

Background: The contribution of phosphatidylethanolamine (PE) to mammalian mitochondrial function was unknown.
Results: A decrease in mitochondrial PE impairs cell growth, respiratory capacity, and ATP production and profoundly alters mitochondrial morphology.
Conclusion: Mitochondrial PE is required for normal morphology and function of mammalian mitochondria.
Significance: Modest reduction of mitochondrial PE might contribute to mitochondrial dysfunction in some disease states.
Keywords: Electron Transport, Mammal, Mitochondria, Mitochondrial Metabolism, Phosphatidylethanolamine, Phosphatidylserine
Abstract
Mitochondrial dysfunction is implicated in neurodegenerative, cardiovascular, and metabolic disorders, but the role of phospholipids, particularly the nonbilayer-forming lipid phosphatidylethanolamine (PE), in mitochondrial function is poorly understood. Elimination of mitochondrial PE (mtPE) synthesis via phosphatidylserine decarboxylase in mice profoundly alters mitochondrial morphology and is embryonic lethal (Steenbergen, R., Nanowski, T. S., Beigneux, A., Kulinski, A., Young, S. G., and Vance, J. E. (2005) J. Biol. Chem. 280, 40032–40040). We now report that moderate <30% depletion of mtPE alters mitochondrial morphology and function and impairs cell growth. Acute reduction of mtPE by RNAi silencing of phosphatidylserine decarboxylase and chronic reduction of mtPE in PSB-2 cells that have only 5% of normal phosphatidylserine synthesis decreased respiratory capacity, ATP production, and activities of electron transport chain complexes (C) I and CIV but not CV. Blue native-PAGE analysis revealed defects in the organization of CI and CIV into supercomplexes in PE-deficient mitochondria, correlated with reduced amounts of CI and CIV proteins. Thus, mtPE deficiency impairs formation and/or membrane integration of respiratory supercomplexes. Despite normal or increased levels of mitochondrial fusion proteins in mtPE-deficient cells, and no reduction in mitochondrial membrane potential, mitochondria were extensively fragmented, and mitochondrial ultrastructure was grossly aberrant. In general, chronic reduction of mtPE caused more pronounced mitochondrial defects than did acute mtPE depletion. The functional and morphological changes in PSB-2 cells were largely reversed by normalization of mtPE content by supplementation with lyso-PE, a mtPE precursor. These studies demonstrate that even a modest reduction of mtPE in mammalian cells profoundly alters mitochondrial functions.
Introduction
Mitochondrial oxidative phosphorylation (OXPHOS)2 generates the majority of ATP in mammalian cells. Mitochondrial dysfunction has been implicated in neurodegenerative disorders (2), cardiovascular disease/metabolic syndrome (3), diabetes (4), and tumor development (5). Mitochondria are highly dynamic organelles that undergo fusion and fission in response to the environment and during the cell cycle, suggesting that mitochondrial dynamics and function are inter-related. Emerging evidence supports a role for mitochondrial phospholipids in key cellular/mitochondrial functions such as programmed cell death (6), respiration/ATP synthesis (7–10), and autophagy/mitophagy (11, 12). For example, reduction in the amount of the mitochondrion-specific phospholipid cardiolipin (CL), which comprises ∼10% of mitochondrial phospholipids, alters mitochondrial membrane potential (13), OXPHOS (8, 14), organization of electron transport chain (ETC) components into supercomplexes (8, 9, 15), mitochondrial matrix osmolarity (16), and cristae morphology (17, 18). Furthermore, the cardiomyopathy Barth syndrome is caused by defective CL biosynthesis (19).
Phosphatidylethanolamine (PE) is more abundant in mammalian mitochondria than in other organelles and contributes ∼30% of mitochondrial phospholipids. PE participates in contractile ring disassembly during cytokinesis (20, 21) and regulates the topology of integral membrane proteins in Escherichia coli (22, 23). In addition, PE is a precursor of the ∼30% of hepatic phosphatidylcholine made via PE N-methyltransferase (24). PE is also a precursor of anandamide (N-arachidonoylethanolamine) (25, 26), the ethanolamine moiety of glycosylphosphatidylinositol anchors of many cell surface signaling proteins (27), and the ethanolamine linked to eukaryotic elongation factor eEF1A (28). Furthermore, PE appears to be required for autophagosome formation (12, 29) and prion propagation in the brain (30). Although recent in vitro studies suggest that mitochondrial PE (mtPE) regulates mitochondrial outer membrane permeability (31), the role of mtPE in mammalian mitochondrial function is poorly understood.
The majority of PE in mammalian cells is synthesized by two spatially separated pathways (32, 33) as follows: the CDP-ethanolamine pathway (the final step of which occurs on the endoplasmic reticulum (ER) (34, 35)), and the phosphatidylserine (PS) decarboxylase (PSD) pathway in mitochondrial inner membranes (36, 37). In addition, small amounts of PE are made by base-exchange between ethanolamine and PS (38). In yeast, PE can also be made by acylation of lyso-PE by the acyltransferase, Ale1p (39, 40). Although a family of genes related to Ale1 is present in mammals, the contribution of this pathway to PE synthesis in mammalian cells is unknown. Importantly, the majority of mtPE in mammalian and yeast cells is made in mitochondria from PSD, whereas only a small fraction of mtPE is imported from the ER (41–44). For PS to be decarboxylated to PE, PS is imported into mitochondria from its site of synthesis in mitochondrion-associated membranes (MAM) (45), an ER domain that becomes transiently tethered to mitochondria (35, 46–48).
In mammalian cells, a single gene (Pisd) encodes PSD activity (1, 49), whereas yeast PSD activity is encoded by two distinct genes (50, 51). Reduction of PE synthesis via PSD alters mitochondrial morphology in yeast (52) and Trypanosoma brucei (53), and depletion of PE to <4% of yeast phospholipids impairs survival (51, 54). PE synthesis from PSD is essential in yeast mutants lacking CL synthase, suggesting that CL and PE have overlapping functions (55). Remarkably, however, E. coli cells survive when PE, normally the most abundant phospholipid, is reduced to 0.007% of total phospholipids (56). PSD is required for mouse development because disruption of the mouse Pisd gene was embryonic lethal and profoundly altered mitochondrial morphology, even when PE was actively made from CDP-ethanolamine (1, 57). Moreover, global disruption of the CDP-ethanolamine pathway for PE synthesis in mice is also embryonic lethal (58) demonstrating that both major PE biosynthetic pathways are required for mouse viability.
We have now investigated mechanisms underlying mitochondrial defects in mammalian cell models in which mtPE was reduced either chronically or acutely. Depletion of mtPE by only 20–30% reduced cell growth, respiratory capacity, and ATP production, consistent with altered respirasome organization and defects in ETC complexes I and IV. In addition, mitochondrial ultrastructure was profoundly aberrant, and mitochondria were extensively fragmented. Supplementation of mtPE-deficient cells with lyso-PE normalized mtPE content and mitochondrial morphology, promoted cell growth, and increased ATP production. Thus, even a modest decrease in mtPE is detrimental for mitochondrial function and dynamics.
EXPERIMENTAL PROCEDURES
Cell Culture
CHO-K1 cells were purchased from the American Type Tissue Culture Collection (Manassas, VA). The mutant CHO cell line PSB-2 (59) was a gift from Dr. M. Nishijima (Tokyo, Japan). All cells were cultured at 37 °C in Ham's F-12 medium (Invitrogen) containing 10% fetal bovine serum (FBS; Invitrogen).
Gene Silencing and Quantitative Real Time PCR
Dicer-substrate siRNAs (DsiRNAs) were purchased from Integrated DNA Technologies (Coralville). For silencing hamster (Cricetulus criseus) Pisd, three duplexes were designed (Pisd KD#1, 5′-CCC AGA UAC AGC AAG GGU UCC UAC A-3′; Pisd KD#2, 5′-GCC UCA AAG AUG AGC ACA AUA GUA GAG-3′; and Pisd KD#3, 5′-CUC GCU GAG GUU UCG GUA GUG GUG CAG-3′). The sequence of the nontargeting (NegCtrl) duplex was 5′-AUA CGC GUA UUA UAC GCG AUU AAC GAC-3′. CHO cells (0.6 × 104 cells/cm2) were grown for 24 h and then transfected for 6 h with 10 nm DsiRNAs and 2 μl/ml Lipofectamine 2000 in serum-free Opti-MEM I medium (Invitrogen). Subsequently, the medium was changed to F-12 + 10% heat-inactivated FBS. mRNA was quantified by real time PCR. Total RNA was extracted with TRIzol (Invitrogen) and digested with amplification-grade RNase-free DNase (Invitrogen). RNA (2 μg) was reverse-transcribed in a 20-μl volume containing oligo(dT) and Superscript II reverse transcriptase. Platinum SYBR Green quantitative PCR Supermix-uracil-DNA glycosylase was used to amplify genes. Each sample was analyzed in triplicate using a standard curve and normalized to cyclophilin A. Primers used were as follows: hamster Pisd, forward 5′-CAC TCC CCT ACT GAC TGG AC-3′ and reverse 5′-CAC GCT CAT TGT GAC AGA AGA-3′; cyclophilin A, forward 5′-TCC AAA GAC AGC AGA AAA CTT TCG-3′ and reverse 5′-TCT TCT TGC TGG TCT TGC CAT TCC-3′.
PS Decarboxylase (PSD) Activity
PSD activity (disintegrations/min in PE/h/mg of protein) was quantified in cell lysates using [14C]PS (Amersham Biosciences) (1).
Mitochondrial DNA
Genomic DNA was isolated using the DNeasy kit (Qiagen). Mitochondrial DNA was amplified with primers complementary to ATP synthase subunit ATP6 as follows: forward 5′-ACC GCA GGA CAT CTG CTA AT-3′ and reverse 5′-GGT AGG CTG ATT GAG GTT AGG A-3′. Nuclear DNA was amplified with primers complementary to Pisd as follows: forward 5′-CTG GAC CTT TGG GGT AAA CA-3′ and reverse 5′-AGG TTT CGG TAG TGG TGC AG-3′.
Metabolic Labeling of PS and PE
Cells were grown to 80% confluence in 100-mm dishes and then incubated in Ham's F-12 medium containing 5 μCi/ml l-[G-3H]serine (PerkinElmer Life Sciences). Lipids were extracted (60) and separated by thin layer chromatography with the developing solvent chloroform/methanol/acetic acid/formic acid/water (70:30:12:4:2, v/v). Phospholipids were visualized by exposure to iodine vapor and PS and PE identified by comparison with authentic PS and PE (Avanti Polar Lipids). Incorporation of [3H] into PS and PE was quantified by scintillation counting.
Isolation of Mitochondria, MAM, and Microsomes
Subcellular fractions were isolated as described previously (35, 41). Briefly, cells were homogenized in isolation medium (225 mm mannitol, 75 mm sucrose, 10 mm Tris-HCl (pH 7.2), 1 mm EGTA, 0.1% fatty acid-free bovine serum albumin) with 15 strokes in a Balch homogenizer (Isobiotec Precision Engineering) followed by 20 strokes in a glass-Teflon homogenizer. The homogenate was centrifuged at 600 × g for 5 min at 4 °C, and the supernatant was centrifuged at 10,300 × g for 10 min to pellet crude mitochondria. Microsomes were pelleted by centrifugation of the resultant supernatant at 100,000 × g for 1 h. Crude mitochondria were applied to the top of tubes containing 30% Percoll (v/v) in isolation medium and centrifuged at 95,000 × g for 30 min. Mitochondria were removed and washed with isolation medium (without albumin). MAM were removed from the Percoll gradient, diluted with isolation medium, and centrifuged for 10 min at 6,300 × g. The supernatant was centrifuged at 100,000 × g for 1 h to pellet MAM.
Measurement of Phospholipid Mass
Phospholipids were extracted from cell lysates and subcellular fractions (60), isolated by thin layer chromatography as described above, and quantified as Pi (61). CL was isolated by thin layer chromatography in the developing solvent chloroform/hexane/methanol/acetic acid (50:30:10:5, v/v).
Cell Growth
Cell number was determined by trypan blue exclusion, and growth rate was calculated according to an exponential growth model (62). Nuclei were stained with 100 nm 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI; Molecular Probes). 100–500 DAPI-stained nuclei were counted.
Confocal Microscopy
Cells were imaged using an LSM 510 multiphoton confocal microscope (Zeiss) and analyzed with Zeiss software. Fixed cells were plated on coverslips coated with poly-d-lysine (Sigma). For visualization of mitochondria, cells were stained for 30 min with 100 nm MitoTracker Red CMX (Molecular Probes), fixed for 15 min at room temperature in 4% paraformaldehyde, and then washed with phosphate-buffered saline. Nuclei were stained with DAPI and cells mounted on slides using ProlongGold (Molecular Probes). For immunocytochemistry, cells were fixed for 15 min at room temperature in 4% paraformaldehyde and then permeabilized for 5 min with 0.25% (v/v) Triton X-100 in phosphate-buffered saline. After blocking for 30 min with Image-iT FX Signal Enhancer (Molecular Probes), cells were incubated overnight at 4 °C with mouse monoclonal primary antibodies in blocking solution (Dako) as follows: anti-Drp1 (catalog no. 61112, dilution 1:200; BD Transduction Laboratories); anti-Mfn1 (catalog no. ab57602, dilution 1:500; Abcam); anti-Mfn2 (catalog no. ab56889, dilution 1:500; Abcam), and subsequently with FITC-conjugated secondary antibody (dilution 1:1000; Molecular Probes). All experiments included negative controls with secondary antibody alone.
Mitochondrial membrane potential was measured in live cells using tetramethylrhodamine methyl ester (TMRM) (10 nm, 30 min at 37 °C). Mitochondrial mass was assessed by staining live cells for 30 min at 37 °C with 100 nm 10-N-nonylacridine orange (membrane potential-insensitive dye; Molecular Probes). Fluorescence intensities were measured in five random fields/cell line. Nuclei were stained for 10 min with 500 nm Hoechst 33342 (Molecular Probes).
Electron Microscopy
Cells were fixed in phosphate-buffered saline containing 4% paraformaldehyde and 2% glutaraldehyde. Ultra-thin sections were stained with uranyl acetate and osmium tetroxide. Transmission electron microscopy images were obtained with a JEOL 1200 EX microscope and analyzed by ImageJ software.
Respirometry
Oxygen uptake was measured at 37 °C in suspensions of cells grown to 70–80% confluence with a Clark-type oxygen electrode (Warner Instruments, Hamden, CT) connected to a Strathkelvin 782 oxygen meter. Intact cells were collected by trypsin treatment, and the suspension was diluted to 4 × 106 cells/ml. Carbonyl cyanide 4-trifluoromethoxyphenylhydrazone (10 μm) was used as an uncoupling agent. Substrate-driven respiration was measured in cells permeabilized for 5 min with 20 μg of digitonin/ml/2 × 106 cells on ice and then resuspended in respiration buffer (4 × 106 cells/ml) containing 80 mm KCl, 25 mm Tris-HCl (pH 7.5), 1 mm EDTA, 5 mm KH2PO4, 3 mm MgCl2, 0.01% bovine serum albumin, 2 mm ADP. Respiratory substrates were as follows: 1.25 mm pyruvate, 1.25 mm malate (complex I); 5 mm succinate (complex II) + 2 μm rotenone (complex I inhibitor); 100 μm N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride (electron donor for complex IV) + 0.6 mm ascorbate (reducing agent).
Cellular ATP and ATP Production
Trichloroacetic acid (2.5%) was added to cells, and the mixture was neutralized with NaHCO3/Tris (pH 7.0). ATP was quantified with an ATP bioluminescence kit (Molecular Probes) and normalized to protein concentration.
For measurement of the rate of ATP synthesis, 1.85 × 106 wild-type cells/15-cm dish and 3.7 × 106 PSB-2 cells/15-cm dish were plated 48 h prior to experiments so that 70–80% confluency was attained. Pisd KD#1 and NegCtrl cells were cultured as above. Cells were washed with 25 mm Tris-HCl (pH 7.4), 140 mm KCl, 10 mm NaCl, 2.5 mm MgCl2, 0.1 μm CaCl2 and then permeabilized for 5 min at 37 °C in medium containing 40 μg/ml digitonin, 150 mm KCl, 25 mm Tris-HCl (pH 7.4), 2 mm EDTA, 10 mm KH2PO4, and 0.1% fatty acid-free bovine serum albumin. The cells were then incubated for 30 min at 37 °C in the same medium containing 1 mm ADP ± respiratory substrates. Reactions were terminated with 1% trichloroacetic acid and neutralized with NaHCO3. Samples were centrifuged for 10 min at 13,000 × g, and nanomoles of ATP/min/mg of protein were quantified in supernatants.
ETC Enzymatic Activities
Specific activities of complex I (NADH-ubiquinone oxidoreductase), complex II (succinate dehydrogenase), and complex IV (cytochrome c oxidase) were measured spectrophotometrically in mitochondria exposed to one freeze-thaw cycle to permeabilize inner membranes to NADH. Mitochondria (20 μg of protein/ml) were incubated in buffer containing 10 mm Tris-HCl (pH 7.4), 50 mm KCl, 1 mm EDTA, 2 mm KCN, 2 μm antimycin A, and 100 μm NADH. The reaction was started with 50 μm coenzyme Q1. Complex I activity (NADH oxidation) was measured by decreased absorbance at 340 nm. Complex II activity was measured with 30 μg of protein/ml in 10 mm Tris-HCl (pH 7.2), 50 mm KCl, 2 mm KCN, and 32 mm succinate as reduction of 2,6-dichloroindophenol (absorbance at 600 nm). The sample was preincubated for 20 min at room temperature with 8 μm dichloroindophenol, and then the reaction was started with 50 μm dichloroindophenol and 100 μm coenzyme Q1. Complex IV activity was measured as oxidation of reduced cytochrome c (at 550 nm) in buffer containing 2–5 μg of protein/ml, 10 mm KH2PO4 (pH 7.2), and 0.015% Triton X-100. Mitochondrial membranes were permeabilized by incubation for 5 min at 37 °C, and the reaction was started with 0.04% (w/v) reduced cytochrome c.
Immunoblotting
Proteins in 62.5 mm Tris-HCl (pH 6.8), 10% glycerol, 2% SDS, and 5% β-mercaptoethanol were electrophoresed on 10% polyacrylamide gels containing 0.1% SDS. Proteins were transferred to polyvinylidene difluoride membranes and incubated in 10 mm Tris-HCl (pH 7.4), 100 mm NaCl, 0.1% Tween 20 for 1 h at room temperature with primary antibodies as follows: mouse monoclonal anti-OPA1 (catalog no. 612606, dilution 1:1000; BD Transduction Laboratories); total OXPHOS rodent WB antibody mixture (catalog no. MS604, dilution 1:2000; Mitosciences); mouse monoclonal anti-Mfn1 (catalog no. ab57602, dilution 1:1000) or anti-Mfn-2 (catalog no. ab56889, dilution 1:1000; Abcam); mouse monoclonal anti-cytochrome c oxidase (anti-COX4, catalog no. sc-58348, dilution 1:250); goat polyclonal anti-TOM20 (catalog no. sc-11021, dilution 1:250; Santa Cruz Biotechnology); mouse monoclonal anti-outer mitochondrial membrane voltage-dependent anion channel (anti-VDAC1, catalog no. ab14734, dilution 1:1000; Mitosciences). Subsequently, peroxidase-conjugated secondary antibodies (dilution 1:5000; Pierce) were added for 1 h at room temperature, and immunoreactivity was detected by enhanced chemiluminescence (Pierce).
Blue Native-PAGE (BN-PAGE)
Percoll-purified mitochondria were solubilized in digitonin (4%, w/v) in buffer containing 750 mm aminocaproic acid, 50 mm BisTris (pH 7.0), 1 mm EDTA. Samples were centrifuged at 100,000 × g for 20 min at 4 °C, and then Coomassie Blue G-250 (8 g of digitonin/g of dye) and 10% glycerol were added to the supernatant. Mitochondrial complexes (75 μg of protein/lane) were separated overnight at 40 V at 4 °C on NativePAGE Novex 3–12% BisTris gels (Invitrogen); complexes/supercomplexes were identified using NativeMark unstained protein standards (Invitrogen). For immunoblotting, the gel was shaken in 20 mm Tris-HCl (pH 8.3), 0.15 m glycine, 20% methanol, and 0.02% SDS for 30 min at room temperature, and then proteins were transferred to polyvinylidene difluoride membranes (150 mA for 90 min, 4 °C). Membranes were blocked with 5% skimmed milk in 50 mm Tris-HCl (pH 7.5) and 150 mm NaCl for 1 h at room temperature and then incubated with the indicated antibodies.
In-gel Assay of OXPHOS Activities
Duplicate BN-polyacrylamide gels were used for immunoblotting and in-gel assays. For complex I activity (NADH dehydrogenase), the gel was incubated for 1 h at room temperature with 5 mm Tris-HCl (pH 7.4), 0.1 mg/ml NADH + 2.5 mg/ml nitrotetrazolium blue (Sigma), then stained overnight at 4 °C, and washed, and band intensity was quantified. For complex II, the gel was incubated for 1 h at room temperature with 5 mm Tris-HCl (pH 7.4), 0.25 mm phenazine methosulfate, 0.5 m succinate, and 2.5 mg/ml nitrotetrazolium blue and then washed, and band intensity was immediately quantified. For complex IV, the gel was incubated with 1 mg/ml 3,3′-diaminobenzidine, 50 mm phosphate buffer (pH 7.0), 75 mg/ml sucrose, 34 mg/ml catalase, and 10 mg/ml cytochrome c. For complex V, the gel was incubated for 3 h in 270 mm glycine, 35 mm Tris (pH 8.4). ATP hydrolysis was measured by incubation of the gel overnight at room temperature in 270 mm glycine, 35 mm Tris (pH 7.8), 8 mm ATP, 14 mm MgSO4, 0.2% Pb(NO3)2 to generate lead-phosphate precipitates.
Lyso-PE Supplementation
Cells were incubated for 96 h with an ethanolic solution of 100 μm lyso-PE (18:1; Avanti Polar Lipids). An equivalent volume of ethanol was added to cells not supplemented with lyso-PE.
Other Methods
Protein concentrations were determined by the BCA method (Pierce) with bovine serum albumin as standard.
Statistical Analysis
Data are means ± S.D. or S.E. as indicated. Differences between means were evaluated by the two-tailed Student's t test. p values <0.05 were considered significant.
RESULTS
Cells with Reduced mtPE
The requirement of mtPE for mitochondrial function was investigated in two cell models with reduced mtPE synthesis as follows: (i) PSB-2 cells, mutant CHO cells in which PS synthesis was only 5% of that in wild-type (WT) cells (chronically reduced mtPE synthesis) (63), and (ii) CHO cells in which PSD expression was silenced with DsiRNA (acutely reduced mtPE synthesis). Duplexes targeting distinct regions of hamster Pisd mRNA were transiently transfected into CHO cells to generate Pisd knockdown (KD) cells (Pisd KD#1, KD#2, and KD#3) in which Pisd transcript levels 48 h after transfection were 80–94% lower than in cells transfected with nontargeting DsiRNA (NegCtrl) (Fig. 1A). Transfection with fluorophore-labeled DsiRNA (Cy5-KD#1) for 48 h yielded a signal in 80% of cells, consistent with 78% reduction of Pisd mRNA and confirming efficient transfection (Fig. 1A). In vitro PSD activity in Pisd KD#1 and KD#2 cells 48 h after transfection was 58 and 44% lower, respectively, than in NegCtrl cells (Fig. 1B); 72 h after transfection, PSD activity was reduced in Pisd KD#1 and KD#2 cells by 62 and 44%, respectively (Fig. 1B). In all experiments Pisd KD#1 cells were used 72 h after transfection unless otherwise stated.
FIGURE 1.

mtPE synthesis in Pisd KD cells and PSB-2 cells. A, quantitative PCR analysis of Pisd mRNA relative to cyclophilin mRNA in CHO cells transiently transfected with DsiRNAs targeted to distinct regions of hamster Pisd (KD#1, KD#2, and KD#3) or with nontargeting DsiRNA (NegCtrl); Cy5-KD#1 = fluorophore-labeled KD#1. Data are means ± S.D. of triplicate analyses from two independent transfections (*, p < 0.05 versus NegCtrl). B, PSD activity in Pisd KD#1, KD#2, KD#3, and NegCtrl cells 48 and 72 h after transfection. Data are means ± S.D. from triplicate analyses of two independent experiments (*, p < 0.0001 versus NegCtrl). C–F, [3H]serine incorporation into PE and PS in Pisd KD#1 and NegCtrl cells transfected for 72 h (C and D) and PSB-2 and WT cells (E and F). Data are means ± S.D. of triplicate analyses of three independent experiments (p < 0.05 versus control at 2, 4, and 6 h); some error bars are too small to be visible.
PE synthesis from PS was further assessed by comparing [3H]serine incorporation into PE in Pisd KD#1 cells and PSB-2 cells with that in respective control cells. The amount of [3H]PE was ∼80% lower in Pisd KD#1 cells than in NegCtrl cells (Fig. 1C), and [3H]PS was reduced by ∼40% (Fig. 1D). Thus, when [3H]PE was calculated as a fraction of total [3H]PS + [3H]PE, the conversion of PS to PE was ∼50% lower in Pisd KD#1 cells than in NegCtrl cells. PSB-2 cells, in which PS synthesis but not PSD activity was reduced, contained ∼90% less [3H]PE than did WT cells (Fig. 1E) corresponding to a similar reduction in [3H]PS (Fig. 1F). Thus, PE production from PS was diminished in both PSB-2 cells (chronically reduced PS supply) and Pisd KD#1 cells (acutely reduced PSD activity).
Consistent with attenuated production of PE from PS in Pisd KD#1 and PSB-2 cells, the mass of mtPE was 21.5 and 27% lower than in NegCtrl cells and WT cells, respectively (Fig. 2A), with no significant change in mitochondrial PS (Fig. 2B). The amount of mtPE as percent of total mitochondrial phospholipids was 14 and 24% lower in Pisd KD#1 and PSB-2 cells, respectively, than in control cells (Fig. 2C). Interestingly, the PS content of MAM from PSB-2 cells was 50% lower than in WT MAM with no reduction in PE (Fig. 2D). The PE content of cell lysates and microsomes was not reduced by mtPE deficiency; PSB-2 and WT cell lysates contained 68.3 and 71.4 nmol of PE/mg of protein, respectively, and lysates of Pisd KD#1 and NegCtrl cells contained 69.8 and 71.4 nmol of PE/mg of protein, respectively. Amounts of PS and phosphatidylcholine were not lower in lysates or microsomes of Pisd KD#1 cells than in NegCtrl cells, nor was phosphatidylcholine reduced in PSB-2 lysates or microsomes (data not shown). Thus, decreased mtPE production reduced the PE content of mitochondria but not microsomes or cell lysates. Importantly, the amount of cardiolipin (CL), a mitochondrion-specific phospholipid required for normal mitochondrial functions (52, 64, 65), was not reduced in either PSB-2 or Pisd KD#1 cells (Fig. 2E).
FIGURE 2.
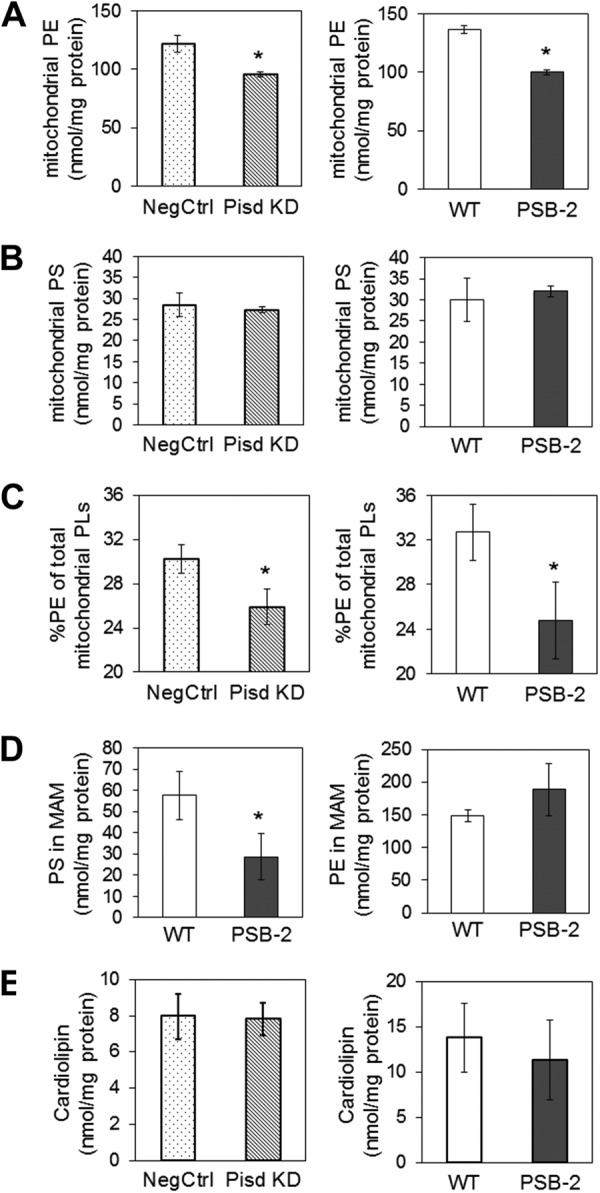
Mitochondrial phospholipids. PE (A) and PS (B) were quantified (nmol/mg protein) in Percoll-purified mitochondria from Pisd KD#1, NegCtrl, PSB-2, and WT cells. C, amount of mtPE as % of mitochondrial phospholipids (PLs). D, amount of PS and PE in MAM from PSB-2 and WT cells. E, cardiolipin content of lysates of mtPE-deficient and control cells. Data are means ± S.D. from three independent experiments (*, p < 0.05 versus control).
MtPE Deficiency Impairs Cell Growth
Whereas NegCtrl cells attained 85–90% confluency 72 h after transfection, the number of Pisd KD#1 and KD#2 cells was 66 and 55% lower, respectively, compared with NegCtrl cells (Fig. 3A). The reduction in Pisd KD#1 cells is consistent with slower growth (by 23%, 72 and 96 h after transfection) (Fig. 3B). PSB-2 cells also grew more slowly (by ∼40%) than WT cells (Fig. 3C). However, no apoptotic cells were detected upon TUNEL staining of any of the cell lines (data not shown). Thus, mtPE-deficiency impairs cell growth.
FIGURE 3.
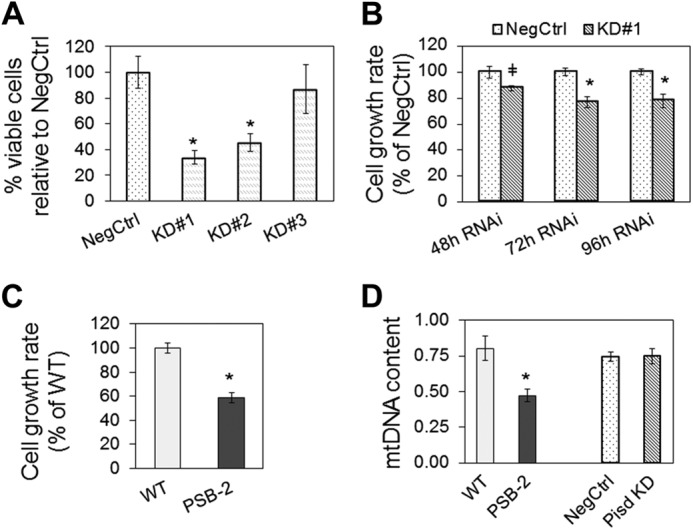
Cell growth and mitochondrial DNA. A, number of Pisd KD#1, #2, and #3 cells relative to NegCtrl cells measured in 100–500 nuclei from five fields. B, growth rate of Pisd KD#1 cells and NegCtrl cells 48, 72, and 96 h after transfection according to a model of exponential growth. Data are means ± S.D. from three independent experiments (*, p < 0.05; ‡, p < 0.1). C, growth rate of PSB-2 cells and WT cells. Data are means ± S.D. from three independent experiments (* p < 0.05). D, ratio of mtDNA to nuclear DNA. Data are means ± S.D. from three independent experiments (*, p < 0.05 versus control).
Mitochondrial biogenesis was assessed by quantification of mtDNA. The mtDNA content was equivalent in Pisd KD#1 and NegCtrl cells 72 h after transfection, whereas PSB-2 cells contained 41% less mtDNA than did WT cells (Fig. 3D). Thus, chronic, but not acute, mtPE depletion decreases mitochondrial biogenesis. In addition, as an indicator of mitochondrial mass, cells were stained with 10-N-nonylacridine orange, a mitochondrial membrane potential-insensitive dye. According to this indicator, mitochondrial mass was similar in PSB-2 and WT cells and was also similar in Pisd KD#1 and NegCtrl cells both 48 and 72 h after transfection (Fig. 4, A and B).
FIGURE 4.

Mitochondrial mass and membrane potential. A, representative confocal images of live cells stained with the membrane potential-insensitive dye nonylacridine orange (NAO; green); nuclei stained with Hoechst (blue). B, quantification of nonylacridine orange staining in arbitrary fluorescence intensity units (AFU) normalized to cell area. C, representative confocal images of live cells stained with TMRM (red); nuclei stained with Hoechst (blue). D, quantification of fluorescence intensity from images in C normalized to cell area. Data in B and D are means ± S.E. from three independent experiments (*, p < 0.05 versus control). Size bar in A and B, 20 μm.
Mitochondrial membrane potential, a key indicator of mitochondrial function and cell viability, was assessed with the membrane potential-sensitive dye TMRM. Mitochondria in PSB-2 and Pisd KD#1 cells (48 and 72 h after transfection) were significantly hyperpolarized (increased fluorescence) compared with control mitochondria (Fig. 4, C and D). Thus, mitochondrial membrane potential was increased, not decreased, by mtPE deficiency.
Cell Size and Mitochondrial Morphology of mtPE-deficient Cells
The average area of Pisd KD#1 cells was 22% larger than that of NegCtrl cells (337 μm2 versus 278 μm2, p < 0.05) (Fig. 5, A and B). Fig. 5B shows enlarged Pisd KD cells in a population of cells of different sizes. Approximately 18% of Pisd KD cells were 3–4-fold larger than NegCtrl cells and contained enlarged (Fig. 5B, upper panel) and/or multiple (Fig. 5B, lower panel) nuclei. Moreover, PSB-2 cells were markedly more elongated than WT cells (Figs. 4C and 5C).
FIGURE 5.

Mitochondrial fragmentation. Representative confocal images of cells with mitochondria stained with MitoTracker Red (red) and nuclei with DAPI (blue). A, Pisd KD and NegCtrl cells. Arrows indicate punctate mitochondria in Pisd KD cells. B, representative images of mixed populations of Pisd KD#1 cells. Arrows show cells with enlarged (top) and multiple (lower) nuclei. Size bar in A and B, 20 μm. C, PSB-2 and WT cells; size bar, 10 μm.
The balance between fusion and fission of mitochondria regulates mitochondrial morphology and function (66, 67). Because mitochondria in Pisd−/− mouse embryos are highly fragmented (1), we hypothesized that mtPE deficiency would increase mitochondrial fragmentation. Confocal microscopy using MitoTracker Red revealed that filamentous mitochondria were present in >95% of WT and NegCtrl cells (Fig. 5, A and C), whereas >80% of Pisd KD#1 cells contained many small, fragmented mitochondria (Fig. 5, A and B). No tubular mitochondria were detected in >95% of PSB-2 cells (Fig. 5C). These observations indicate that reduction in mtPE induces mitochondrial fragmentation. Mitochondrial fusion is promoted by two dynamin-related, outer mitochondrial membrane proteins, mitofusin 1 and 2 (Mfn1 and Mfn2) (66, 68). To determine whether reduction in the mitofusins could have contributed to mitochondrial fragmentation, we compared amounts of Mfn1 and Mfn2 by immunoblotting of mitochondria from mtPE-deficient cells and control cells. Mfn1 and Mfn2 levels were not lower in Pisd KD#1 cells than NegCtrl cells, and Mfn1 was not reduced in PSB-2 cells (Fig. 6A). The small (22%) increase in Mfn2 in PSB-2 cells is consistent with greater immunofluorescence staining for Mfn2 in PSB-2 cells, compared with WT cells (data not shown). Opa1 (optic atrophy protein 1) is another protein required for mitochondrial fusion (69–71). The amount of Opa1 in Pisd KD#1 mitochondria was the same as in NegCtrl cells. Thus, increased mitochondrial fragmentation in Pisd KD#1 cells did not correlate with reduced expression of Mfn1, Mfn2, or Opa1. However, PSB-2 cells contained 25% less Opa1 than did WT cells (Fig. 6A) possibly contributing to increased mitochondrial fragmentation.
FIGURE 6.

Mitochondrial fusion and fission proteins. A, representative immunoblot of fusion factors in mitochondria from Pisd KD#1, NegCtrl, PSB-2, and WT cells using monoclonal antibodies against mitofusin1 (Mfn1), mitofusin2 (Mfn2), and optic atrophy protein1 (Opa1). 20 μg of protein/lane. Amounts of proteins relative to TOM20 are means ± S.D. from three independent experiments (*, p < 0.05 versus control). B and C, confocal immunofluorescence images of Drp1 (green), MitoTracker Red (red), and DAPI (blue) in NegCtrl and Pisd KD#1 cells (B) and WT and PSB2 cells (C). D, co-localization of Drp1 with mitochondria (Mito-Drp1) quantified as fraction of total green signal (Drp1) co-localizing with mitochondria (red) analyzed in >100 cells. Data are means ± S.E. from 3 to 4 independent experiments (*, p < 0.05 versus control).
Mitochondrial morphology is also regulated by mitochondrial fission. Recruitment of the dynamin-related protein 1 (Drp1) to mitochondria enhances mitochondrial fission (72, 73). Confocal immunofluorescence microscopy showed no significant difference in co-localization of Drp1 with mitochondria between Pisd KD#1 and NegCtrl cells (Fig. 6, B and D), whereas more Drp1 co-localized with mitochondria in PSB-2 than in WT cells (Fig. 6, C and D). Thus, increased Drp1-mediated fission might contribute to increased mitochondrial fragmentation in PSB-2 cells.
Mitochondrial morphology and ultrastructure were also examined by electron microscopy. Mitochondria in NegCtrl and WT cells contained intact cristae and had a normal elongated shape (Fig. 7, A and B) with shape factors (ratio of major and minor axes) of 2.7 ± 0.1 and 3.2 ± 0.2, respectively (Fig. 7C). In contrast, mitochondria in Pisd KD#1 and PSB-2 cells were more rounded (Fig. 7, A and B) with reduced shape factors (1.9 ± 0.1 and 2.0 ± 0.1, respectively) (Fig. 7C). Correspondingly, mean mitochondrial circularity was significantly higher in Pisd KD#1 and PSB-2 cells than control cells (Fig. 7C). Abnormal, swollen mitochondria were abundant in Pisd KD#1 and PSB-2 cells but not in NegCtrl or WT cells (Fig. 7, A and B), and mitochondrial matrix density in mtPE-deficient cells was less than in control cells (Fig. 7, A and B). The abundance of small, rounded mitochondria in Pisd KD#1 cells was consistent with an ∼50% reduction in mean area/mitochondrion compared with that in NegCtrl cells (data not shown). Opa1 deficiency is associated with cristae disorganization (69, 74, 75) so that the small reduction in Opa1 in PSB-2 mitochondria (Fig. 6A) is consistent with the higher proportion of PSB-2 mitochondria lacking distinct cristae (Fig. 7). Thus, even a modest reduction of mtPE in Pisd KD#1 and PSB-2 cells profoundly alters mitochondrial morphology and ultrastructure.
FIGURE 7.

Electron microscopy. Electron micrographs of NegCtrl and Pisd KD#1 cells (A) and WT and PSB-2 cells (B) are shown. Scale bar, 1 μm in upper micrographs and 0.5 μm in lower micrographs. ER, endoplasmic reticulum; m, mitochondrion; N, nucleus. Arrows indicate swollen mitochondria. C, mitochondrial shape factors (length/width) and circularity calculated from electron micrographs of >500 mitochondria. (*, p < 0.05 versus control).
MtPE Deficiency Decreases Mitochondrial Respiration and ATP Synthesis
Mitochondria generate ATP by OXPHOS coupled to O2 consumption via the ETC. In intact PSB-2 cells, basal respiration (O2 consumption from endogenous substrates) was 33% lower than in WT cells, and maximal O2 consumption (in the presence of an uncoupling agent) was similarly reduced (Fig. 8A). In Pisd KD#1 cells maximum, but not basal, O2 consumption was significantly lower than in NegCtrl cells (Fig. 8B). Rotenone (a CI inhibitor) decreased oxygen consumption in both mtPE-deficient cell lines by 80% (data not shown) confirming that CHO cells rely primarily on CI for basal respiration. Furthermore, the rate of substrate-driven respiration through ETC CI and CII (from pyruvate/malate or succinate, respectively) was markedly attenuated in digitonin-permeabilized PSB-2 cells compared with WT cells, and respiration with CI, but not CII, was lower in Pisd KD#1 than in NegCtrl cells (Fig. 8, C and D). Oxygen consumption via CIV from an artificial electron donor was not reduced by mtPE deficiency (Fig. 8, C and D). These observations indicate that mtPE deficiency inhibits respiration and ETC activity.
FIGURE 8.
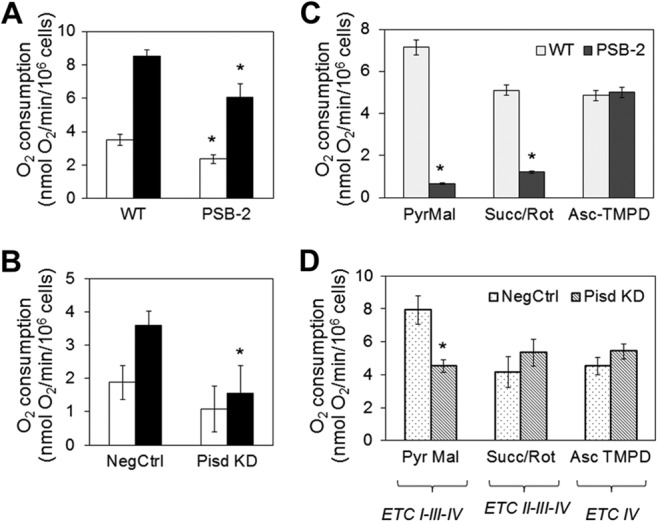
Oxygen consumption in mtPE-deficient cells. A and B, basal rate of O2 consumption in intact cells from endogenous substrates (white bars) and maximal rate of O2 consumption (black bars) measured after addition of the uncoupling agent carbonylcyanide 4-trifluoromethoxyphenylhydrazone (10 μm). C and D, substrate-driven O2 consumption in digitonin-permeabilized cells supplied with ETC substrates: PyrMal, pyruvate + malate (complex I); Succ/Rot, succinate (complex II) + rotenone (Rot, inhibitor of complex I); Asc-TMPD, ascorbate (reducing agent) + tetramethylphenylenediamine dihydrochloride (TMPD, electron donor for complex IV). Data are means ± S.D. from three experiments (*, p < 0.05 versus control).
In PSB-2 cells, cellular ATP was >2-fold higher than in WT cells (Fig. 9A), whereas in Pisd KD#1 cells (after 72 h but not 48 h) ATP was reduced by 80% (Fig. 9A). The reason for the divergent responses of ATP levels to acute and chronic mtPE deficiency is unclear, but long term compensatory mechanisms might have been induced in PSB-2 cells. Because the steady-state level of ATP reflects a balance between ATP synthesis and degradation, we measured the rate of ATP synthesis driven by respiratory substrates. In Pisd KD#1 and PSB-2 cells, the rate of ATP production via CI was 33 and 53% lower, respectively, than in control cells (Fig. 9B) and was 40 and 46% lower via CII (Fig. 9B). Furthermore, ATP production through CIV, from the artificial electron donor N,N,N′,N′-tetramethyl-p-phenylenediamine in the presence of ascorbate, was also lower in PSB-2 cells than in control cells (Fig. 9B). Thus, multiple ETC components are dysfunctional in mtPE-deficient cells, leading to diminished ATP production.
FIGURE 9.
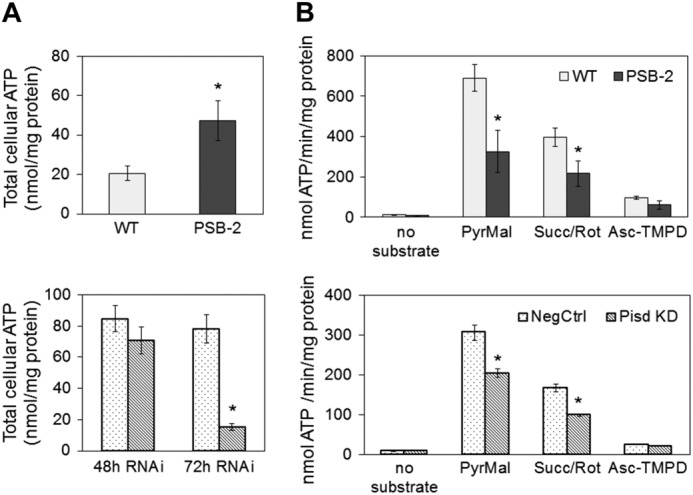
ATP production. A, cellular ATP was measured by luciferase assays in WT and PSB-2 cells and in NegCtrl and Pisd KD#1 cells 48 and 72 h after transfection. Data are means ± S.D. from triplicate analyses of one experiment representative of three experiments with similar results (*, p < 0.05 versus control). B, ATP synthesis in cells supplied with PyrMal, pyruvate + malate (for CI); Succ/Rot, succinate (for CII) + rotenone (rot, inhibitor of complex I); Asc-TMPD, ascorbate (reducing agent) + tetramethylphenylenediamine dihydrochloride (TMPD; electron donor for CIV). Data are means ± S.D. from triplicate analyses of one experiment from three experiments with similar results (*, p < 0.05 versus control).
MtPE Deficiency Inhibits ETC Activity and Supercomplex Formation
As additional evidence that ETC activity is compromised by mtPE deficiency, we measured enzymatic activities of individual ETC components. The specific activities of CI (NADH dehydrogenase) and CIV (cytochrome c oxidase) were significantly lower in mitochondria from both mtPE-deficient cell lines than in control cells (Fig. 10, A and B). In addition, CII activity (succinate dehydrogenase) was 45% lower in PSB-2 cells than in WT cells (Fig. 10B). Consistent with these observations, immunoblotting of Percoll-purified mitochondria showed that Pisd KD#1, KD#2, and PSB-2 cells contained less CIV protein than did NegCtrl cells (Fig. 11). Moreover, amounts of CI and CII were significantly reduced in PSB-2 cells but not in Pisd KD#1 cells (Fig. 11). However, the amount of CV (ATPase) was not changed by mtPE deficiency (Fig. 11). Although we detected CIII in rat heart mitochondria by immunoblotting, we did not detect CIII in CHO cells using the same antibodies. Consequently, we could not assess amounts of CIII. These data show that the abundance and activities of multiple ETC proteins, but not CV, were reduced by mtPE deficiency.
FIGURE 10.
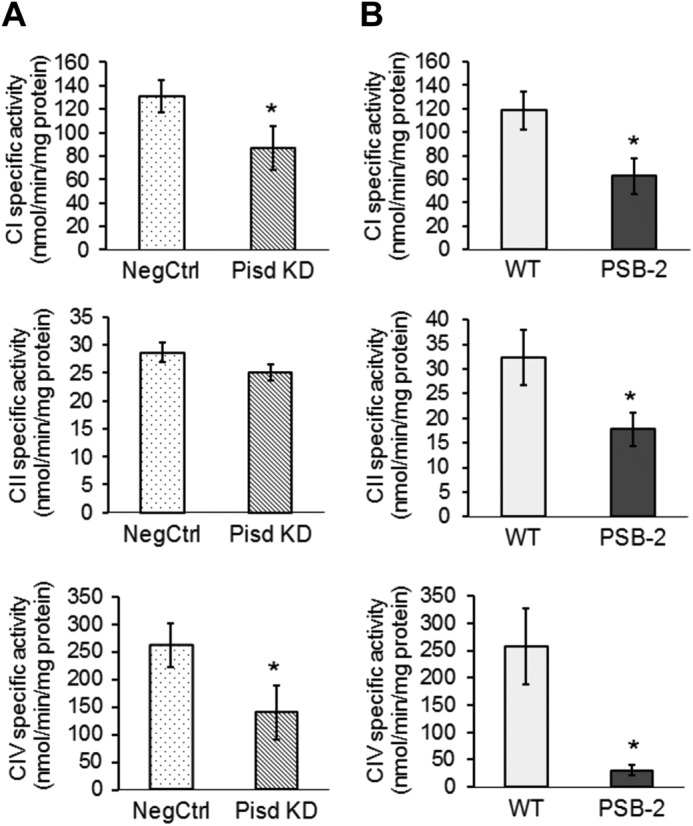
Enzymatic activities of OXPHOS complexes. Activities of complexes I, II, and IV (CI, CII, and CIV) measured spectrophotometrically in mitochondria from Pisd KD#1 and NegCtrl cells (A) and PSB-2 and WT cells (B). Data are means ± S.D. from 3 to 4 independent experiments (*, p < 0.05 versus control).
FIGURE 11.
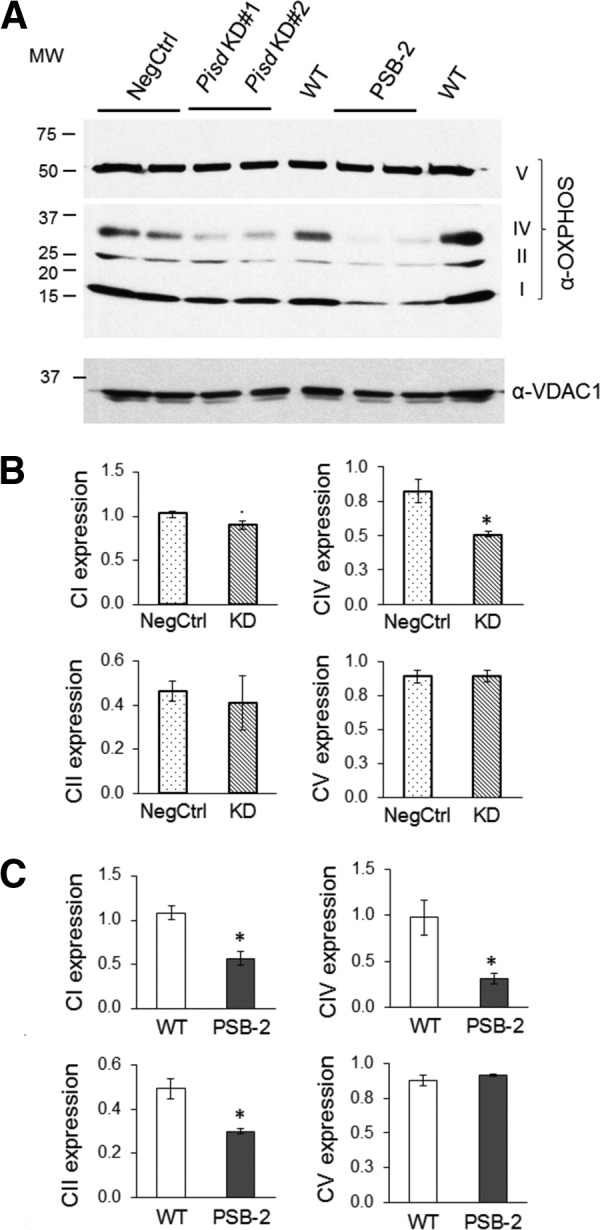
OXPHOS complexes I, II, and IV. A, representative immunoblot of Percoll-purified mitochondria from Pisd KD#1, KD#2, and NegCtrl cells 72 h after transfection, and PSB-2 and WT cells, using antibodies against OXPHOS complexes I–V (CI–CV). Locations of complexes are indicated at right of gel and molecular weights at left. B and C, quantification of OXPHOS proteins relative to voltage-dependent anion channel-1 (VDAC1). Data are means from three independent experiments (*, p < 0.05 versus control).
Decreased levels of ETC proteins can be associated with impaired assembly and/or stability of ETC supercomplexes. In mammals, these supercomplexes contain CI monomers, CIII dimers, and up to four copies of CIV; association with CIV enhances the activity of CI and CIII (76). We assessed amounts of mitochondrial ETC supercomplexes by BN-PAGE analysis (76). Location of complexes on the gel was established by immunoblotting with a mixture of CI-CV antibodies (Fig. 12A). Identity of CI, CII, CIV, and CV was confirmed by in-gel assays (Fig. 12, B–D). CI is present in a lower molecular mass complex in CHO mitochondria (750–800 kDa, Fig. 12A) than in rat/bovine heart mitochondria (∼1000 kDa) (77) but is similar in size to rat muscle CI (78). The strong in-gel activity of this band (I1 in Fig. 12B) confirmed its presence as functional CI. Higher molecular mass assemblies of CI were observed only in control cells (arrow, Fig. 12B). CII was also identified by in-gel assays (data not shown) as a rapidly migrating band on BN-polyacrylamide gels (II1, Fig. 12A). PSB-2 cells contained less II1 than WT cells, but the amount of II1 was only slightly lower in Pisd KD#1 cells than NegCtrl cells (Fig 12A). Several CIV oligomers and complexes with dimeric CIII are present in rat heart mitochondria (77); CHO cells also contain CIV oligomers (IVn) and complexes of CIV with CIII and CI (Fig. 12, A and C). Immunoblotting of CIV (data not shown) and in-gel assays (Fig. 12C) revealed fewer CIV oligomers and supercomplexes in Pisd KD#1 and PSB-2 cells than in control cells. In-gel assays showed that, as in rat and bovine heart (77), CV was present in an ∼700-kDa complex corresponding to CV monomers (V1, Fig. 12D). Monomeric ATP synthase is active (77, 79), whereas dimeric CV has been reported to be either active (80) or inactive (79); no CV dimers were detected in CHO cells. Mitochondria from Pisd KD#1 and NegCtrl cells had similar in-gel CV activity, but this activity was significantly lower in PSB-2 cells than WT cells (Fig. 12D). Thus, immunoblotting and in-gel assays indicate that mtPE plays an important role in formation and/or stability of multiple ETC proteins and supercomplexes.
FIGURE 12.
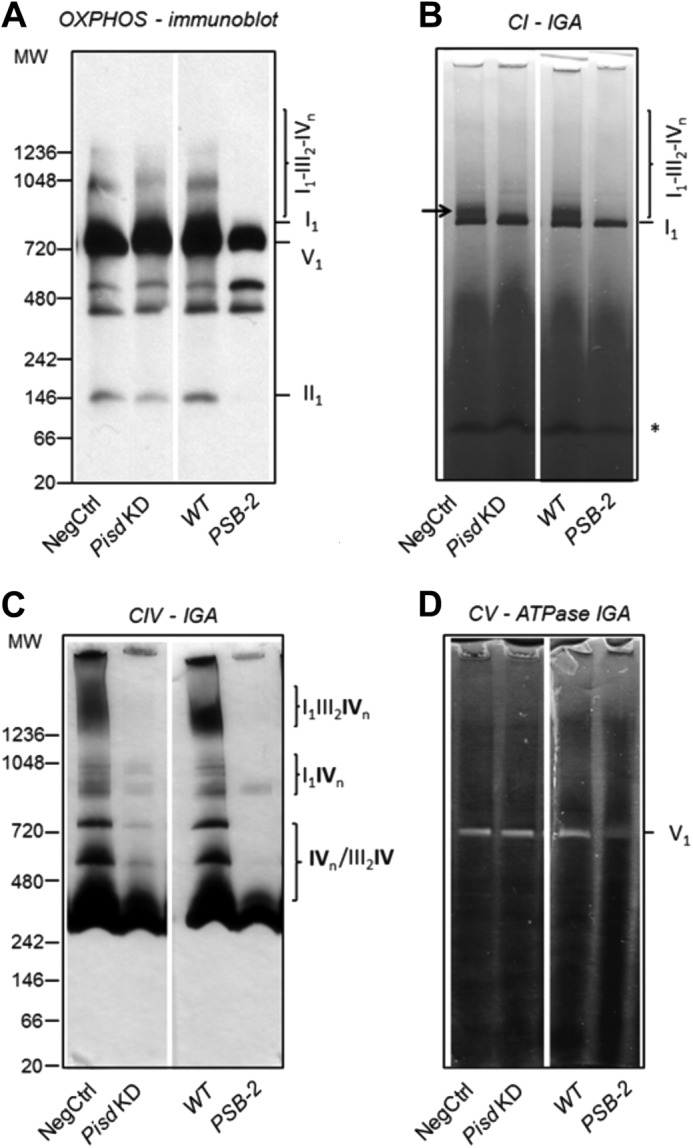
OXPHOS supercomplexes. Digitonin-solubilized mitochondrial complexes (75 μg of protein/lane) from NegCtrl, Pisd KD#1, WT, and PSB-2 cells were separated by one-dimensional BN-PAGE. A, immunoblotting of CI–CV proteins with anti-OXPHOS antibodies. Individual complexes and supercomplexes are indicated at right and molecular weights at left. B, in-gel activity (IGA) of CI (NADH dehydrogenase); *, nonspecific activity; arrow indicates supercomplex of CI with CIV. C, IGA of CIV (cytochrome c oxidase). D, IGA of CV (ATPase) shown as white, lead phosphate precipitate. The following complexes and supercomplexes were identified: CI monomer (I1); CI associated with CIII and/or CIV (I1III2; I1IVn; I1III2IVn); CIV monomer or multimer and associated with CIII (III2IV1) and CI; supercomplexes of I1III2IVn, with monomeric CI (I1), dimeric CIII (III2), and n copies of CIV (IVn, n = 1–4); CV monomer (V1). CII was identified as a complex (II1) by immunoblotting and in-gel activity (data not shown). Data are representative of 3–4 independent experiments with similar results.
Restoration of mtPE with Exogenous Lyso-PE
To confirm that mitochondrial defects in PSB-2 cells were specifically due to reduction of mtPE rather than reduction of PS (63), we supplemented the cells with 100 μm lyso-PE, which in Saccharomyces cerevisiae is preferentially converted to mtPE (39, 40). Lyso-PE supplementation of PSB-2 cells for 96 h increased mtPE by 45% (Fig. 13A) to approximately the WT level (Fig. 2A) without significantly altering the amount of PS or other phospholipids in mitochondria or microsomes (Fig. 13A). The mitochondrial phospholipid composition of WT cells was unaltered by lyso-PE supplementation (data not shown). Lyso-PE supplementation increased the rate of cell growth and approximately doubled the number of PSB-2 cells (Fig. 13B), and it also increased the rate of ATP synthesis via CI and CII (Fig. 13C). Furthermore, the number of punctate mitochondria was reduced, and filamentous mitochondria were present in >80% of lyso-PE-supplemented PSB-2 cells (Fig. 13D). Thus, replenishment of mtPE in PSB-2 cells markedly improved the defects in cell growth, ATP production, and mitochondrial morphology, without normalizing the PS content of mitochondria or microsomes. We conclude that these mitochondrial functions critically depend on the level of mtPE.
FIGURE 13.

Lyso-PE supplementation of PSB-2 cells. A, mass (nmol/mg protein) of PE, phosphatidylcholine (PC), phosphatidylinositol (PI), and PS in Percoll-purified mitochondria (MITO, upper panel) and microsomes (MIC) from PSB-2 cells grown for 96 h in medium containing 100 μm lyso-PE (LPE; light bars) or vehicle (dark bars). B, representative images of PSB-2 cells cultured for 96 h ± 100 μm lyso-PE. Graph shows growth rate of PSB-2 cells ± LPE. C, rate of ATP synthesis (from substrates in Fig. 9) in PSB-2 cells cultured with (light bars) or without (dark bars) lyso-PE. D, confocal images of MitoTracker Red-stained PSB-2 cells grown for 96 h ± lyso-PE; nuclei stained with DAPI. Arrows indicate filamentous mitochondria in cells supplemented with LPE. Data in graphs are means ± S.D. of triplicate analyses from two independent experiments (*, p < 0.05 versus control). Succ, succinate; Rot, rotenone; substr, substrate; PyrMal, pyruvate + malate; AscTMPD, ascorbate (reducing agent) + tetramethylphenylenediamine dihydrochloride.
In conclusion, cell growth, mitochondrial morphology, respiratory capacity, ATP production, and ETC activity are impaired in mammalian cells with chronic (PSB-2 cells) or acute (Pisd KD cells) mtPE deficiency. Importantly, mitochondrial defects in PSB-2 cells were largely reversed when mtPE content was normalized by lyso-PE supplementation.
DISCUSSION
Previous studies showed that in CHO cells (41) and yeast (42, 43) most mtPE is synthesized in mitochondria from PSD and that import of PE synthesized from CDP-ethanolamine cannot maintain a normal complement of mtPE. Moreover, elimination of PSD in mice, in which PE synthesis from CDP-ethanolamine was active, was embryonic lethal establishing an absolute requirement for PSD in mice (1). However, Pisd+/− mice that expressed 50% of normal Pisd mRNA and PSD activity were viable, and the mtPE content of tissues was normal (1, 57). We therefore tested the hypothesis that reduction of mtPE synthesis by >50% in CHO cells would decrease mtPE content and impair mitochondrial morphology and function.
mtPE Deficiency Reduces Cell Growth
In PSB-2 cells (chronically reduced mtPE) and Pisd KD cells (acutely reduced mtPE), mtPE mass was reduced by 22–27% with no reduction in CL, a lipid required for normal mitochondrial function. Although mtPE was only modestly reduced, the growth of PSB-2 and Pisd KD cells was compromised, and attenuation of growth of Pisd KD cells correlated with the extent of Pisd knockdown. Because PE was reduced in mitochondria but not lysates, we conclude that the mitochondrial abnormalities and growth defects in the mtPE-deficient cells are caused by depletion of PE specifically in mitochondria. Interestingly, extra-mitochondrial PE levels were normal but did not compensate for decreased mtPE synthesis and did not normalize mtPE content. These observations are consistent with the embryonic lethality of Pisd knock-out mice in which PE synthesis from CDP-ethanolamine was normal or increased (1).
Reduction of mtPE and the ETC
Respiratory capacity, ETC enzymatic activities, and ATP synthesis are compromised in cells with reduced mtPE content. The rate of ATP synthesis was attenuated in mtPE-deficient cells despite normal amount and enzymatic activity of CV (ATP synthase) probably because the assembly/stability of several supercomplexes, including CI and CIV, were reduced. We could not discriminate between altered formation and stability of these complexes using BN-PAGE because Coomassie Blue disrupts protein interactions. However, because the amounts of CI and CIV in purified mitochondria were reduced, integration of supercomplexes into the mitochondrial inner membrane was probably impaired. We therefore attribute the decreased activities of CI and CIV primarily to reduced amounts of normally active CI and CIV rather than decreased intrinsic activity of these complexes. Similarly, the reduced activity of CII in PSB-2 cells appears to be due to lower abundance of normally active CII. Consistent with these conclusions, x-ray crystallography revealed that PE is tightly associated with bovine heart mitochondrial CIV (81) and that CL and PE are bound to yeast CIII (8). In addition, the activity of CI depends on bound PE (82). Moreover, inclusion of PE in proteoliposomes reconstituted with CIII increased the respiratory control ratio (83). Thus, a threshold level of mtPE appears to be crucial for formation and/or integration of supercomplexes into mitochondrial membranes.
Inhibition of the ETC often correlates with decreased mitochondrial membrane potential. However, although OXPHOS activity and ATP production were decreased in Pisd KD#1 cells, the mitochondria were more highly polarized than those of NegCtrl cells, according to TMRM staining. The enhanced membrane potential was not due to increased mitochondrial mass because nonylacridine orange staining and quantification of mtDNA indicated that mitochondrial mass and biogenesis were not increased in Pisd KD#1 or PSB-2 cells. These data suggest that ETC function is uncoupled from ATP synthesis in Pisd KD#1 and PSB-2 cells probably because of decreased phosphorylation rather than increased proton leakage. Thus, protons would accumulate in the inter-membrane space and membrane potential would rise. The idea that mtPE deficiency uncouples ETC function and ATP synthesis is consistent with the lower maximum rate of oxygen consumption in mtPE-depleted cells compared with control cells. Thus, organization of ETC complexes and ATP generation are both compromised, perhaps because of limited availability of ADP for ATP production or because CV is bound to an inhibitor (84).
Mitochondrial Morphology in mtPE-deficient Cells
Electron microscopy highlighted the grossly aberrant morphology of PE-deficient mitochondria. Furthermore, confocal fluorescence microscopy showed that mitochondrial fragmentation was greatly increased in mtPE-deficient cells compared with control cells, as was previously observed in Pisd−/− embryonic fibroblasts (1). In yeast, elimination of mtPE synthesis (85) or concomitant loss of synthesis of both CL and PE also promotes mitochondrial fragmentation (52). Mitochondrial morphology and function are closely linked, and mitochondrial dysfunction can induce autophagy/mitophagy and mitochondrial fragmentation (86–88). Defects in mitochondrial fusion, ATP synthesis, and cell growth occur in mammalian cells lacking the mitochondrial fusion factors Mfn1 and Mfn2, and in cells with reduced amounts of the fusion factor Opa1 (66, 85). It is unlikely, however, that the extensive mitochondrial fragmentation in Pisd KD#1 cells is primarily due to deficiency of fusion factors because amounts of Mfn1, Mfn2, and Opa1 are equivalent in Pisd KD#1 and NegCtrl cells, nor is it likely that the widespread mitochondrial fragmentation in PSB-2 cells is due to reduced amounts of Mfn1 or Mfn2 because Mfn1 is equally abundant in PSB-2 and WT cells, and Mfn2 is slightly increased. Mfn2 is required for tethering mitochondria to ER/MAM (48) and mediating PS import from the ER to mitochondrial PSD (12). Thus, the small increase in Mfn2 in PSB-2 cells might reflect induction of a long term compensatory mechanism for promoting PS import into the mitochondria, particularly because the PS content of MAM was reduced by ∼50%. PSB-2 cells also contained less Opa1 than did WT cells, which might contribute to the increased mitochondrial fragmentation in PSB-2 cells.
Mitochondrial fusion is required for inheritance and maintenance of mtDNA (66, 89–91). Thus, the reduction in mtDNA content in PSB-2 cells might be caused by defective mitochondrial fusion. Moreover, our finding that mitochondrial mass was not decreased in either model of mtPE deficiency, despite increased mitochondrial fragmentation, is consistent with the report that hyperpolarized mitochondria fail to be eliminated by autophagy (88, 92).
When mitochondrial fusion is inhibited, unopposed fission can lead to increased mitochondrial fragmentation. Drp1-dependent mitochondrial fission can be enhanced by inhibition of either the ETC or the permeability transition pore (93). The increased co-localization of the fission protein Drp1 with mitochondria of PSB2, compared with WT, cells suggests that the enhanced fragmentation of PSB-2 mitochondria might be partially due to increased Drp1-mediated fission. However, because mitochondrial fragmentation was similarly increased in both PSB-2 cells and Pisd KD cells (in which Opa1 was not reduced and Drp1 was not increased), it is unlikely that the small changes in these factors in PSB-2 cells are primarily responsible for the increased mitochondrial fragmentation. We speculate that a change in physical properties of mitochondrial membrane, as a result of mtPE deficiency (a feature of both PSB-2 and Pisd KD cells), is more likely the primary cause of morphological changes in mitochondria. PE is a cone-shaped lipid that can generate nonlamellar membrane structures and induce negative membrane curvature and membrane fusion (85, 94, 95). Thus, mtPE deficiency might inhibit mitochondrial fusion (85). In general, defects in mitochondrial function and morphology were more pronounced in PSB-2 cells (chronic mtPE depletion) than in cells with acute reduction in mtPE (Pisd KD cells). In addition, some long term compensatory changes appear to have been induced in PSB-2 cells.
Reversal of mtPE Deficiency with Lyso-PE
To confirm that mitochondrial defects in PSB-2 cells could be ascribed specifically to reduction in mtPE, we supplemented PSB-2 cells with lyso-PE thereby restoring a normal mtPE content without increasing PE, or normalizing the PS content, of cell lysates or microsomes. Thus, lyso-PE is efficiently used for mtPE synthesis in mammalian cells as well as yeast (39). Normalization of mtPE in PSB-2 cells largely restored cell growth, increased ATP synthesis, and re-generated a filamentous mitochondrial network. In cell lysates, the content of neither PE nor PS content was increased by lyso-PE supplementation confirming that the mitochondrial defects in PSB-2 cells are caused specifically by mtPE depletion.
A limitation of the models used in these studies is that the sequence of events by which depletion of mtPE impairs mitochondrial properties remains unclear. A graded reduction in mtPE might establish the sequence in which mitochondrial targets (bioenergetics, cell growth, and morphology) are affected. Our results suggest, however, that altered mitochondrial morphology in mtPE-deficient cells is not a consequence of impaired mitochondrial function. These studies represent the first demonstration that attenuation of mtPE biosynthesis can reversibly and profoundly alter mitochondrial morphology and function in mammalian cells. Manipulation of the mtPE content could therefore be useful for controlling mitochondrial dynamics and bioenergetics. The findings raise the possibility that mitochondrial defects in some common human disorders might, at least in part, be attributable to mtPE deficiency.
Acknowledgments
We thank Russ Watts and Randy Nelson for excellent technical assistance and Dr. Moira Glerum for crucial discussions on assay of ETC enzymes.
This work was supported by the Natural Sciences and Engineering Research Council of Canada (to J. E. V.).
- OXPHOS
- oxidative phosphorylation
- BN
- blue-native
- CL
- cardiolipin
- CI-V
- complex I–V
- ER
- endoplasmic reticulum
- ETC
- electron transport chain
- KD
- RNAi knockdown
- MAM
- mitochondrion-associated membrane
- mtPE
- mitochondrial phosphatidylethanolamine
- PE
- phosphatidylethanolamine
- Pisd
- phosphatidylserine decarboxylase gene
- PS
- phosphatidylserine
- PSD
- PS decarboxylase
- TMRM
- tetramethylrhodamine methyl ester
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- DsiRNA
- Dicer-substrate RNA
- NegCtrl
- negative control.
REFERENCES
- 1. Steenbergen R., Nanowski T. S., Beigneux A., Kulinski A., Young S. G., Vance J. E. (2005) Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 280, 40032–40040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johi A., Beal M. F. (2012) Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 342, 619–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ren J., Pulakat L., Whaley-Connell A., Sowers J. R. (2010) Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. 88, 993–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Supale S., Li N., Brun T., Maechler P. (2012) Mitochondrial dysfunction in pancreatic beta cells. Trends Endocrinol. Metab. 23, 477–487 [DOI] [PubMed] [Google Scholar]
- 5. Baysal B. E., Ferrell R. E., Willett-Brozick J. E., Lawrence E. C., Myssiorek D., Bosch A., van der Mey A., Taschner P. E., Rubinstein W. S., Myers E. N., Richard C. W., 3rd, Cornelisse C. J., Devilee P., Devlin B. (2000) Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287, 848–851 [DOI] [PubMed] [Google Scholar]
- 6. Choi S. Y., Gonzalvez F., Jenkins G. M., Slomianny C., Chretien D., Arnoult D., Petit P. X., Frohman M. A. (2007) Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ. 14, 597–606 [DOI] [PubMed] [Google Scholar]
- 7. Koshkin V., Greenberg M. L. (2000) Oxidative phosphorylation in cardiolipin-lacking yeast mitochondria. Biochem. J. 347, 687–691 [PMC free article] [PubMed] [Google Scholar]
- 8. Pfeiffer K., Gohil V., Stuart R. A., Hunte C., Brandt U., Greenberg M. L., Schägger H. (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880 [DOI] [PubMed] [Google Scholar]
- 9. Claypool S. M., Oktay Y., Boontheung P., Loo J. A., Koehler C. M. (2008) Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 182, 937–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang J., Guan Z., Murphy A. N., Wiley S. E., Perkins G. A., Worby C. A., Engel J. L., Heacock P., Nguyen O. K., Wang J. H., Raetz C. R., Dowhan W., Dixon J. E. (2011) Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 13, 690–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fadok V. A., de Cathelineau A., Daleke D. L., Henson P. M., Bratton D. L. (2001) Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J. Biol. Chem. 276, 1071–1077 [DOI] [PubMed] [Google Scholar]
- 12. Hailey D. W., Rambold A. S., Satpute-Krishnan P., Mitra K., Sougrat R., Kim P. K., Lippincott-Schwartz J. (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang F., Ryan M. T., Schlame M., Zhao M., Gu Z., Klingenberg M., Pfanner N., Greenberg M. L. (2000) Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 275, 22387–22394 [DOI] [PubMed] [Google Scholar]
- 14. McKenzie M., Lazarou M., Thorburn D. R., Ryan M. T. (2006) Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J. Mol. Biol. 361, 462–469 [DOI] [PubMed] [Google Scholar]
- 15. Mileykovskaya E., Penczek P. A., Fang J., Mallampalli V. K., Sparagna G. C., Dowhan W. (2012) Arrangement of the respiratory chain complexes in Saccharomyces cerevisiae supercomplex III2IV2 revealed by single particle cryo-electron microscopy. J. Biol. Chem. 287, 23095–23103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koshkin V., Greenberg M. L. (2002) Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem. J. 364, 317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu Y., Condell M., Plesken H., Edelman-Novemsky I., Ma J., Ren M., Schlame M. (2006) A Drosophila model of Barth syndrome. Proc. Natl. Acad. Sci. U.S.A. 103, 11584–11588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ohtsuka T., Nishijima M., Akamatsu Y. (1993) A somatic cell mutant defective in phosphatidylglycerophosphate synthase, with impaired phosphatidylglycerol and cardiolipin biosynthesis. J. Biol. Chem. 268, 22908–22913 [PubMed] [Google Scholar]
- 19. Vreken P., Valianpour F., Nijtmans L. G., Grivell L. A., Plecko B., Wanders R. J., Barth P. G. (2000) Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem. Biophys. Res. Commun. 279, 378–382 [DOI] [PubMed] [Google Scholar]
- 20. Emoto K., Kobayashi T., Yamaji A., Aizawa H., Yahara I., Inoue K., Umeda M. (1996) Redistribution of phosphatidylethanolamine at the cleavage furrow of dividing cells during cytokinesis. Proc. Natl. Acad. Sci. U.S.A. 93, 12867–12872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Emoto K., Umeda M. (2000) An essential role for a membrane lipid in cytokinesis: regulation of contractile ring disassembly by redistribution of phosphatidylethanolamine. J. Cell Biol. 149, 1215–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bogdanov M., Sun J., Kaback H. R., Dowhan W. (1996) A phospholipid acts as a chaperone in assembly of a membrane transport protein. J. Biol. Chem. 271, 11615–11618 [DOI] [PubMed] [Google Scholar]
- 23. Bogdanov M., Umeda M., Dowhan W. (1999) Phospholipid-assisted refolding of an integral membrane protein. Minimum structural features for phosphatidylethanolamine to act as a molecular chaperone. J. Biol. Chem. 274, 12339–12345 [DOI] [PubMed] [Google Scholar]
- 24. Walkey C. J., Donohue L. R., Bronson R., Agellon L. B., Vance D. E. (1997) Disruption of the murine gene encoding phosphatidylethanolamine N-methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 94, 12880–12885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Devane W. A., Hanus L., Breuer A., Pertwee R. G., Stevenson L. A., Griffin G., Gibson D., Mandelbaum A., Etinger A., Mechoulam R. (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258, 1946–1949 [DOI] [PubMed] [Google Scholar]
- 26. Jin X. H., Okamoto Y., Morishita J., Tsuboi K., Tonai T., Ueda N. (2007) Discovery and characterization of a Ca2+-independent phosphatidylethanolamine N-acyltransferase generating the anandamide precursor and its congeners. J. Biol. Chem. 282, 3614–3623 [DOI] [PubMed] [Google Scholar]
- 27. Menon A. K., Stevens V. L. (1992) Phosphatidylethanolamine is the donor of the ethanolamine residue linking a glycosylphosphatidylinositol anchor to protein. J. Biol. Chem. 267, 15277–15280 [PubMed] [Google Scholar]
- 28. Signorell A., Jelk J., Rauch M., Bütikofer P. (2008) Phosphatidylethanolamine is the precursor of the ethanolamine phosphoglycerol moiety bound to eukaryotic elongation factor 1A. J. Biol. Chem. 283, 20320–20329 [DOI] [PubMed] [Google Scholar]
- 29. Nebauer R., Rosenberger S., Daum G. (2007) Phosphatidylethanolamine, a limiting factor of autophagy in yeast strains bearing a defect in the carboxypeptidase Y pathway of vacuolar targeting. J. Biol. Chem. 282, 16736–16743 [DOI] [PubMed] [Google Scholar]
- 30. Deleault N. R., Piro J. R., Walsh D. J., Wang F., Ma J., Geoghegan J. C., Supattapone S. (2012) Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. U.S.A. 109, 8546–8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rostovtseva T. K., Gurnev P. A., Chen M. Y., Bezrukov S. M. (2012) Membrane lipid composition regulates tubulin interaction with mitochondrial voltage-dependent anion channel. J. Biol. Chem. 287, 29589–29598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vance J. E. (2008) Phosphatidylserine and phosphatidylethanolamine in mammalian cells. Two metabolically related aminophospholipids. J. Lipid Res. 49, 1337–1387 [DOI] [PubMed] [Google Scholar]
- 33. Vance J. E., Tasseva G. (2013) Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim. Biophys. Acta, in press [DOI] [PubMed] [Google Scholar]
- 34. Kennedy E. P., Weiss S. B. (1956) The function of cytidine coenzymes in the biosynthesis of phospholipids. J. Biol. Chem. 222, 193–214 [PubMed] [Google Scholar]
- 35. Vance J. E. (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256 [PubMed] [Google Scholar]
- 36. Borkenhagen L. F., Kennedy E. P., Fielding L. (1961) Enzymatic formation and decarboxylation of phosphatidylserine. J. Biol. Chem. 236, PC28–PC32 [Google Scholar]
- 37. Zborowski J., Dygas A., Wojtczak L. (1983) Phosphatidylserine decarboxylase is located on the external side of the inner mitochondrial membrane. FEBS Lett. 157, 179–182 [DOI] [PubMed] [Google Scholar]
- 38. Sundler R., Akesson B., Nilsson A. (1974) Quantitative role of base exchange in phosphatidylethanolamine synthesis in isolated rat hepatocytes. FEBS Lett. 43, 303–307 [DOI] [PubMed] [Google Scholar]
- 39. Riekhof W. R., Voelker D. R. (2006) Uptake and utilization of lyso-phosphatidylethanolamine by Saccharomyces cerevisiae. J. Biol. Chem. 281, 36588–36596 [DOI] [PubMed] [Google Scholar]
- 40. Riekhof W. R., Wu J., Jones J. L., Voelker D. R. (2007) Identification and characterization of the major lysophosphatidylethanolamine acyltransferase in Saccharomyces cerevisiae. J. Biol. Chem. 282, 28344–28352 [DOI] [PubMed] [Google Scholar]
- 41. Shiao Y.-J., Lupo G., Vance J. E. (1995) Evidence that phosphatidylserine is imported into mitochondria via a mitochondria-associated membrane and that the majority of mitochondrial phosphatidylethanolamine is derived from decarboxylation of phosphatidylserine. J. Biol. Chem. 270, 11190–11198 [DOI] [PubMed] [Google Scholar]
- 42. Birner R., Bürgermeister M., Schneiter R., Daum G. (2001) Roles of phosphatidylethanolamine and of its several biosynthetic pathways in Saccharomyces cerevisiae. Mol. Biol. Cell 12, 997–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bürgermeister M., Birner-Grünberger R., Nebauer R., Daum G. (2004) Contribution of different pathways to the supply of phosphatidylethanolamine and phosphatidylcholine to mitochondrial membranes of the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 1686, 161–168 [DOI] [PubMed] [Google Scholar]
- 44. Kainu V., Hermansson M., Hanninen S., Hokynar K., Somerharju P. (2012) Import of phosphatidylserine to and export of phosphatidylethanolamine molecular species from mitochondria. Biochim. Biophys. Acta 1831, 429–437 [DOI] [PubMed] [Google Scholar]
- 45. Stone S. J., Vance J. E. (2000) Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 275, 34534–34540 [DOI] [PubMed] [Google Scholar]
- 46. Vance J. E. (1991) Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J. Biol. Chem. 266, 89–97 [PubMed] [Google Scholar]
- 47. Rizzuto R., Pinton P., Carrington W., Fay F. S., Fogarty K. E., Lifshitz L. M., Tuft R. A., Pozzan T. (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766 [DOI] [PubMed] [Google Scholar]
- 48. de Brito O. M., Scorrano L. (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610 [DOI] [PubMed] [Google Scholar]
- 49. Kuge O., Nishijima M., Akamatsu Y. (1991) A cloned gene encoding phosphatidylserine decarboxylase complements the phosphatidylserine biosynthetic defect of a Chinese hamster ovary cell mutant. J. Biol. Chem. 266, 6370–6376 [PubMed] [Google Scholar]
- 50. Trotter P. J., Pedretti J., Voelker D. R. (1993) Phosphatidylserine decarboxylase from Saccharomyces cerevisiae: isolation of mutants, cloning of the gene, and creation of a null allele. J. Biol. Chem. 268, 21416–21424 [PubMed] [Google Scholar]
- 51. Trotter P. J., Pedretti J., Yates R., Voelker D. R. (1995) Phosphatidylserine decarboxylase 2 of Saccharomyces cerevisiae. Cloning and mapping of the gene, heterologous expression and creation of the null allele. J. Biol. Chem. 270, 6071–6080 [DOI] [PubMed] [Google Scholar]
- 52. Joshi A. S., Thompson M. N., Fei N., Hüttemann M., Greenberg M. L. (2012) Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J. Biol. Chem. 287, 17589–17597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Signorell A., Gluenz E., Rettig J., Schneider A., Shaw M. K., Gull K., Bütikofer P. (2009) Perturbation of phosphatidylethanolamine synthesis affects mitochondrial morphology and cell-cycle progression in procyclic-form Trypanosoma brucei. Mol. Microbiol. 72, 1068–1079 [DOI] [PubMed] [Google Scholar]
- 54. Storey M. K., Clay K. L., Kutateladze T., Murphy R. C., Overduin M., Voelker D. R. (2001) Phosphatidylethanolamine has an essential role in Saccharomyces cerevisiae that is independent of its ability to form hexagonal phase structures. J. Biol. Chem. 276, 48539–48548 [DOI] [PubMed] [Google Scholar]
- 55. Gohil V. M., Thompson M. N., Greenberg M. L. (2005) Synthetic lethal interaction of the mitochondrial phosphatidylethanolamine and cardiolipin biosynthetic pathways in Saccharomyces cerevisiae. J. Biol. Chem. 280, 35410–35416 [DOI] [PubMed] [Google Scholar]
- 56. DeChavigny A., Heacock P. N., Dowhan W. (1991) Sequence and inactivation of the pss gene of Escherichia coli. Phosphatidylethanolamine may not be essential for cell viability. J. Biol. Chem. 266, 5323–5332 [PubMed] [Google Scholar]
- 57. Steenbergen R., Nanowski T. S., Nelson R., Young S. G., Vance J. E. (2006) Phospholipid homeostasis in phosphatidylserine synthase-2-deficient mice. Biochim. Biophys. Acta 1761, 313–323 [DOI] [PubMed] [Google Scholar]
- 58. Fullerton M. D., Hakimuddin F., Bakovic M. (2007) Developmental and metabolic effects of disruption of the mouse CTP:phosphoethanolamine cytidylyltransferase gene (Pcyt2). Mol. Cell. Biol. 27, 3327–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Saito S., Goto K., Tonosaki A., Kondo H. (1997) Gene cloning and characterization of CDP-diacylglycerol synthase from rat brain. J. Biol. Chem. 272, 9503–9509 [DOI] [PubMed] [Google Scholar]
- 60. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 61. Rouser G., Siakotos A. N., Fleischer S. (1966) Quantitative analysis of phospholipids by thin layer chromatography and phosphorus analysis of spots. Lipids 1, 85–86 [DOI] [PubMed] [Google Scholar]
- 62. Gottesman M. M. (1987) Chinese hamster ovary cells. Methods Enzymol. 151, 3–8 [DOI] [PubMed] [Google Scholar]
- 63. Saito K., Nishijima M., Kuge O. (1998) Genetic evidence that phosphatidylserine synthase II catalyzes the conversion of phosphatidylethanolamine to phosphatidylserine in Chinese hamster ovary cells. J. Biol. Chem. 273, 17199–17205 [DOI] [PubMed] [Google Scholar]
- 64. Chang S.-C., Heacock P. N., Mileykovskaya E., Voelker D. R., Dowhan W. (1998) Isolation and characterization of the gene (CLS1) encoding cardiolipin synthase in Saccharomyces cerevisiae. J. Biol. Chem. 273, 14933–14941 [DOI] [PubMed] [Google Scholar]
- 65. Zhang M., Mileykovskaya E., Dowhan W. (2005) Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J. Biol. Chem. 280, 29403–29408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen H., Chomyn A., Chan D. C. (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 280, 26185–26192 [DOI] [PubMed] [Google Scholar]
- 67. Parone P. A., Da Cruz S., Tondera D., Mattenberger Y., James D. I., Maechler P., Barja F., Martinou J. C. (2008) Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PloS ONE 3, e3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen H., Detmer S. A., Ewald A. J., Griffin E. E., Fraser S. E., Chan D. C. (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Frezza C., Cipolat S., Martins de Brito O., Micaroni M., Beznoussenko G. V., Rudka T., Bartoli D., Polishuck R. S., Danial N. N., De Strooper B., Scorrano L. (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189 [DOI] [PubMed] [Google Scholar]
- 70. Ishihara N., Fujita Y., Oka T., Mihara K. (2006) Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 25, 2966–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ehses S., Raschke I., Mancuso G., Bernacchia A., Geimer S., Tondera D., Martinou J. C., Westermann B., Rugarli E. I., Langer T. (2009) Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 187, 1023–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Smirnova E., Griparic L., Shurland D. L., van der Bliek A. M. (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ishihara N., Nomura M., Jofuku A., Kato H., Suzuki S. O., Masuda K., Otera H., Nakanishi Y., Nonaka I., Goto Y., Taguchi N., Morinaga H., Maeda M., Takayanagi R., Yokota S., Mihara K. (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966 [DOI] [PubMed] [Google Scholar]
- 74. Olichon A., Baricault L., Gas N., Guillou E., Valette A., Belenguer P., Lenaers G. (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 278, 7743–7746 [DOI] [PubMed] [Google Scholar]
- 75. Griparic L., Kanazawa T., van der Bliek A. M. (2007) Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 178, 757–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wittig I., Braun H. P., Schägger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428 [DOI] [PubMed] [Google Scholar]
- 77. Wittig I., Schägger H. (2005) Advantages and limitations of clear-native PAGE. Proteomics 5, 4338–4346 [DOI] [PubMed] [Google Scholar]
- 78. da Cunha E. S., Domingues C. C., de Paula E. (2011) Modified native electrophoresis protocol for the solubilization and separation of mitochondrial protein complexes. Anal. Biochem. 418, 158–160 [DOI] [PubMed] [Google Scholar]
- 79. Tomasetig L., Di Pancrazio F., Harris D. A., Mavelli I., Lippe G. (2002) Dimerization of F0F1ATP synthase from bovine heart is independent from the binding of the inhibitor protein IF1. Biochim. Biophys. Acta 1556, 133–141 [DOI] [PubMed] [Google Scholar]
- 80. Seelert H., Dencher N. A. (2011) ATP synthase superassemblies in animals and plants: two or more are better. Biochim. Biophys. Acta 1807, 1185–1197 [DOI] [PubMed] [Google Scholar]
- 81. Shinzawa-Itoh K., Aoyama H., Muramoto K., Terada H., Kurauchi T., Tadehara Y., Yamasaki A., Sugimura T., Kurono S., Tsujimoto K., Mizushima T., Yamashita E., Tsukihara T., Yoshikawa S. (2007) Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 26, 1713–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sharpley M. S., Shannon R. J., Draghi F., Hirst J. (2006) Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry 45, 241–248 [DOI] [PubMed] [Google Scholar]
- 83. Hayer-Hartl M., Schägger H., von Jagow G., Beyer K. (1992) Interactions of phospholipids with the mitochondrial cytochrome-c reductase studied by spin-label ESR and NMR spectroscopy. Eur. J. Biochem. 209, 423–430 [DOI] [PubMed] [Google Scholar]
- 84. Sánchez-Cenizo L., Formentini L., Aldea M., Ortega A. D., García-Huerta P., Sánchez-Aragó M., Cuezva J. M. (2010) Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. J. Biol. Chem. 285, 25308–25313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chan E. Y., McQuibban G. A. (2012) Phosphatidylserine decarboxylase 1 (Psd1) promotes mitochondrial membrane fusion by regulating the biophysical properties of the mitochondrial membrane and alternative topogenesis of mitochondrial genome maintenance protein 1 (Mgm1). J. Biol. Chem. 287, 40131–40139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Elmore S. P., Qian T., Grissom S. F., Lemasters J. J. (2001) The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 15, 2286–2287 [DOI] [PubMed] [Google Scholar]
- 87. Priault M., Salin B., Schaeffer J., Vallette F. M., di Rago J. P., Martinou J. C. (2005) Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ. 12, 1613–1621 [DOI] [PubMed] [Google Scholar]
- 88. Twig G., Elorza A., Molina A. J., Mohamed H., Wikstrom J. D., Walzer G., Stiles L., Haigh S. E., Katz S., Las G., Alroy J., Wu M., Py B. F., Yuan J., Deeney J. T., Corkey B. E., Shirihai O. S. (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nakada K., Inoue K., Ono T., Isobe K., Ogura A., Goto Y. I., Nonaka I., Hayashi J. I. (2001) Inter-mitochondrial complementation. Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat. Med. 7, 934–940 [DOI] [PubMed] [Google Scholar]
- 90. Chen H., McCaffery J. M., Chan D. C. (2007) Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130, 548–562 [DOI] [PubMed] [Google Scholar]
- 91. Chen H., Vermulst M., Wang Y. E., Chomyn A., Prolla T. A., McCaffery J. M., Chan D. C. (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Narendra D., Tanaka A., Suen D. F., Youle R. J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. De Vos K. J., Allan V. J., Grierson A. J., Sheetz M. P. (2005) Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Curr. Biol. 15, 678–683 [DOI] [PubMed] [Google Scholar]
- 94. Cullis P. R., de Kruijff B. (1978) The polymorphic phase behaviour of phosphatidylethanolamines of natural and synthetic origin. A 31P NMR study. Biochim. Biophys. Acta 513, 31–42 [DOI] [PubMed] [Google Scholar]
- 95. van den Brink-van der Laan E., Killian J. A., de Kruijff B. (2004) Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim. Biophys. Acta 1666, 275–288 [DOI] [PubMed] [Google Scholar]