cayman Chemicals), 10 mm in DMSO.
Membrane Biotinylation and Biotin Feeding
In both surface expression and internalization (biotin feeding (7)) assays we used the biotin derivative sulfo-NHS-SS-biotin, which can be removed upon application of impermeable glutathione (therefore only internalized, biotin-labeled proteins are protected from biotin cleavage). Thus, 24 h after transfection, N2A cells were incubated with 1 mg/ml sulfo-NHS-SS-biotin (Pierce) at 4 °C for 1 h and washed three times with ice-cold PBS containing 100 mm glycine. The cells were oxidized with 25 μm dTDP in DMEM, serum-free, at 37 °C for 5 min and then incubated for 5, 25, or 55 min in normal DMEM. Then, for surface expression measurements, cells were washed three times in ice-cold PBS and harvested. For internalization measurements, cells were washed three times with PBS and immediately subjected to three cycles of biotin cleavage by 10-min incubation in ice-cold PBS containing 75 mm reduced glutathione. Cells were harvested in radioimmune precipitation assay buffer (1% Nonidet P-40, 50 mm Tris (pH 7.4), 150 mm NaCl, 1 mm EDTA, 0.5% (w/v) deoxycholate, 0.1% (w/v) SDS). We then freshly added 1 mm PMSF and protease inhibitors mixture set I (Calbiochem) for 30 min at 4 °C. Cell lysates were centrifuged at 16,000 × g for 15 min at 4 °C, and the supernatant was mixed with (SA)-linked streptavidin-agarose beads (Pierce) and incubated overnight at 4 °C. The samples were centrifuged at 8000 × g for 10 min and washed four times with immunoprecipitation buffer. Samples were incubated in SDS/PAGE sample buffer containing 10% (w/v) 2-mercaptoethanol at 90–100 °C for 15 min and centrifuged at 8000 × g for 5 min. The supernatant containing the eluted KCNB1-HA protein was immunoblotted with monoclonal anti-HA antibody (Roche).
Immunofluorescence
24 h after transfection, cells seeded on coverslips were biotinylated as described above. Then cells were incubated in the presence or absence of 25 μm dTDP in DMEM without serum at 37 °C for 7 min. Cells were washed three times with PBS, fixed with 4% paraformaldehyde in PBS (pH 7.4) for 15 min, and washed twice with PBS. Then cells were permeabilized with 1% Triton X-100 in PBS for 20 min, washed twice with PBS, and blocked with 1% BSA in PBS for 30 min at room temperature. After blocking, cells were incubated with streptavidin-FITC (Invitrogen) in blocking buffer for 45 min and washed three times with PBS. Cells were analyzed with a Zeiss LSM 510 laser scanning confocal microscope.
Annexin V Apoptosis Assays
Apoptosis measurements were performed as described before (6). Briefly, cells positive to annexin V were computed manually (using the Annexin V/PI kit, Roche) in 25 μm2 areas (typically, four to six measurements). Experiments were performed blindly. Thus, N2A cells were plated on 12-mm glass coverslips. Twenty-four hours after transfection, the cells were washed with PBS, incubated for 15–30 min with inhibitors, and exposed to 25 μm dTDP or 1.0 mm hydrogen peroxide (H2O2) for 5 min and then washed twice with PBS. The cells were incubated for 6 h in fresh DMEM. The cells were washed with PBS and incubated with annexin V labeling buffer solution (20 μl of annexin V-Fluos and 20 μl of propidium iodide in 1 ml of incubation buffer). Propidium iodide detects necrotic cells provided by the kit for 15 min according to the instructions of the manufacturer and mounted on an Olympus BX61 microscope equipped with Nomarski optics and a digital camera.
DCF Fluorimetry
Measurements were performed as described before (6). Briefly, N2A cells were incubated with 5 μm 2′,7′-dichlorofluorescein diacetate, DCFH-DA, for 30 min. 2′,7′-dichlorofluorescein diacetate permeates the cell membrane and becomes hydrolyzed by intracellular esterases to nonfluorescent dichlorofluorescein, DCFH. In the presence of ROS, DCFH is oxidized to fluorescent dichlorofluorescein, DCF. Cells were washed twice with PBS and analyzed with an Olympus BX61 microscope equipped with Nomarski optics and a digital camera. Digitally acquired pictures were subsequently analyzed with ImageJ 1.44 software (National Institutes of Health).
Statistical Analysis
Quantitative data are presented as mean ± S.E. The level of significance was calculated using Student's t test for single comparisons and analysis of variance for multiple comparisons. Normality was assessed by the F-test, and corrections were performed using a Tukey means comparison test with a 95% confidence level using OriginPro 7.5 software. Statistical significance was assumed at the 95% confidence limit or greater (p < 0.05).
RESULTS
Oxidized KCNB1 Channels Exhibit Defective Endocytosis
Oxidants promote the formation of KCNB1 oligomers (held together by disulfide bridges between an N-terminal cysteine, Cys-73, and a C-terminal cysteine, Cys-710) that exacerbate apoptosis in mammalian cells (6, 8). Here we determined the fate of oxidized KCNB1 channels. In a first approach, we evaluated surface expression of KCNB1 oligomers by fluorescence. Fig. 1A shows representative confocal images of undifferentiated mouse neuroblastoma (N2A) cells labeled with FITC-conjugated biotin. Protein labeling qualitatively increased in oxidized cells expressing KCNB1. This suggested that oxidative conditions may lead to the accumulation of KCNB1 channels in the plasma membrane by impairing their endocytosis. To test this possibility, we determined the fraction of internalized KCNB1 channels by biotin feeding (7). KCNB1 channels epitope-tagged to the HA tag in the C terminus (KCNB1-HA) were labeled with glutathione-cleavable sulfo-NHS-SS-biotin, oxidized, treated in the absence/presence of glutathione (75 mm), streptavidin-immunoprecipitated, and immunoblotted with anti-HA antibody. Representative results of experiments carried out with wild-type and C73A KCNB1, respectively, are shown in Fig. 1B. The bands in the two left lanes of the immunoblots in the figure (− glutathione, Surface) correspond to the amounts of channels in the membrane of cells incubated in the absence/presence of 25 μm dTDP. Densitometry analysis of five experiments such as the one shown in Fig. 1B indicated that wild-type channel surface expression increased in cells subjected to an oxidative challenge by +18 ± 1% (p < 0.013), whereas C73A expression did not change significantly (+4 ± 0.1%). These results were consistent with confocal data (Fig. 1A) and with a previous report (9). The bands in the two right lanes of the immunoblots (+ glutathione, Internalized) correspond to the fractions of internalized channels in control and oxidative conditions. Accordingly, wild-type internalization was impaired in response to an oxidative insult. In contrast, C73A-mutant channels did not show accumulation in the membrane nor did they exhibit internalization defects. The time course of KCNB1 internalization, expressed as the fraction of internalized channels in the presence of dTDP normalized to the control, 10, 30, and 60 min post-oxidation is shown in Fig. 1C. Wild-type internalization was impaired markedly within the first 10 min after the oxidative insult, but within 1 h it had recovered by more than 70%. In contrast, C73A channels did not exhibit internalization defects. We showed previously that in cells expressing the wild type, ROS levels increased post-oxidation (6). This increase was likely the culprit of the incomplete recovery of wild-type channels 1 h post-oxidation. As a means of control, endocytosis was inhibited by maintaining the temperature at 4 °C (n = 4 experiments). As the representative experiment in Fig. 1D illustrates, under these conditions internalization of KCNB1 channels was inhibited irrespective of the redox status of the cell. Together, these results indicate that oxidative conditions leads to the accumulation of wild-type KCNB1 channels in the plasma membrane by impairing their endocytosis.
FIGURE 1.

Oxidized KCNB1 channels exhibit defective internalization. A, representative confocal images of N2A cells transfected with wild-type KCNB1-HA in control cells or after 5-min incubation with 25 μm dTDP. Proteins were labeled with biotin and with a streptavidin-FITC-conjugated secondary antibody. Scale bar = 5 μm. B, − glutathione (Glut.) lanes: representative Western blot analysis visualization of wild-type and C73A KCNB1-HA channels in the plasma membrane of N2A cells in control cells or subjected to an oxidative insult. + glutathione lanes, fraction of internalized wild-type and C73A KCNB1-HA channels in a 10-min interval. Channels were labeled with impermeant biotin derivative sulfo-NHS-SS-biotin and incubated for 5 min in DMEM containing 25 μm dTDP and for an additional 5 min in normal DMEM. Then the cells were either washed with PBS (− glutathione) or 75 mm reduced glutathione in PBS (+ glutathione) to remove surface biotin, immunoprecipitated with streptavidin-agarose beads, and immunoblotted with monoclonal anti-HA antibody. C, fraction of internalized wild-type or C73A channels 10, 30, and 60 min after an oxidative insult (DTDP) normalized to the fraction of internalized channels in the control. Densitometry analysis was performed using ImageJ 1.44 software (National Institutes of Health). n = 3–5 experiments/point. D, as in B in cells maintained at 4 °C. *, p < 0.05; **, p < 0.01.
KCNB1 Endocytosis Requires Dynamin 2
KCNB1 localizes in glycolipid rafts (10), whose most common form of endocytosis proceeds through clathrin-coated pits. This mechanism of internalization requires the GTPase dynamin 2 (11). Dynamin-independent internalization is also thought to occur but only in caveolar lipid rafts (12). To distinguish these possibilities, we employed a dominant-negative dynamin 2 mutant bearing a Lys-to-Ala replacement at position 44 (K44A) (13). As expected, the K44A mutant markedly inhibited the internalization of KCNB1 channels (Fig. 2A). Densitometry analysis of three experiments revealed a decrease of ∼75% in the fraction of internalized channels in cells cotransfected with K44A compared with cells cotransfected with wild-type dynamin 2 (Fig. 2B). Thus, KCNB1 endocytosis requires dynamin 2, consistent with the notion that the channel localizes in planar, non-caveolar lipid rafts.
FIGURE 2.
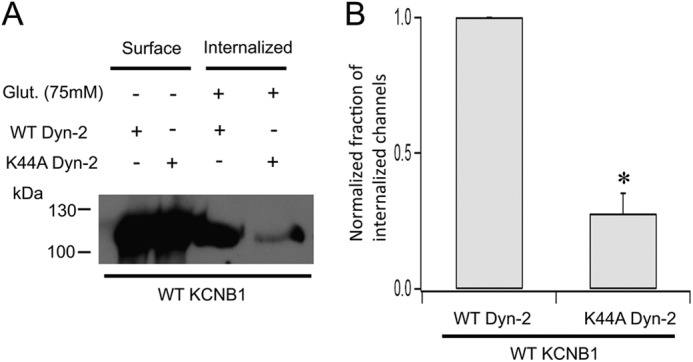
KCNB1 endocytosis is dynamin 2-dependent. A, representative Western blot showing surface expression and internalization of wild-type KCNB1 channels cotransfected with wild-type or dominant-negative K44A dynamin 2. Glut., glutathione. B, normalized fraction of internalized wild-type KCNB1 channels in N2A cells cotransfected with wild-type or K44A dynamin 2. n = 3 experiments. *, p < 0.05.
KCNB1 Oligomerization Perturbs Lipid Raft Structure
Lipid raft microdomains contain stress receptors which can initiate cellular apoptosis in conjunction with elevated ROS (reviewed in Ref. 14). This argues that KCNB1 aggregates may cause raft disturbance and, consequently, activate an apoptotic cascade. To test this idea, we determined whether reinforcement of raft integrity reduced apoptosis under oxidative conditions. We preincubated cells with 30 μg/ml cholesterol, typically for 2 h, before the application of 25 μm dTDP. Six hours post-oxidation, cells were scored for apoptosis by annexin V staining. In agreement with previous reports (6, 8), dTDP treatment increased apoptotic death by more than 2-fold in cells expressing wild-type KCNB1 (Fig. 3A). Cells preincubated with cholesterol exhibited markedly lower apoptotic rates. Coupling cholesterol with 1 mm MβCD-cholesterol complex, which favors adsorption of free cholesterol, further lessened apoptosis. Moreover, cholesterol supplementation had negligible effect on the levels of apoptosis, which remained fairly low, in cells transfected with C73A (Fig. 3B). This result was expected because the mutant does not enhance apoptosis in cells challenged with oxidants. Cholesterol depletion by incubation with 3.0 mm MβCD for 2 h promoted apoptosis as expected (Fig. 3C). However, apoptotic levels were comparable in cells transfected with either the wild type or C73A (Fig. 3, C and D). This result corroborated the notion that oxidation of KCNB1 promotes lipid raft disruption because if the two mechanisms were distinct, apoptotic rates should have been lower in cells transfected with C73A. Furthermore, the number of cells undergoing necrosis, which did not depend on dTDP treatment, was low and fairly stable in all conditions tested (≤ 5 ± 1%). The anti-apoptotic effect of cholesterol could stem from the reinforcement of the structure of the raft or, alternatively, rescue of defective KCNB1 internalization. To distinguish these possibilities, we carried out biotin feeding experiments in the presence of cholesterol (Fig. 4). In three experiments, pretreatment with cholesterol did not prevent nor rescue defective internalization of oxidized KCNB1 channels (Fig. 4B). We conclude that cholesterol suppresses apoptosis by acting to restore raft organization.
FIGURE 3.

Cholesterol suppresses KCNB1 oxidation-induced apoptosis. A, percentage of N2A cells expressing wild-type KCNB1, positive to annexin V in the absence/presence of 30 μg/ml cholesterol ± 1 mm MβCD-cholesterol complex. Cells were preincubated for 2 h in DMEM containing cholesterol ± MβCD-cholesterol complex. n = 9 experiments. B, as in A in cells transfected with C73A. n = 5 experiments. C, percentage of N2A cells expressing wild-type KCNB1 positive to annexin V in the absence/presence of 3.0 mm MβCD. Cells were preincubated for 2 h in DMEM supplemented with MβCD. n = 6 experiments. D, as in C for N2A cells transfected with C73A. n = 4 experiments. In all experiments, cells were oxidized by 5-min exposure to 25 μm dTDP. Annexin V staining was performed 6 h post-oxidation. *, p < 0.05; **, p < 0.01.
FIGURE 4.
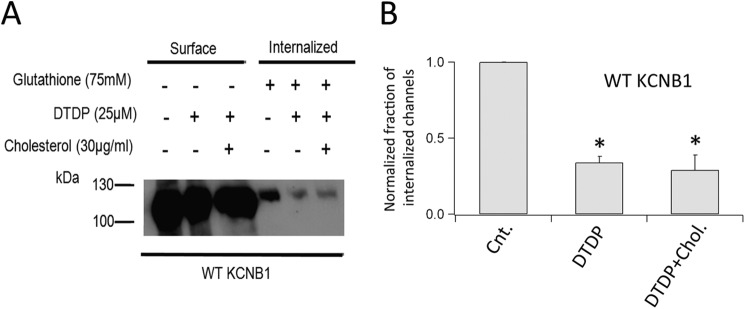
Cholesterol does not affect KCNB1 endocytosis. A, Western blot analysis visualization of a representative biotin feeding experiment in N2A cells expressing wild-type KCNB1 channels. Cells were preincubated (2 h) and maintained in 30 μg/ml cholesterol until oxidation with 25 μm dTDP. B, densitometry analysis of KCNB1 internalization in control cells and in the absence/presence of dTDP + cholesterol. n = 5 experiments. *, p < 0.05.
KCNB1 Oligomerization Activates c-Src Kinases
Glycolipid rafts contain several stress receptors that could be activated in response to KCNB1 aggregation. We focused on raft-associating Src tyrosine kinases because they have been shown to interact with the channel (15). Thus, in a first set of experiments, we inhibited Src activity with selective inhibitor PP2. Cells were incubated for 30 min with the compound, oxidized with dTDP, and scored for apoptosis. PP2 treatment completely suppressed apoptosis caused by oxidation of wild-type KCNB1 (Fig. 5A). In contrast, the compound did not alter apoptotic rates in cells expressing C73A, which remained fairly low (Fig. 5B). Moreover, inhibition of caspase 8, which is activated in response to Fas signaling, another well characterized apoptotic pathway triggered by raft disruption, did not ameliorate apoptosis (Fig. 5C). This result suggests that KCNB1 oligomers specifically act to recruit/activate Src tyrosine kinase. This enzyme can be constitutively active (v-Src) as opposed to normal c-Src (16). Therefore, to ascertain whether KCNB1 activated v-Src, c-Src, or both, we employed a c-Src dominant-negative mutant in which a lysine residue at position 295 is replaced with an arginine (K295R) (17). Coexpression of the wild type or K295R c-Src with wild-type KCNB1 did not significantly increase apoptotic levels in the absence of oxidative insults (Fig. 5D). In contrast, when cells were exposed to dTDP, apoptosis was augmented in cells expressing the wild-type c-Src, whereas cells transfected with K295R exhibited levels of apoptosis comparable with the control. Moreover, inhibition of c-Src by either PP2 or K295R expression did not affect necrotic death, which remained fairly low and constant in all conditions (≤ 6 ± 2%).
FIGURE 5.

KCNB1 oligomerization activates c-Src tyrosine kinase. A, percentage of N2A cells expressing wild-type KCNB1 positive to annexin V in the absence/presence of PP2 in the indicated concentrations. Cells were preincubated for 30 min in DMEM containing PP2. n = 8 experiments. **, p < 0.01. B, as in A in cells transfected with C73A. n = 5 experiments. C, percentage of N2A cells expressing wild-type KCNB1 positive to annexin V in the absence/presence of caspase 8 inhibitor IETD-CHO in the indicated concentrations. Cells were preincubated for 15 min in DMEM containing IETD-CHO. n = 4 experiments. *, p < 0.05. D, percentage of N2A cells cotransfected with wild-type KCNB1 and the indicated c-Src variants positive to annexin V. n = 9 experiments. *, p < 0.05. In all experiments, cells were oxidized by 5-min exposure to 25 μm dTDP. Annexin V staining was performed 6 h post-oxidation.
Inhibition of JNK Kinase Activity Suppresses KCNB1-mediated Apoptosis
c-Src kinases promote the downstream activation of the stress-activated JNKs, which initiate apoptotic signaling through multiple mechanisms, including oxidative stress (18). Therefore, we probed the role of JNK in the mechanism of KCNB1 oxidation-mediated toxicity by inhibiting the activity of the kinase by selective inhibitor SP600125. Fig. 6A illustrates the levels of apoptosis in cells expressing wild-type KCNB1 in control cells or cells preincubated with SP600125 prior the oxidative insult. At all concentrations tested (0.5–5 μm), the compound suppressed the pro-apoptotic effect of the channel in a slight concentration-dependent fashion.
FIGURE 6.

KCNB1 oligomerization results in JNK activation. A, percentage of N2A cells expressing wild-type KCNB1 positive to annexin V in the absence/presence of specific JNK inhibitor SP600125 in the indicated concentrations. Cells were preincubated for 15 min in DMEM containing SP600125. Cells were oxidized by 5-min exposure to 25 μm dTDP. Annexin V staining was performed 6 h post-oxidation. n = 7 experiments. *, p < 0.05. B, normalized number of N2A cells expressing DCF fluorescence in the absence/presence of PP2 at the indicated conditions. Cells were preincubated for 30 min in DMEM containing PP2. Cells were oxidized by 5-min exposure to 1 mm H2O2. DCF-fluorescent cells were counted 3 h post-oxidation. n = 3–4 experiments with cells transfected with wild-type and C73A, respectively.
c-Src Activity Coupled to KCNB1 Oligomerization Increases ROS
High levels of ROS under steady-state conditions were observed previously in cells expressing wild-type KCNB1 exposed to an oxidative insult (6). Destabilization of active mitochondria, with consequent ROS release and apoptosis, is a distinctive feature of death signaling pathways such as the Src/JNK pathway. Therefore, we determined whether oxidative stress was dependent on c-Src activity. Toward this end, we quantified ROS levels by DCF (the oxidized product of 2′,7′-dichlorofluorescein) fluorimetry as done before (6). In these experiments, we replaced dTDP with H2O2, which is rapidly metabolized by the cell (19) and therefore only marginally contributed to DCF fluorescence, which was measured 3 h post-oxidation. Accordingly, in both mock or C73A-transfected cells, exposure to H2O2 produced low levels of DCF fluorescence (Fig. 6B). In contrast, DCF signals markedly increased in cells expressing the wild-type channel (Fig. 6C), but inhibition of c-Src activity, by preincubation with PP2, reduced DCF signals in a dose-dependent fashion.
DISCUSSION
In this study, we investigated the molecular mechanisms underlying the cytotoxic effect of KCNB1 oxidation. Our results show that KCNB1 oligomers, which are formed as a result of oxidation of Cys-73 (6), were poorly endocytosed. This led to accumulation of oxidized KCNB1 channels in the plasma membrane. In contrast, C73A mutant channels, which do not form oligomers, did not exhibit internalization defects and, therefore, did not build up in the membrane. KCNB1 channels localize in planar lipid rafts, microdomains enriched in cholesterol, and sphingolipids (20). Rafts are thought to contain signaling molecules capable of activating pro-apoptotic and anti-apoptotic cascades. Changes that result in glycolipid raft disruption, such as accumulation of KCNB1 oligomers, may activate these death receptors and initiate apoptosis. Accordingly, we found that supplementing cholesterol to the cells markedly reduced apoptotic death. The mechanism through which cholesterol protects cells against KCNB1 oxidation-induced apoptosis is not completely understood, but biochemical evidence showed that cholesterol supplementation did not rescue defective internalization of KCNB1 oligomers. More likely, cholesterol acted to restore and/or maintain lipid raft organization and, consequently, prevent the activation of death receptors. Furthermore, pharmacological, molecular, and fluorimetric evidence indicated that the formation of KCNB1 aggregates was associated with activation of c-Src tyrosine kinase along with oxidative stress. ROS and c-Src act in concert to activate JNK (reviewed in Ref. 14). Therefore, it appears likely that c-Src activation, along with elevated ROS levels, promoted sustained activation of JNK which, in turn, initiated apoptosis. In conclusion, these data lead us to formulate a model that predicts that the formation of KCNB1 aggregates in the membrane disrupts lipid raft integrity, causing apoptosis via the activation of the c-Src/JNK pathway.
Pharmacological inhibition of c-Src activity partially decreased oxidative stress associated with KCNB1 oxidation (6). This observation suggests that multiple pathways act to increase ROS levels in the cell. c-Src has been shown to interact with NADPH oxidase and to stimulate the production and release of ROS (21). In addition, oxidative stress is a natural consequence of apoptotic cascades such as the one triggered by c-Src activation. The molecular nature of c-Src-independent pathways is not known, but we can reasonably assume that they are initiated in the membrane because C73A did not induce oxidative stress (6). This leads us to speculate that KCNB1 oligomers may act to activate the ROS-generating machinery present in lipid raft, such as NADPH-oxidase. Future studies will elucidate these mechanisms.
Src tyrosine kinases have been shown to modulate KCNB1 through phosphorylation of Tyr-124, Tyr-686, and Tyr-810 (22, 23). Although the physiological relevance of Tyr-686 and Tyr-810 phosphorylation by Src is not known, Tyr-124 plays an important role in mouse Schwann cells, where its phosphorylation constitutes an important step during proliferation and myelination (15). It has also been suggested that oxidative conditions induce phosphorylation of Tyr-124 in concert with phosphorylation of Ser-800 by p38 MAPK. This activity leads to insertion of de novo synthesized KCNB1 channels in the membrane, causing apoptosis through a robust K+ current surge (9, 24). However, the C73A variant, which is likely to be a substrate for the c-Src kinase, did not cause apoptosis under oxidative conditions nor accumulated in the membrane. In addition, oxidized KCNB1 channels do not conduct current (6). Overall, this evidence argues that phosphorylation of KCNB1 by c-Src and p38 it is neither sufficient nor necessary for apoptosis, even though it is likely to occur under oxidative conditions. Furthermore, the requirement of newly exocytosed KCNB1 channels to explain their plasma accumulation seems unnecessary in light of our findings, which show their impaired internalization. The two mechanisms do not exclude each other, though, and future studies will elucidate this specific issue.
Oxidation of K+ channels by ROS is a general mechanism of toxicity in the aging nervous system. In the invertebrate C. elegans, oxidative modification of voltage-gated K+ channel KVS-1 leads to progressive neurodegeneration (5). In mice, oxidized KCNB1 channels were found to be ∼10-fold more abundant in old rather than young animals and were hyperoxidized (∼200-fold) in the brain of the 3xTg-AD model of Alzheimer's disease (6), a condition characterized by extensive oxidative damage. Hence, considering the impact of neuronal aging on global human health issues, these findings may contribute to a better understanding of the cellular and molecular basis of nervous system aging.
Acknowledgments
We thank Dr. Frank Suprynowicz for the K295R Src-kinase mutant, Dr. Geoffrey Abbott for the K44A dynamin 2 construct, and Dr. Shuang Liu for critical reading of the manuscript.
This work was supported by National Science Foundation Grants 0842708 and 1026958 and an American Heart Association Grant 09GRNT2250529 (to F. S.). This work was also supported by the RISE Research in Science and Engineering Rutgers-University of Medicine and Dentistry of New Jersey joint program (to M. B.).
- ROS
- reactive oxygen species
- MβCD
- methyl-β-cyclodextrin
- DMSO
- dimethyl sulfoxide
- DCF
- dichlorofluorescein
- PP2
- 4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrmidine.
REFERENCES
- 1. Valko M., Leibfritz D., Moncol J., Cronin M. T., Mazur M., Telser J. (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84 [DOI] [PubMed] [Google Scholar]
- 2. Stadtman E. R. (1992) Protein oxidation and aging. Science 257, 1220–1224 [DOI] [PubMed] [Google Scholar]
- 3. Harman D. (1972) The biologic clock. The mitochondria? J. Am. Geriatr. Soc. 20, 145–147 [DOI] [PubMed] [Google Scholar]
- 4. Sesti F., Liu S., Cai S. Q. (2010) Oxidation of potassium channels by ROS. A general mechanism of aging and neurodegeneration? Trends Cell Biol. 20, 45–51 [DOI] [PubMed] [Google Scholar]
- 5. Cai S. Q., Sesti F. (2009) Oxidation of a potassium channel causes progressive sensory function loss during aging. Nat. Neurosci. 12, 611–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cotella D., Hernandez-Enriquez B., Wu X., Li R., Pan Z., Leveille J., Link C. D., Oddo S., Sesti F. (2012) Toxic role of K+ channel oxidation in mammalian brain. J. Neurosci. 32, 4133–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu X., Kanda V. A., Choi E., Panaghie G., Roepke T. K., Gaeta S. A., Christini D. J., Lerner D. J., Abbott G. W. (2009) Mink-dependent internalization of the IKs potassium channel. Cardiovasc. Res. 82, 430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pal S., Hartnett K. A., Nerbonne J. M., Levitan E. S., Aizenman E. (2003) Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J. Neurosci. 23, 4798–4802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pal S. K., Takimoto K., Aizenman E., Levitan E. S. (2006) Apoptotic surface delivery of K+ channels. Cell Death Differ. 13, 661–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martens J. R., Navarro-Polanco R., Coppock E. A., Nishiyama A., Parshley L., Grobaski T. D., Tamkun M. M. (2000) Differential targeting of Shaker-like potassium channels to lipid rafts. J. Biol. Chem. 275, 7443–7446 [DOI] [PubMed] [Google Scholar]
- 11. Robinson M. S. (1994) The role of clathrin, adaptors and dynamin in endocytosis. Curr. Opin. Cell Biol. 6, 538–544 [DOI] [PubMed] [Google Scholar]
- 12. Nabi I. R., Le P. U. (2003) Caveolae/raft-dependent endocytosis. J. Cell Biol. 161, 673–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van der Bliek A. M., Redelmeier T. E., Damke H., Tisdale E. J., Meyerowitz E. M., Schmid S. L. (1993) Mutations in human dynamin block an intermediate stage in coated vesicle formation. J. Cell Biol. 122, 553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morgan M. J., Kim Y. S., Liu Z. (2007) Lipid rafts and oxidative stress-induced cell death. Antioxid. Redox Signal. 9, 1471–1483 [DOI] [PubMed] [Google Scholar]
- 15. Sobko A., Peretz A., Attali B. (1998) Constitutive activation of delayed-rectifier potassium channels by a Src family tyrosine kinase in Schwann cells. EMBO J. 17, 4723–4734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anderson S. K., Gibbs C. P., Tanaka A., Kung H. J., Fujita D. J. (1985) Human cellular Src gene. Nucleotide sequence and derived amino acid sequence of the region coding for the carboxy-terminal two-thirds of pp60c-Src. Mol. Cell. Biol. 5, 1122–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suprynowicz F. A., Baege A., Sunitha I., Schlegel R. (2002) c-Src activation by the E5 oncoprotein enables transformation independently of PDGF receptor activation. Oncogene 21, 1695–1706 [DOI] [PubMed] [Google Scholar]
- 18. Dhanasekaran D. N., Reddy E. P. (2008) JNK signaling in apoptosis. Oncogene 27, 6245–6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dringen R. (2000) Metabolism and functions of glutathione in brain. Progress Neurobiol. 62, 649–671 [DOI] [PubMed] [Google Scholar]
- 20. George K. S., Wu S. (2012) Lipid raft. A floating island of death or survival. Toxicol. Appl. Pharmacol. 259, 311–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Catarzi S., Biagioni C., Giannoni E., Favilli F., Marcucci T., Iantomasi T., Vincenzini M. T. (2005) Redox regulation of platelet-derived-growth-factor-receptor. Role of NADPH-oxidase and c-Src tyrosine kinase. Biochim. Biophys. Acta 1745, 166–175 [DOI] [PubMed] [Google Scholar]
- 22. Song M. Y., Hong C., Bae S. H., So I., Park K. S. (2012) Dynamic modulation of the kv2.1 channel by SRC-dependent tyrosine phosphorylation. J. Proteome Res. 11, 1018–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tiran Z., Peretz A., Attali B., Elson A. (2003) Phosphorylation-dependent regulation of Kv2.1 channel activity at tyrosine 124 by Src and by protein-tyrosine phosphatase ϵ. J. Biol. Chem. 278, 17509–17514 [DOI] [PubMed] [Google Scholar]
- 24. Redman P. T., He K., Hartnett K. A., Jefferson B. S., Hu L., Rosenberg P. A., Levitan E. S., Aizenman E. (2007) Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc. Natl. Acad. Sci. U.S.A. 104, 3568–3573 [DOI] [PMC free article] [PubMed] [Google Scholar]