

Background: Mechanisms linking cortical actin with adherens junctions (AJs) required for forming restrictive endothelial barrier remain unclear.
Results: We show that neural Wiskott-Aldrich syndrome protein (N-WASP) binds AJ constituent, p120-catenin, forms cortical actin, and links AJs with actin.
Conclusion: N-WASP stabilizes AJs and thereby maintains endothelial barrier function.
Significance: N-WASP represents a novel target for preventing leaky endothelial barrier syndrome.
Keywords: Adherens Junction, Cadherins, Endothelial Cell, Permeability, Vascular Biology, Adherens Junctions, Cortical Actin, Endothelial Barrier, Focal Adhesion Kinase (FAK), Neural Wiskott-Aldrich Syndrome Protein (N-WASP)
Abstract
Stable adherens junctions (AJs) are required for formation of restrictive endothelial barrier. Vascular endothelial cadherin from contiguous endothelial cells forms AJs, which are stabilized intracellularly by binding of p120-catenin and cortical actin. Mechanisms inducing cortical actin formation and enabling its linkage with p120-catenin remain enigmatic. We altered the function of neural Wiskott-Aldrich syndrome protein (N-WASP), which induces actin polymerization through actin-related protein 2/3 complex (Arp2/3), to address the role of N-WASP in regulating AJ stability and thereby endothelial permeability. We show that depletion of N-WASP in endothelial cells impaired AJ adhesion and favored the organization of actin from cortical actin to stress fibers, resulting thereby in formation of leaky endothelial barrier. Exposure of the N-WASP-depleted endothelial cell monolayer to the permeability-increasing mediator, thrombin, exaggerated AJ disruption and stress fiber formation, leading to an irreversible increase in endothelial permeability. We show that N-WASP binds p120-catenin through its verprolin cofilin acid (VCA) domain, induces cortical actin formation through Arp2, and links p120-catenin with cortical actin. The interaction of N-WASP with p120-catenin, actin, and Arp2 requires phosphorylation of N-WASP at the Tyr-256 residue by focal adhesion kinase. Expression of the VCA domain of N-WASP or phosphomimicking (Y256D)-N-WASP mutant in endothelial cells stabilizes AJs and facilitates barrier recovery after thrombin stimulation. Our study demonstrates that N-WASP, by mediating p120-catenin interaction with actin-polymerizing machinery, maintains AJs and mitigates disruption of endothelial barrier function by edemagenic agents, therefore representing a novel target for preventing leaky endothelial barrier syndrome.
Introduction
Stable adherens junctions (AJs)3 primarily restrict passage of plasma proteins and larger solutes across endothelial barrier (1, 2) and thereby play a vital role in maintaining tissue-fluid homeostasis (2, 3). Homotypic adhesion between vascular endothelial (VE)-cadherin from contiguous cells forms AJs (4, 5). Intracellularly, VE-cadherin associates with the β- and p120-catenins (5, 6). β-catenin in turn binds α-catenin. Additionally, the cortical actin, the actin ring near the plasma membrane, is required for stabilizing AJs (7–10). Edemagenic agonists such as thrombin rapidly induce actin reorganization from cortical actin into actin stress fibers, which induces AJ disruption and interendothelial gap formation, leading to increased endothelial permeability that can result in protein-rich tissue edema and increased transendothelial migration of inflammatory cells (2, 8–11). α-catenin performs the vital task of stabilizing AJ interaction with cortical actin by binding to actin (12). Intriguingly, findings demonstrate that p120-catenin is a critical regulator of VE-cadherin cell surface retention in endothelial cells (13–17). Depletion of p120-catenin or post-translational modification of VE-cadherin in endothelial cells interfered with its association with VE-cadherin, thereby weakening AJs (13, 17). However, p120-catenin does not bind α-catenin or actin, indicating that additional mechanisms may be required for mediating p120-catenin stabilization of AJs.
The actin-related protein 2/3 (Arp2/3) complex generates actin filament-driven protrusive force (18, 19), which may stabilize AJs. Interestingly, Arp2 was shown to interact with E-cadherin (20), indicating that protein regulating Arp2/3 complex function may contribute to AJ stability. Neural Wiskott-Aldrich syndrome protein (N-WASP) belongs to the WASP family of actin-polymerizing proteins that regulate actin polymerization by activating Arp2/3 complex (21, 22). N-WASP contains an NH2-terminal WASP homology domain (WH), a central GTPase binding domain (GBD), and a C terminus verprolin central acidic (VCA) domain (22). In resting cells, N-WASP is autoinhibited by an intramolecular interaction between the VCA and GBD domains. Several mechanisms have been implicated in facilitating the unfolding of the VCA domain from GBD domain, including WASP phosphorylation by tyrosine kinases and interaction with phosphatidylinositol (3,4,5)-triphosphate or Cdc42 GTPases (21, 23). Focal adhesion kinase (FAK), a nonreceptor tyrosine kinase, was shown to phosphorylate N-WASP at Tyr-256, leading to its activation (24). FAK and Cdc42 are known to maintain AJ function in part by interacting with p120-catenin (25–27). It remains unknown whether N-WASP regulates cortical actin formation during alteration of AJ function and hence contributes to attainment of basal endothelial barrier function.
In this study, we depleted N-WASP or used N-WASP mutant that cannot be phosphorylated at Tyr-256 (Y256F-N-WASP) to test the hypothesis that activated N-WASP stabilizes AJs and thereby maintains endothelial barrier function. Intriguingly, we find that N-WASP, besides inducing cortical actin formation, binds p120-catenin, thereby linking p120-catenin with cortical actin, which strengthens AJs. We further show that FAK-mediated N-WASP phosphorylation is the critical step in inducing N-WASP interaction with p120-catenin.
EXPERIMENTAL PROCEDURES
Materials
Human pulmonary arterial endothelial cells (HPAEC) and endothelial growth medium were obtained from Lonza (Walkersville, MD). COS7 cells were obtained from ATCC (Manassas, VA). Human α-thrombin was obtained from Enzyme Research Laboratories (South Bend, IN). The Nucleofector human coronary arterial endothelial cells kit and electroporation system were from Amaxa (Gaithersburg, MD). Lipofectamine 2000 transfection reagent was from Invitrogen. N-WASP, Arp2, actin, FAK, phosphotyrosine, p120-catenin, horseradish peroxidase-conjugated anti-mouse immunoglobulin G (IgG) antibodies (Abs), and protein A/G beads were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). GFP and anti-phospho-Tyr-256-N-WASP antibodies were purchased from Roche Applied Science (Mannheim, Germany) and Abcam (Cambridge, MA), respectively. Alexa Fluor-labeled secondary antibodies, 4′, 6′-diamidino-2-phenylindole (DAPI), rhodamine phalloidin, and ProLong Gold antifade were from Molecular Probes (Eugene, OR). N-WASP, FAK, and Arp2 SMARTpool siRNA were purchased from Dharmacon (Lafayette, CO). p120-catenin siRNA was custom-ordered from Dharmacon (14). Scrambled siRNA with no sequence homology to human genome were purchased from Ambion (Applied Biosystems). N-WASP mutants Y256D, Y256F, CA, and VCA-N-WASP were a gift from Dr. Takenawa (Department of Biochemistry, University of Tokyo, Tokyo, Japan).
Cell Culture
HPAEC and COS7 cells were cultured as described previously (27, 28). Briefly, HPAEC were cultured in 10% fetal bovine serum containing endothelial growth medium 2, whereas COS7 cells were cultured in 10% fetal bovine serum containing DMEM. Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air until confluent. Cells from each primary flask were detached with 0.025% trypsin/EDTA and plated on either 60-mm dishes or coverslips for further studies. In all experiments, HPAEC between passages 6 and 8 were used.
Cell Transfection
Indicated siRNA (2.4 μg) or cDNA (3.5 μg) were transfected in HPAEC using Amaxa electroporation system as described (28). In parallel, cells were transfected with control siRNA that has no sequence homology to any known mammalian gene. Cells were used after 72 h of transfection when there was clear evidence of protein suppression. For transduction of indicated cDNA, 3.5 μg of cDNA was electroporated in endothelial cells using Amaxa electroporation system as described (28, 29). COS7 cells were transfected with the indicated cDNA using Lipofectamine 2000 as described (30).
Transendothelial Electrical Resistance
The time course of endothelial cell retraction in real time, a measurement of increased endothelial permeability via the paracellular pathway, was measured as described (29, 31).
Immunoprecipitation
Lysates were immunoprecipitated using previously published protocol (32). Briefly, HPAEC or COS7 cells lysed in radioimmunoprecipitation buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 0.25 mm EDTA, pH 8.0, 1% deoxycholic acid, 0.5% Nonidet P-40, 0.1% SDS, 1 mm NaF, 1 mm sodium orthovanadate, 1 mm phenylmethylsulfonyl fluoride, and 2 μg/ml each of leupeptin, aprotinin, and pepstatin A) were incubated with the indicated antibodies overnight at 4 °C followed by the addition of protein A/G-agarose beads for 4 h at 4 °C. The beads were then collected by centrifugation and washed three times using detergent-free radioimmunoprecipitation buffer.
Immunofluorescence
Cells stimulated with thrombin for the specified period of time were fixed with 2% paraformaldehyde for 20 min. Cells were then permeabilized and immunostained using the appropriate primary antibodies followed by incubation with Alexa Fluor-labeled secondary antibodies, phalloidin, and DAPI for staining actin filaments and nucleus, respectively.
Statistical Analysis
Comparisons between experimental groups were made by one-way analysis of variance and post hoc test. Interendothelial gap area and co-localization between proteins were quantified using Image J software (National Institutes of Health). Differences in mean values were considered significant at p < 0.05.
RESULTS
Depletion of N-WASP Impairs AJ Adhesion and Cortical Actin Formation, Leading to Persistent Increase in Endothelial Permeability by Thrombin
To address the role of N-WASP in regulating AJ stability and endothelial barrier function, we first assessed whether N-WASP is required for formation of stable AJs. Endothelial cells (EC) were transfected with scrambled or N-WASP siRNA, after which cells were fixed at 24, 48, and 72 h followed by immunostaining with anti-p120-catenin antibody to assess AJs, whereas phalloidin was used to determine actin organization. At 24 h, EC transfected with scrambled siRNA (referred to as control cells) or N-WASP siRNA showed patchy cell surface p120-catenin staining, less defined cortical actin ring, and visible interendothelial gap area, but these responses were more pronounced in N-WASP-depleted cells (Fig. 1, A and B). In control cells, both p120-catenin and actin staining began to organize in a discrete pattern at 48 h such that at 72 h, p120-catenin was uniformly expressed at cell surface with juxtaposed cortical actin, thereby sealing interendothelial gaps (Fig. 1, A and B). However, time-dependent depletion of N-WASP expression further impaired AJ adhesion and cortical actin formation, leading to significant increase in the interendothelial gap area (Fig. 1, A and B). To corroborate these findings, we also determined the effect of N-WASP depletion at 24 and 72 h on the alteration of the endothelial barrier by determining transendothelial electrical resistance (TER), which measures AJ adhesion in real time (29, 31). We show that TER remained significantly lower at each time point in N-WASP-depleted EC, recapitulating the above findings (Fig. 1C). Thus, N-WASP plays a key role in stabilizing AJs and thereby in forming restrictive endothelial barrier.
FIGURE 1.

N-WASP promotes the formation of AJs. A, B, D, and E, HPAEC transfected with N-WASP (siN-WASP) or scrambled (siSc) siRNA were fixed at the indicated time points after stimulation without (A and B) or with thrombin (D and E). The cells were stained with rhodamine phalloidin or anti-p120-catenin antibody followed by Alexa Fluor-labeled secondary antibodies and visualized by confocal microscope. Scale bars, 10 μm. Inset, immunoblot shows the knockdown of N-WASP at indicated times. Actin was used as loading control. Efficiency of knockdown was quantified using densitometric analysis. B and E, the plot shows mean ± S.D. of interendothelial gaps in siSc and siN-WASP transfected cells at the indicated time points from three individual experiments. * indicates significant increase in the gap area compared to scrambled siRNA transfected cells (p < 0.05); # indicates significant increase in the gap area in monolayers transfected with siN-WASP when compared with monolayers transfected with siSc at each time point after thrombin stimulation (p < 0.05). C and F, HPAEC plated on gold electrodes were transfected with siSc or siN-WASP, and changes in TER were determined in real time at the indicated time points. Data represent mean ± S.D. from three experiments performed in triplicates. * indicates significance from siSc transfected monolayer (p < 0.05). Ω indicates ohms.
Permeability-increasing mediators such as thrombin rapidly destabilize AJs and induce actin stress fiber formation (2, 8–11); this is followed by a lag phase of 1–2 h, during which AJs reanneal and stress fibers reorganize into the cortical actin-restoring basal endothelial barrier (2). Because the above findings showed that N-WASP depletion impaired AJ stability, we therefore stimulated the N-WASP-depleted monolayer with thrombin to assess its role in regulating reannealing of AJs and restoration of basal endothelial barrier. We found that thrombin exaggerated the disruption of AJs and actin stress fiber formation in N-WASP-depleted monolayer (Fig. 1D), resulting in the formation of numerous intercellular gaps, which persisted even after 2 h (Fig. 1, D–F). As expected, AJs reannealed and actin reorganized into cortical actin within 2 h in control cells, thereby resealing the intercellular gaps (Fig. 1, D–F). These findings demonstrate that N-WASP is also required for inducing reannealing of AJs and cortical actin formation promoting the recovery of basal endothelial barrier function following the increase in endothelial permeability by thrombin.
N-WASP Interacts with p120-Catenin
N-WASP interacts with Arp2/3 complex and actin to mediate actin polymerization (21, 22). As the above findings show that knockdown of N-WASP impaired p120-catenin localization at the surface and induced actin stress fiber formation, we addressed the possibility that N-WASP interacts with p120-catenin during alteration in endothelial barrier function and thereby serves to link p120-catenin with cortical actin. Endothelial cell lysates were immunoprecipitated with anti-N-WASP antibody followed by immunoblotting with anti-p120-catenin, anti-Arp2, or anti-actin antibodies to assess their interactions. We found that N-WASP interacted with p120-catenin basally, but crucially, the interaction increased to ∼5-fold at 30 min after thrombin challenge and persisted to 2-fold at 2 h (Fig. 2, right). As expected, N-WASP also pulled down actin and Arp2 under basal conditions (Fig. 2, left). Thrombin further increased Arp2 and actin interaction with N-WASP in association with p120-catenin interaction with N-WASP (Fig. 2, right). These findings suggest that N-WASP integrates actin-polymerizing machinery with p120-catenin to stabilize AJs.
FIGURE 2.
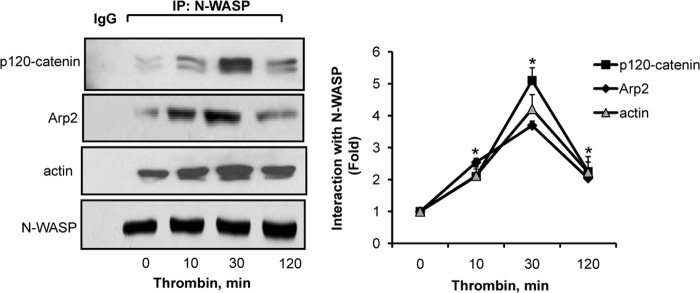
N-WASP interacts with p120-catenin, Arp2, and actin. Left, HPAEC were stimulated with 50 nm thrombin for the indicated time points, after which cell lysates were immunoprecipitated (IP) using N-WASP antibody followed by immunoblotting with the indicated antibodies. Right, the plot shows densitometric analysis of N-WASP interaction with p120-catenin, Arp2, and actin basally and following thrombin stimulation. Data represent mean ± S.D. from three individual experiments. * indicates significance from unstimulated cells (p < 0.05).
Interaction of N-WASP with p120-Catenin Does Not Require Arp2
Arp2 could mediate N-WASP interaction with p120-catenin and actin (33). Thus, we immunoprecipitated N-WASP from Arp2-depleted monolayer (Fig. 3, inset) to assess the role of Arp2 in mediating N-WASP interaction with p120-catenin. Although depletion of Arp2 by 70% had no effect on interaction between N-WASP and p120-catenin (Fig. 3A), it markedly reduced N-WASP association with actin (Fig. 3A). On the other hand, similar depletion of N-WASP (Fig. 3B, inset) significantly reduced the interaction of p120-catenin with actin and Arp2 (Fig. 3B), thereby identifying N-WASP as a key molecule required for mediating complex formation between p120-catenin, actin, and Arp2.
FIGURE 3.

N-WASP interaction with p120-catenin occurs independent of Arp2. HPAEC transfected with Arp2 (siArp2), N-WASP (siN-WASP), or scrambled (siSc) siRNA were stimulated with thrombin for 30 min, after which the cell lysates were immunoprecipitated using N-WASP or p120-catenin antibodies and immunoblotted with p120-catenin, actin, Arp2, or N-WASP antibodies. A, top panels, representative immunoblot showing the effect of Arp2 knockdown (inset) on N-WASP interaction with p120-catenin and actin. B, top panels, representative immunoblot showing the effect of N-WASP knockdown (inset) on interaction of p120-catenin with Arp2 and actin. A and B, bottom panels, the plot shows mean ± S.D. of -fold increase in interaction between indicated proteins from three individual experiments. * indicates significance from unstimulated siSc transfected cells (p < 0.05). # indicates significance from the scrambled siRNA transfected cells at the respective time point (p < 0.05).
Thrombin Induces N-WASP Phosphorylation in a FAK-dependent Manner That Restores Barrier Function
Phosphorylation of N-WASP by tyrosine kinases such as FAK has been shown to induce its activation (24). We therefore addressed whether thrombin induces the phosphorylation of N-WASP in a FAK-dependent manner. N-WASP was immunoprecipitated from EC following stimulation with thrombin for the indicated times followed by immunoblotting with anti-phosphotyrosine antibodies to determine N-WASP phosphorylation. We observed that thrombin increased N-WASP phosphorylation by 2-fold within 10 min, which increased to 4-fold at 30 min and remained elevated at 2-fold until 120 min (Fig. 4A). Similar results were obtained using Tyr-256 residue-specific N-WASP phospho-antibody (Fig. 4B). We observed that depletion of FAK markedly decreased Tyr-256 phosphorylation after thrombin treatment (Fig. 4C), indicating that FAK is required for inducing N-WASP phosphorylation.
FIGURE 4.

FAK induces N-WASP phosphorylation that restores endothelial barrier function. A and B, time course of tyrosine phosphorylation of N-WASP following thrombin stimulation. HPAEC were stimulated with 50 nm thrombin for the indicated time points, after which lysates were either immunoprecipitated with N-WASP antibody and immunoblotted with total phospho-Tyr (pTyr) antibodies (A) or phospho-Tyr-256 (pTyr256) N-WASP antibody (B). The plot in A and B shows mean ± S.D. of increase in N-WASP phosphorylation from three individual experiments. * indicates significance from control unstimulated condition (p < 0.05). C, effect of FAK knockdown on N-WASP phosphorylation. HPAEC transfected with scrambled (siSc) or FAK siRNA (siFAK) were stimulated with 50 nm thrombin for 30 min. Cell lysates were electrophoresed and immunoblotted with FAK or N-WASP phospho-Tyr-256 antibodies. The plot represents mean ± S.D. of -fold increase in phosphorylation from three individual experiments. * indicates significance from scrambled siRNA transfected cells after thrombin treatment (p < 0.05). D, effect of N-WASP phosphorylation on endothelial barrier function. HPAEC plated on gold electrodes were transfected either with phosphospecific N-WASP mutants Y256D and Y256F or with GFP alone. Cells were stimulated with 50 nm thrombin, and changes in TER were determined. Data represent mean ± S.D. from three individual experiments performed in triplicates. * indicates significance from cells expressing Y256D mutant or control vector (GFP) (p < 0.05).
To determine the causal role of Tyr-256 phosphorylation of N-WASP in regulating endothelial barrier recovery, we measured the TER of endothelial monolayer transducing control vector (GFP), phosphodefective (GFP-Y256F), or phosphomimicking (GFP-Y256D) N-WASP mutants. We observed that transduction of Y256F-N-WASP mutant increased endothelial permeability under basal condition because these cells showed lower TER (Fig. 4D). Thrombin led to irreversible increase in endothelial permeability in Y256F-N-WASP transducing mutant monolayer. In contrast, Y256D-N-WASP transducing cells showed enhanced barrier function (high TER) and recovered faster after thrombin challenge (Fig. 4D).
Depletion of p120-Catenin Does Not Affect N-WASP Phosphorylation and Interaction with Arp2 and Actin
To address the possibility that disruption of AJs per se induces N-WASP activation, we immunoprecipitated N-WASP from p120-catenin-depleted monolayers following thrombin stimulation. We observed that thrombin induced N-WASP phosphorylation to a similar level in p120-catenin-depleted cells as in control cells (Fig. 5). Furthermore, p120-catenin depletion had no effect on N-WASP association with Arp2 and actin (Fig. 5).
FIGURE 5.

p120-catenin does not mediate N-WASP activation. HPAEC transfected with scrambled (siSc) siRNA or p120-catenin siRNA (sip120-catenin) were stimulated with thrombin for 30 min, after which lysates were immunoprecipitated with N-WASP antibody followed by immunoblotting with the indicated antibodies to determine N-WASP activity. The inset shows an immunoblot of p120-catenin depletion in endothelial cells. Plot shows mean ± S.D. of fold increase in interaction between indicated proteins from three individual experiments. * indicates significance from control unstimulated monolayer within each group (p < 0.05). pTyr, total phospho-Tyr antibody; p-N-WASP, N-WASP antibody.
N-WASP Interacts with p120-Catenin through VCA Domain
N-WASP interacts with Arp2/3 complex and actin through the VCA domain (22). Also, binding of the VCA domain with GBD of N-WASP, which contains the Tyr-256 residue, limits its activity (22). Thus, we determined whether N-WASP interacts with p120-catenin through the VCA domain and whether the interaction requires N-WASP phosphorylation. Because COS7 cells express N-WASP at low levels (34), we transfected these cells with either control vector (GFP) or various GFP-tagged N-WASP mutants (Fig. 6, left), and cell lysates were then immunoprecipitated using anti-GFP antibody followed by immunoblotting with p120-catenin and Arp2. We found that Y256D-N-WASP and GFP-VCA mutants efficiently interacted with p120-catenin and Arp2 (Fig. 6, right). However, p120-catenin interaction was barely detectable in lysates transfected with Y256F-N-WASP or CA mutants (Fig. 6, right). These findings demonstrate that the VCA domain of N-WASP interacts with p120-catenin.
FIGURE 6.

VCA domain of N-WASP interacts with p120-catenin. Left, COS7 cells were transfected with the indicated constructs. Forty-eight h later, the lysates were immunoprecipitated with anti-GFP antibodies followed by immunoblotting with the indicated antibodies to assess interaction with p120-catenin or Arp2. Right, the top panel shows representative immunoblots, whereas the bottom panel shows the mean ± S.D. of densitometric analysis of interaction between WASP, Arp2, and p120-catenin from three individual experiments. * indicates significance when compared with Y256F, GFP, GFP-N-WASP, and CA (p < 0.05).
N-WASP VCA Domain Induces AJ Formation and Organizes Cortical Actin
We transfected endothelial cells with GFP-VCA mutant and assessed AJ formation and actin reorganization to address the functional relevance of N-WASP-VCA domain interaction with p120-catenin (Fig. 7). In parallel, we used EC expressing GFP-CA mutant, which cannot bind p120-catenin. Confocal imaging analysis showed that VCA mutant co-localized with p120-catenin and induced AJ formation under basal conditions and after thrombin stimulation (Fig. 7, A and B). However, the CA mutant did not co-localize with p120-catenin and resulted in patchy p120-catenin staining that became distorted further following thrombin challenge (Fig. 7, A and B). VCA mutant transducing cells also showed cortical actin ring basally, which did not alter after thrombin stimulation (Fig. 7C). In contrast, CA mutant transducing cells showed actin stress fibers basally, which increased further upon thrombin treatment (Fig. 7C).
FIGURE 7.

VCA domain of N-WASP co-localizes with p120-catenin and induces cortical actin formation. A–C, HPAEC transfected with the GFP-VCA or GFP-CA mutants were stimulated with 50 nm thrombin for 30 min, after which cells were fixed and stained with rhodamine-phalloidin and p120-catenin antibody to determine N-WASP localization with p120-catenin (A and B) and actin reorganization (C) by confocal imaging. The region of interest is outlined in white, and the inset highlights the co-localization. Scale bars, 10 μm. Data represent mean ± S.D. * indicates significance when compared with GFP-CA (p < 0.05).
DISCUSSION
We have identified p120-catenin as a novel effector of N-WASP in endothelial cells. We also show that N-WASP induces cortical actin formation by Arp2/3 complex. Thus, N-WASP links p120-catenin with cortical actin, enabling AJ stabilization required for formation of restrictive endothelial barrier. Additionally, N-WASP promotes reannealing of AJs and thereby recovery of endothelial barrier formation following the increase in endothelial permeability by thrombin. We further showed that FAK phosphorylation of N-WASP at Tyr-256 was required for attainment of stable AJs.
Homotypic adhesion between VE-cadherin from adjacent endothelial cells constitutes AJs, which form the primary barrier in endothelial cells (4). Stable AJs require interaction of VE-cadherin with catenins as well as the cortical actin ring (5–10). β-catenin, through α-catenin, is believed to induce VE-cadherin interaction with actin (5, 6). However, α-catenin fails to interact with actin filaments and the cadherin-β-catenin complex simultaneously, even in the presence of the actin-binding proteins vinculin and α-actinin (35, 36). Thus, α-catenin association with actin and VE-cadherin-β-catenin complex appear to be a mutually exclusive and dynamic events rather than static, as was previously assumed (35, 36). An important question therefore has been whether this transient association of α-catenin with actin is sufficient to maintain restrictive AJs and thereby the endothelial barrier. We showed that depletion of N-WASP impaired AJ adhesion and favored actin organization into stress fiber formation, leading to formation of a leaky endothelial barrier. Thrombin exacerbated AJ disruption and actin stress fiber formation in N-WASP-depleted EC, leading to an irreversible increase in endothelial permeability. These findings identify N-WASP as a key cellular mechanism that ensures AJ stability and thereby helps to preserve restrictive endothelial barrier.
Because N-WASP activates Arp2/3 complex to induce actin polymerization, we focused on the possibility of whether N-WASP facilitates actin linkage with AJs and the mechanism as to how this might occur. As expected, we showed that N-WASP formed complexes with Arp2 and actin. Interestingly, we showed that these complexes also contained the AJ component, p120-catenin, basally and that the interaction markedly increased following disruption of AJs with thrombin. p120-catenin has emerged as a master regulator of VE-cadherin stability as it prevents VE-cadherin endocytosis by clathrin-coated adaptor AP1 (17, 37, 39) and restricts RhoA activity (40, 41). A recent study showed that in fibroblasts, p120-catenin also induces the activation of cortactin, a well known partner of N-WASP (42), to induce lamellipodia formation for their migratory events (33). We ruled out the role of p120-catenin as the activator of N-WASP because knockdown of p120-catenin had no effect on N-WASP activity or the interaction of N-WASP with Arp2 and actin. Knockdown of Arp2 also did not interfere with interaction of N-WASP with p120-catenin. Thus, we conclude that the N-WASP function in endothelial cells is to bind p120-catenin and induce cortical actin formation by Arp2/3 complex. Hence, N-WASP induces p120-catenin anchorage with actin, thereby stabilizing AJs in naive monolayer and facilitating their stability following disruption of barrier function by thrombin.
N-WASP activity is regulated by an intramolecular interaction between the VCA and GBD domains. In this regard, Cdc42 and phosphatidylinositol (3,4,5)-triphosphate binding with N-WASP as well as the tyrosine phosphorylation of N-WASP has been shown to induce N-WASP activity (23, 24). We previously showed that both Cdc42 and FAK maintain AJs and restore basal endothelial permeability following challenge with edemagenic agents (25, 26). It is possible that N-WASP functions as a node downstream of Cdc42 and FAK to maintain endothelial barrier. We showed that thrombin induces N-WASP phosphorylation at Tyr-256 residue in a FAK-dependent manner, consistent with previous studies (24). The phosphorylation of N-WASP correlated with reformation of cortical actin and p120-catenin interaction. Expression of phosphodefective N-WASP mutant impaired its interaction with p120-catenin and Arp2, leading to disruption of AJs and loss of endothelial barrier function. On the other hand, overexpression of phosphomimicking mutant of N-WASP enhanced basal barrier function and facilitated reannealing of AJs and barrier restoration. These findings suggest that FAK-mediated N-WASP phosphorylation is a critical event in maintaining AJs and in restoration of endothelial barrier.
The VCA domain of N-WASP serves as a constitutively active N-WASP because it can freely bind actin and Arp2/3 complex (22). The CA domain acts as a dominant negative N-WASP because it cannot bind actin. Using COS7 cells, which minimally express N-WASP, we showed that p120-catenin interacted with the VCA domain, whereas it interacted barely with CA domain. We showed that phosphomimicking N-WASP also binds p120-catenin and Arp2. Because the Tyr-256 residue lies within the GBD domain, we interpret from these findings that the verprolin domain in N-WASP primarily interacts with p120-catenin and that phosphorylation serves as a means to unmask the verprolin domain from GBD domain. Actin also binds verprolin region (22), raising the possibility that p120-catenin and actin may share the same verprolin domain, thereby inducing AJ anchorage with actin. Consistently, we showed that expression of the VCA domain in endothelial cells induced the formation of cortical actin and AJs and markedly inhibited interendothelial gap formation after thrombin challenge.
Thrombin, by binding to its receptor, protease-activating receptor 1 (PAR1), induces RhoA activity, which increases endothelial permeability by inducing stress fiber formation (2, 8–11). AJ-localized p120-catenin limits RhoA activity by inducing the activity of p190RhoGAP, a GTPase that specifically inactivates RhoA (38, 40). Our results suggest that N-WASP acts as a critical mechanism for stabilizing p120-catenin at AJs and for suppressing actin stress fiber formation and hence RhoA activity.
In conclusion, our findings have identified a novel role of N-WASP in mediating p120-catenin interaction with actin-polymerizing machinery Arp2/3 complex and thereby cortical actin to stabilize AJs and induce AJ reformation following disruption of endothelial barrier function by edemagenic agents such as thrombin. Loss of endothelial barrier function leads to several pathologic disorders such as lung injury and pulmonary hypertension. Therefore, our results suggest that N-WASP represents a novel target for preventing diseases associated with leaky blood vessels.
Acknowledgments
We acknowledge Dr. Asrar B. Malik (University of Illinois at Chicago, IL) for constructive criticisms and thank Dr. Jun-Lin Guan (University of Michigan, Ann Arbor, MI) and Dr. Takenawa (University of Tokyo, Tokyo, Japan) for the N-WASP constructs.
This work was supported, in whole or in part, by National Institutes of Health Grants HL71794, PO1 HL060678, and HL84153 (to D. M.).
This article was selected as a Paper of the Week.
- AJ
- adherens junction
- VE-cadherin
- vascular endothelial cadherin
- N-WASP
- neural Wiskott-Aldrich syndrome protein
- Arp2/3 complex
- actin-related protein 2/3 complex
- Arp2
- actin-related protein 2
- GBD
- GTPase binding domain
- VCA
- verprolin central acidic
- CA
- central acidic
- FAK
- focal adhesion kinase
- EC
- endothelial cells
- HPAEC
- human pulmonary arterial endothelial cells
- TER
- transendothelial electrical resistance.
REFERENCES
- 1. Bazzoni G., Dejana E. (2004) Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol. Rev. 84, 869–901 [DOI] [PubMed] [Google Scholar]
- 2. Mehta D., Malik A. B. (2006) Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86, 279–367 [DOI] [PubMed] [Google Scholar]
- 3. Liu K. D., Matthay M. A. (2008) Advances in critical care for the nephrologist: acute lung injury/ARDS. Clin. J. Am. Soc. Nephrol. 3, 578–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taveau J. C., Dubois M., Le Bihan O., Trépout S., Almagro S., Hewat E., Durmort C., Heyraud S., Gulino-Debrac D., Lambert O. (2008) Structure of artificial and natural VE-cadherin-based adherens junctions. Biochem. Soc. Trans. 36, 189–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vincent P. A., Xiao K., Buckley K. M., Kowalczyk A. P. (2004) VE-cadherin: adhesion at arm's length. Am. J. Physiol. Cell Physiol. 286, C987–997 [DOI] [PubMed] [Google Scholar]
- 6. Niessen C. M., Gottardi C. J. (2008) Molecular components of the adherens junction. Biochim. Biophys. Acta 1778, 562–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Green K. J., Getsios S., Troyanovsky S., Godsel L. M. (2010) Intercellular junction assembly, dynamics, and homeostasis. Cold Spring Harb. Perspect. Biol. 2, a000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dudek S. M., Garcia J. G. (2001) Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 91, 1487–1500 [DOI] [PubMed] [Google Scholar]
- 9. Fukuhara S., Sakurai A., Sano H., Yamagishi A., Somekawa S., Takakura N., Saito Y., Kangawa K., Mochizuki N. (2005) Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol. Cell Biol. 25, 136–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stelzner T. J., Weil J. V., O'Brien R. F. (1989) Role of cyclic adenosine monophosphate in the induction of endothelial barrier properties. J. Cell Physiol. 139, 157–166 [DOI] [PubMed] [Google Scholar]
- 11. Phillips P. G. (1994) Thrombin-induced alterations in endothelial cell cytoarchitectural and functional properties. Semin. Thromb. Hemost. 20, 417–425 [DOI] [PubMed] [Google Scholar]
- 12. Kobielak A., Fuchs E. (2004) α-Catenin: at the junction of intercellular adhesion and actin dynamics. Nat. Rev. Mol. Cell Biol. 5, 614–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iyer S., Ferreri D. M., DeCocco N. C., Minnear F. L., Vincent P. A. (2004) VE-cadherin-p120 interaction is required for maintenance of endothelial barrier function. Am. J. Physiol. Lung Cell Mol. Physiol. 286, L1143–L1153 [DOI] [PubMed] [Google Scholar]
- 14. Xiao K., Allison D. F., Kottke M. D., Summers S., Sorescu G. P., Faundez V., Kowalczyk A. P. (2003) Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. J. Biol. Chem. 278, 19199–19208 [DOI] [PubMed] [Google Scholar]
- 15. Chiasson C. M., Wittich K. B., Vincent P. A., Faundez V., Kowalczyk A. P. (2009) p120-catenin inhibits VE-cadherin internalization through a Rho-independent mechanism. Mol. Biol. Cell 20, 1970–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peifer M., Yap A. S. (2003) Traffic control: p120-catenin acts as a gatekeeper to control the fate of classical cadherins in mammalian cells. J. Cell Biol. 163, 437–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ireton R. C., Davis M. A., van Hengel J., Mariner D. J., Barnes K., Thoreson M. A., Anastasiadis P. Z., Matrisian L., Bundy L. M., Sealy L., Gilbert B., van Roy F., Reynolds A. B. (2002) A novel role for p120 catenin in E-cadherin function. J. Cell Biol. 159, 465–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Verma S., Shewan A. M., Scott J. A., Helwani F. M., den Elzen N. R., Miki H., Takenawa T., Yap A. S. (2004) Arp2/3 activity is necessary for efficient formation of E-cadherin adhesive contacts. J. Biol. Chem. 279, 34062–34070 [DOI] [PubMed] [Google Scholar]
- 19. Goley E. D., Welch M. D. (2006) The ARP2/3 complex: an actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 7, 713–726 [DOI] [PubMed] [Google Scholar]
- 20. Kovacs E. M., Goodwin M., Ali R. G., Paterson A. D., Yap A. S. (2002) Cadherin-directed actin assembly: E-cadherin physically associates with the Arp2/3 complex to direct actin assembly in nascent adhesive contacts. Curr. Biol. 12, 379–382 [DOI] [PubMed] [Google Scholar]
- 21. Takenawa T., Suetsugu S. (2007) The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 8, 37–48 [DOI] [PubMed] [Google Scholar]
- 22. Padrick S. B., Rosen M. K. (2010) Physical mechanisms of signal integration by WASP family proteins. Annu. Rev. Biochem. 79, 707–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suetsugu S., Hattori M., Miki H., Tezuka T., Yamamoto T., Mikoshiba K., Takenawa T. (2002) Sustained activation of N-WASP through phosphorylation is essential for neurite extension. Dev. Cell 3, 645–658 [DOI] [PubMed] [Google Scholar]
- 24. Wu X., Suetsugu S., Cooper L. A., Takenawa T., Guan J. L. (2004) Focal adhesion kinase regulation of N-WASP subcellular localization and function. J. Biol. Chem. 279, 9565–9576 [DOI] [PubMed] [Google Scholar]
- 25. Knezevic N., Tauseef M., Thennes T., Mehta D. (2009) The G protein βγ subunit mediates reannealing of adherens junctions to reverse endothelial permeability increase by thrombin. J. Exp. Med. 206, 2761–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramchandran R., Mehta D., Vogel S. M., Mirza M. K., Kouklis P., Malik A. B. (2008) Critical role of Cdc42 in mediating endothelial barrier protection in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 295, L363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holinstat M., Knezevic N., Broman M., Samarel A. M., Malik A. B., Mehta D. (2006) Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J. Biol. Chem. 281, 2296–2305 [DOI] [PubMed] [Google Scholar]
- 28. Knezevic N., Roy A., Timblin B., Konstantoulaki M., Sharma T., Malik A. B., Mehta D. (2007) GDI-1 phosphorylation switch at serine 96 induces RhoA activation and increased endothelial permeability. Mol. Cell. Biol. 27, 6323–6333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Singh I., Knezevic N., Ahmmed G. U., Kini V., Malik A. B., Mehta D. (2007) Gαq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J. Biol. Chem. 282, 7833–7843 [DOI] [PubMed] [Google Scholar]
- 30. Kini V., Chavez A., Mehta D. (2010) A new role for PTEN in regulating transient receptor potential canonical channel 6-mediated Ca2+ entry, endothelial permeability, and angiogenesis. J. Biol. Chem. 285, 33082–33091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tauseef M., Kini V., Knezevic N., Brannan M., Ramchandaran R., Fyrst H., Saba J., Vogel S. M., Malik A. B., Mehta D. (2008) Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ. Res. 103, 1164–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mehta D., Ahmmed G. U., Paria B. C., Holinstat M., Voyno-Yasenetskaya T., Tiruppathi C., Minshall R. D., Malik A. B. (2003) RhoA interaction with inositol 1,4,5-trisphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry: role in signaling increased endothelial permeability. J. Biol. Chem. 278, 33492–33500 [DOI] [PubMed] [Google Scholar]
- 33. Boguslavsky S., Grosheva I., Landau E., Shtutman M., Cohen M., Arnold K., Feinstein E., Geiger B., Bershadsky A. (2007) p120 catenin regulates lamellipodial dynamics and cell adhesion in cooperation with cortactin. Proc. Natl. Acad. Sci. U.S.A. 104, 10882–10887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miki H., Miura K., Takenawa T. (1996) N-WASP, a novel actin-depolymerizing protein, regulates the cortical cytoskeletal rearrangement in a PIP2-dependent manner downstream of tyrosine kinases. EMBO J. 15, 5326–5335 [PMC free article] [PubMed] [Google Scholar]
- 35. Yamada S., Pokutta S., Drees F., Weis W. I., Nelson W. J. (2005) Deconstructing the cadherin-catenin-actin complex. Cell 123, 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Drees F., Pokutta S., Yamada S., Nelson W. J., Weis W. I. (2005) α-Catenin is a molecular switch that binds E-cadherin-β-catenin and regulates actin-filament assembly. Cell 123, 903–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pollitt A. Y., Insall R. H. J. (2009) WASP and SCAR/WAVE proteins: the drivers of actin assembly. J. Cell Sci. 122, 2575–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wildenberg G. A., Dohn M. R., Carnahan R. H., Davis M. A., Lobdell N. A., Settleman J., Reynolds A. B. (2006) p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 127, 1027–1039 [DOI] [PubMed] [Google Scholar]
- 39. Aberle H., Schwartz H., Kemler R. (1996) Cadherin-catenin complex: protein interactions and their implications for cadherin function. J. Cell Biochem. 61, 514–523 [DOI] [PubMed] [Google Scholar]
- 40. Anastasiadis P. Z., Moon S. Y., Thoreson M. A., Mariner D. J., Crawford H. C., Zheng Y., Reynolds A. B. (2000) Inhibition of RhoA by p120 catenin. Nat. Cell Biol. 2, 637–644 [DOI] [PubMed] [Google Scholar]
- 41. Yanagisawa M., Huveldt D., Kreinest P., Lohse C. M., Cheville J. C., Parker A. S., Copland J. A., Anastasiadis P. Z. (2008) A p120 catenin isoform switch affects Rho activity, induces tumor cell invasion, and predicts metastatic disease. J. Biol. Chem. 283, 18344–18354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kowalski J. R., Egile C., Gil S., Snapper S. B., Li R., Thomas S. M. (2005) Cortactin regulates cell migration through activation of N-WASP. J. Cell Sci. 118, 79–87 [DOI] [PubMed] [Google Scholar]