

Background: α-Hemoglobin stabilizing protein (AHSP) modifies the redox properties of bound α-subunits.
Results: Isolated hemoglobin subunits exhibit significantly different redox properties compared with HbA. A significant decrease in the reduction potential of α-subunits bound to AHSP results in preferential binding of ferric α.
Conclusion: AHSP·α-subunit complexes do not participate in ferric-ferryl heme redox cycling.
Significance: AHSP binding to α-subunits inhibits subunit pseudoperoxidase activity.
Keywords: Biophysics, Electron Paramagnetic Resonance (EPR), Hemoglobin, Protein Chemistry, Protein Engineering, Redox, UV Spectroscopy, AHSP, α-Hemoglobin Stabilizing Protein, Hemichrome
Abstract
α-Hemoglobin stabilizing protein (AHSP) is a molecular chaperone that binds monomeric α-subunits of human hemoglobin A (HbA) and modulates heme iron oxidation and subunit folding states. Although AHSP·αHb complexes autoxidize more rapidly than HbA, the redox mechanisms appear to be similar. Both metHbA and isolated met-β-subunits undergo further oxidation in the presence of hydrogen peroxide (H2O2) to form ferryl heme species. Surprisingly, much lower levels of H2O2-induced ferryl heme are produced by free met-α-subunits as compared with met-β-subunits, and no ferryl heme is detected in H2O2-treated AHSP·met-α-complex at pH values from 5.0 to 9.0 at 23 °C. Ferryl heme species were similarly not detected in AHSP·met-α Pro-30 mutants known to exhibit different rates of autoxidation and hemin loss. EPR data suggest that protein-based radicals associated with the ferryl oxidation state exist within HbA α- and β-subunits. In contrast, treatment of free α-subunits with H2O2 yields much smaller radical signals, and no radicals are detected when H2O2 is added to AHSP·α-complexes. AHSP binding also dramatically reduces the redox potential of α-subunits, from +40 to −78 mV in 1 m glycine buffer, pH 6.0, at 8 °C, demonstrating independently that AHSP has a much higher affinity for Fe(III) versus Fe(II) α-subunits. Hexacoordination in the AHSP·met-α complex markedly decreases the rate of the initial H2O2 reaction with iron and thus provides α-subunits protection against damaging oxidative reactions.
Introduction
HbA is a well studied O2 transport protein that is known to participate in several biologically important redox reactions in vivo (1–4). This protein consists of two α-subunits and two β-subunits, with each subunit bearing a single, iron-containing protoporphyrin IX prosthetic group (5). Besides reversibly binding O2, these iron-containing groups and the surrounding residues are major sites of redox reactivity within HbA (6). The redox reactivity of HbA and isolated subunits leads in some cases to adverse effects due to radical-generating reactions, heme loss, and aggregation (7). To avoid these problems, Hb in red blood cells is found in a reducing environment. Furthermore, to deal with heme-related redox cycling outside of red blood cells, molecular chaperone proteins are naturally designed to bind and clear free Hb and its oxidation by-products from circulation (8).
AHSP3 is an erythroid-specific molecular chaperone protein (9–11). It rapidly and reversibly binds free α-subunits, but not β, αβ dimers, or tetrameric HbA (12, 13). Several studies have implicated AHSP as a modulator of α-subunit redox reactivity. For example, disruption of the Ahsp gene in mice leads to evidence of oxidative stress (14), and in vitro studies indicate that AHSP binding inhibits α-subunit reactions with oxidants such as hydrogen peroxide (H2O2) (15). Although these findings suggest that AHSP protects isolated α-subunits from oxidative damage and participation in harmful redox reactions in vivo, AHSP has also been shown to accelerate the rate of αO2 autoxidation to the ferric (met) state (16). MetHbA, isolated met-α-subunits, and isolated met-β-subunits are unstable due to accelerated rates of heme loss, denaturation, aggregation, and precipitation (9). Thus, it is puzzling why a molecular chaperone for α-subunits would accelerate autoxidation. We previously suggested that AHSP stabilizes a hemichrome folding intermediate and prevents its incorporation into HbA until the bound α-subunit can be reduced to the ferrous form (17).
Ferrous and ferric forms of HbA and Mb are known to react with H2O2 to produce ferryl heme (Fe(IV)) species (6) as part of a pseuodoperoxidative cycle,
where •Hb refers to a protein-based radical (6). Hamdane et al. (18) reported that reacting ferric α-subunits with H2O2 in the absence of AHSP yields the formation of a ferryl heme intermediate with a rate constant of 320 m−1 s−1, whereas Feng et al. (15) reported that AHSP binding to ferric α markedly inhibits the formation of H2O2-induced ferryl heme species. However, these events have not been well characterized for isolated α-subunits in either the absence or presence of AHSP, even though both ferryl heme and protein-based radicals are known to initiate a cascade of lipid oxidation reactions, which are thought to be involved in Hb toxicity in vivo (4).
Both Tyr and Trp are capable of acting as redox intermediates by facilitating electron flow pathways in metalloproteins (19). These residues are thought to function as part of a pathway for electron flow between solvent reducing equivalents and the heme iron. Tyr-42 in α-subunits has been shown to play a critical role in reducing ferryl heme through such a mechanism (20). These asymmetric, through protein electron transfer pathways may explain the apparent differences between the pseudo-peroxidase activities of α- and β-subunits observed in intact HbA tetramers (20).
To investigate underlying mechanisms of AHSP action, we studied the formation of ferryl heme intermediates and their protein-based radicals (·HbFe(IV) = O) in isolated HbA subunits and α-chains in the absence and presence of AHSP. Autoxidation rates, redox potentials, and radical formation after addition of H2O2 were measured and compared for isolated α- and β-subunits and AHSP·α-subunit complexes. Significant differences between isolated α-subunits and AHSP·α-subunit complexes were observed. Unlike β-subunits, isolated α-subunits do not form significant amounts of protein radicals after reaction with H2O2, although a small amount of ferryl heme is detected. In contrast, no ferryl heme species or associated protein radicals are observed when α-subunits are bound to AHSP. These findings show that α-subunits become resistant to the generation of protein radicals and ferryl heme species when bound to AHSP, and that hexacoordination of the heme iron within AHSP·met-α-subunit complexes appears to be the physiologically preferred species.
EXPERIMENTAL PROCEDURES
Protein Expression, Purification
Recombinant human AHSP was produced using previously published methods (12, 17). HbA was obtained from expired units of human blood (Gulf Coast Regional Blood Bank, Houston, TX) and was purified using previously published methods (21). HbA subunits were also isolated using previously published methods (22), as modified (17). This method occasionally resulted in catalase contamination in β-subunit samples. Catalase could be removed by size exclusion chromatography using Superdex 200 media (GE Healthcare). In the case of HbA and subunits, previously determined extinction coefficients were used to determine protein concentrations in heme equivalents (23). The AHSP extinction coefficient utilized was 11,460 m−1 cm−1 at 280 nm, and was calculated using ExPASy Proteomics Server Protparam. Purified haptoglobin (Hp) solution was a kind gift from Bio Products Laboratory (Hertfordshire, United Kingdom). The isolation and fractionation of this protein from human plasma was done as previously reported (24). Size exclusion HPLC chromatograms of the Hp samples used in this study show the following molecular weight distribution: 60% αβ dimers (Hp 1–1), 21% αβ trimers (Hp 1–2), and 19% larger polymers (Hp 2–2).
Spectrophotometry
Autoxidation studies were performed at 37 °C using air equilibrated 0.05 m potassium phosphate, pH 7.0, at 37 °C (25, 26). Spectra were recorded every 1 min for 4 h, using an integration time of 0.5 s and an interval of 1 nm. AHSP and α-subunit concentrations were fixed at 10 μm in heme equivalents. Where indicated, 10 μg/ml of superoxide dismutase and 200 units/ml of catalase were added to the cuvettes prior to the start of the reactions (25). Co-oxidation of epinephrine was followed under the same buffer conditions by monitoring absorbance at 475 nm using 600 μm epinephrine and the same concentrations of superoxide dismutase and catalase (25).
To detect the ferryl heme oxidation state, ferric subunits were first generated by addition of a 10-fold molar excess of potassium ferricyanide to O2-bound ferrous proteins, followed by brief incubation and removal of the potassium ferricyanide using Sephadex G-25 chromatography media. These steps were performed as quickly as possible at 4 °C because ferric subunits are highly unstable at room temperature. Materials were used within minutes of preparation. Ferryl heme detection studies were completed by manual mixing and stopped-flow spectrophotometry. In our stopped-flow experiments, 30 μm protein in heme equivalents was mixed with 3 mm H2O2 (post-mixing concentrations given) at 8 °C. In the manual mixing experiments, 60 μm protein in heme equivalents was mixed with 90 μm H2O2 at 22 °C. Both sets of experiments utilized 10 mm potassium phosphate buffer, pH 7.0. In the stopped-flow experiments, higher H2O2 concentrations were chosen to accelerate the time courses and to mitigate sample denaturation stemming from necessarily longer sample preparation times. Lower H2O2 concentrations were used in benchtop spectrophotometer experiments to ensure that the reactions were not too rapid to be measured conventionally. Where indicated, 2 mm sodium sulfide (Na2S) was used to derivatize the ferryl heme oxidation state as sulfhemoglobin using established methods (27). Buffers used for variable pH experiments include 200 mm potassium phosphate (pH 7.0 and 8.0 at 23 °C) and 200 mm sodium acetate (pH 5.0 and 6.0 at 23 °C) (28).
Spectroelectrochemistry
Intrinsic reduction-oxidation potentials were determined under anaerobic condition using a custom-built optically transparent thin layer electrode cell. The path length of this cell is 0.06 cm and [Ru(NH3)6]Cl3 was used as a cationic mediator (29–31). Well resolved differences in Soret band absorption for Fe(II) and Fe(III) heme species were used to follow the extent of reduction at different applied potentials Eapplied, which is defined as follows.
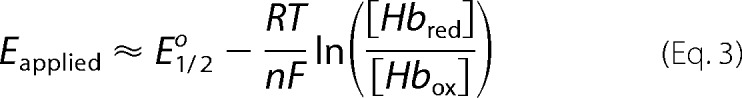 |
Where E½o is the midpoint potential when [Hbred] = [Hbox], R is the universal gas constant, T is the absolute temperature, n is the number of electrons transferred in the redox process for an ideal Nernstian system and is a measure of cooperativity for a non-Nernstian system, and F is the Faraday constant. All potentials are reported with respect to normal hydrogen electrode (31).
In a typical experiment, hemeproteins were placed in 1 m glycine buffer, pH 6.0, at 8 °C, and degassed gently on a salt/ice mixture and then purged using argon gas. β-Subunit concentrations were fixed at 10 μm, and free α-subunit and AHSP·α-subunit complexes were fixed at 80 μm (heme equivalents). The heme to mediator ratio was kept at 1:10 for all reactions. This anaerobic solution was then introduced into an optically transparent thin layer electrode cell fitted with a platinum mesh working electrode, platinum wire auxiliary electrode, and a silver/silver-chloride reference electrode. The potentials were controlled using an EG & G Princeton Applied Research Potentiostat Model 263 (AMETEK, Berwyn, PA) and the Soret bands for oxidized and reduced species were recorded using a Cary 100 spectrophotometer (Varian, Inc., Palo Alto, CA). Heme proteins were oxidized to the Fe(III) state by application of a positive potential, and spectra were recorded after a steady absorbance measurement was obtained at 405 nm for oxidized α-subunits and 413 nm for oxidized AHSP·α-subunit complexes. Once a stable absorbance was achieved for the Soret band at an oxidizing potential, ensuring complete conversion of all the heme groups to the Fe(III) state, the potential of the working electrode was made progressively more negative. At each relatively negative potential the analyte solution was equilibrated for 15 min and the absorbance of both oxidized and reduced species were recorded until at the end point all of the protein samples were converted to reduced heme leading to a stable reduced form Soret band (430 nm for both free reduced α-subunits and reduced AHSP·α-subunit complex). At the end of the experiment, the electrode potential was reversed in polarity to observe the reversibility of the redox process (31).
EPR
EPR spectra were recorded using a Bruker EMX spectrometer equipped with a liquid helium continuous flow cryostat, connected with a GFS600 transfer line and an ITC503 temperature controller (Oxford Instruments, Abingdon, Oxfordshire, UK). The experimental parameters were: frequency, 9.6 GHz; power, 1.0 milliwatts; modulation amplitude, 1 G; modulation frequency, 100 kHz; and temperature, 10 K. Samples were prepared as follows using 0.05 m potassium phosphate buffer, pH 7.0, at 22 °C. In the case of AHSP·α-subunit complexes, stoichiometric amounts of each protein were added to a 1.0-ml volume of buffer prior to oxidation using known molar extinction coefficients (17, 23). In the case of Hp·Hb complexes, excess HbA was added to a small volume of Hp, the sample was allowed to incubate for 15 min on ice, and excess HbA was removed using a Superose 12 10/300 GL column (GE Healthcare). Samples were oxidized to the ferric state by addition of a 10-fold molar excess K3Fe(CN)6 and incubation for ∼3 min on ice. Each sample was then passed through a 10-DG (Bio-Rad) desalting column to remove excess K3Fe(CN)6, after which concentrations were measured. Where indicated, 0.1 m sodium fluoride or a 1.5-fold molar excess of H2O2 were added to each sample. Concentrations in heme equivalents were: 1) α-subunits in the presence of sodium fluoride = 247 μm; 2) α-subunits in the presence of H2O2 = 523 μm; 3) AHSP·α-subunit complexes in the presence of sodium fluoride = 247 μm; 4) AHSP·α-subunit complexes in the presence of H2O2 = 385 μm; 5) β-subunits in the presence of sodium fluoride = 207 μm; 6) β-subunits in the presence of H2O2 = 403 μm; 7) HbA = 1.2 mm for all spectra; 8) Hb·Hp complexes = 0.6 mm for all spectra. In the samples treated with H2O2, incubation occurred for 10 s prior to manual freezing in a dry ice/ethanol bath. Spectra were normalized to heme concentration in Figs. 6 and 7, and in the radical yield calculations given in Table 2.
FIGURE 6.

EPR spectra recorded at 10 K before and after H2O2 treatment. A, spectra of samples in the absence and presence of 0.1 m sodium fluoride. B, spectra of samples prepared with a 1.5-fold excess of H2O2. Sample preparation steps are outlined under ”Experimental Procedures.“ Individual spectra are labeled accordingly. EPR conditions were: microwave power, 1 milliwatt; microwave frequency, 9.3 GHz; scan length, 4000 G; center field, 2200 G; modulation amplitude, 2 G; temperature, 10 K.
FIGURE 7.

Close-up EPR spectra of the radical species before and after H2O2 treatment. Individual spectra are labeled accordingly. EPR conditions were: microwave power, 1 milliwatt; microwave frequency, 9.3 GHz; range 140 G; modulation amplitude, 2 G; temperature, 20 K. Spectra were measured using the same samples as were used to generate the data in Fig. 6.
TABLE 2.
Iron spin states and H2O2-induced protein radical yield in various ferric samples
The percent high spin was calculated from the ratio of the high spin EPR signal at 10 K to the signal recorded in the presence of 0.1 m NaF. The percent low spin was calculated from subtracting this value from 1. Fluoride failed to convert the high-spin heme in AHSP·α. The percent radical intermediate yield was determined based on a Cu(II) standard measured at 115 K. Percentage high-spin heme for AHSP·α was estimated using average signal height at g = 6 for the fluoride complex of both purified α- and β-subunits. These experiments were repeated several times using slightly different experimental conditions (e.g. microwave power, modulation amplitude, etc.).
| Ferric sample | Resting state high spin | Resting state low spin | H2O2-generated radical yield |
|---|---|---|---|
| % | |||
| α | 22.5 | 77.5 | 1.7 |
| AHSP·α | <10 | >90 | 0.078 |
| β | 21.5 | 78.5 | 4.3 |
| HbA | 76.4 | 23.6 | 5.5 |
| Hp·αβ | 83.3 | 16.7 | 4.8 |
Reagents and Instrumentation
Unless otherwise specified, all reagents and chemicals were obtained from Thermo Fisher Scientific (Waltham, MA) or Sigma. Chromatographic media, columns, and chromatography equipment were obtained from GE Healthcare Bio-Sciences Corporation (Piscataway, NJ) and Whatman International Ltd. (Maidstone, Kent, United Kingdom). UV-visible absorbance spectroscopy measurements were made using an Agilent 8453 diode-array spectrophotometer (Agilent Technologies, Inc., Santa Clara, CA) or a Cary 100 Spectrophotometer (Varian, Inc., Palo Alto, CA). In both cases, 1-cm path length cells were used. Stopped-flow measurements were made using an Applied Photophysics SX-18 microvolume stopped-flow spectrophotometer (Leatherhead, Surry, UK). The path length was 10 mm and the entrance and exit slit widths were set to 1 mm each to give 4.8-nm spectral band widths. The volume of the cell was 20 μl. Shot volumes were between 100 and 200 μl, and mixing was performed using equal volumes of reactant solutions.
Data Analysis
Microsoft Excel was used for nonlinear least square data fitting to a single exponent expression to obtain the observed rate constants (Microsoft Corp., Redmond, WA) (32). Fitting routines in Origin were also used to verify the values obtained from Excel (OriginLab Corp., Northampton, MA). Spectroelectrochemical and sulfheme measurements were repeated three times to obtain standard deviations, and autoxidation reactions were repeated four times. Structure images were created using the PyMOL Molecular Graphics System (Schrödinger, LLC, New York).
RESULTS
Autoxidation
HbA oxidation from the ferrous (Fe(II)) to the ferric (Fe(III)) state occurs spontaneously in the presence of air-equilibrated aqueous buffer and produces superoxide anion (O2˙̄) (33, 34). The released superoxide anion then undergoes dismutation to H2O2 and O2, and the H2O2 produced in this step can react with the protein to produce several other species (6). This spontaneous conversion of ferrous to ferric state in hemeproteins under aerobic conditions and the subsequent redox processes involving the superoxide anion, H2O2, and ferric Hb are broadly classified as autoxidation.
Although AHSP has previously been shown to accelerate the initial rate of αΟ2-subunit autoxidation (16), the generation of subsequent ferryl heme and protein radical species (Eqs. 1 and 2) have not been well defined. Initially we examined whether AHSP·αΟ2 autoxidation occurs by mechanisms similar to those reported for native tetrameric HbA.
AHSP·αO2 complexes were allowed to autoxidize in the presence and absence of superoxide dismutase (an O2˙̄ scavenger) and/or catalase (an H2O2 scavenger) in accordance with the methods of Misra and Fridovich (25). The rate of autoxidation was monitored by optical absorbance changes in the visible region. AHSP·αO2 complexes exhibit peaks at ∼541 and ∼576 nm (16, 23), whereas AHSP·met-α complexes exhibit peaks at 535 and 565 nm following oxidation to the ferric state (13, 15, 16). The latter positions are indicative of a hemichrome or bis-histidyl conformation in which the imidazole nitrogen atoms of αHis-58 and αHis-87 both coordinate the ferric iron atom and exclude H2O and other exogenous ligands (15). Representative time courses produced from monitoring absorbance decays at 576 nm are shown in Fig. 1A.
FIGURE 1.

Autooxidation and H2O2 and O2˙̄ production by AHSP·α-subunit complexes. A, representative autooxidation time courses in the absence and presence of catalase or superoxide dismutase. B, time courses for autoxidation of isolated αO2- and βO2-subunits and the AHSP·αO2 complex. C, co-oxidation of epinephrine by AHSP·αO2 in the presence of catalase or superoxide dismutase. Autooxidation was performed at 37 °C using air-equilibrated 0.05 m potassium phosphate, pH 7.0, at 37 °C, and 10 μm protein in heme equivalents. Where indicated, 10 μg/ml of superoxide dismutase (SOD) and 200 units/ml of catalase (CAT) were added prior to data collection. Co-oxidation of epinephrine was followed at 475 nm using 600 μm epinephrine and the same buffer and temperature conditions as were used for autoxidation (25). Complete time courses for isolated α and β could not be obtained due to protein precipitation. Data in panel A were normalized to total absorbance signal changes. Open circles, squares, and triangles represent data points for AHSP·α alone, AHSP·α with superoxide dismutase, and AHSP·α with catalase, respectively. Lines in panel A are theoretical fits generated using the single-exponent expression Yt = Y0 + Y1e−k1*t. Observed rate constants from four replicates performed on two different days are given in the text. Data in panels B and C were normalized to correct for slight concentration differences.
In agreement with previous studies (16, 35), 10 μm AHSP·α-subunit complexes autoxidize with an apparent rate constant of ∼2.3 ± 0.2 h−1 (Fig. 1A). The addition of catalase reduced the apparent rate constant to 1.2 ± 0.2 h−1 under the same experimental conditions. Superoxide dismutase did not appreciably affect the apparent rate constant of autoxidation, which was measured to be 2.4 ± 0.3 h−1 in its presence. For comparison, Fig. 1B depicts the initial phases of the autoxidation plots for isolated α- and β-subunits, which show at least 10-fold slower apparent rate constants (≤0.3 h−1). AHSP binding to α-subunits induces conformational changes that facilitate superoxide dissociation and hemichrome formation. Because catalase converts H2O2 into H2O and O2, these data suggest that the slower rate of autoxidation in the presence of catalase is due to the diminished availability of H2O2, which is capable of reacting with ferrous and ferric HbA to facilitate more rapid oxidation of the protein. Caughey and co-workers (26) observed a similar decrease in the rate of autoxidation of HbAO2 due to addition of catalase. The absence of a significant effect of superoxide dismutase agrees with most previous work (25, 26). In this case, SOD catalyzes the rapid dismutation of O2˙̄ into H2O2 and O2, indicating that H2O2, and not O2˙̄, accelerates autoxidation. Even in the case of catalase, the effect is very small compared with the dramatic effect of AHSP binding on the autoxidation of αO2 (Fig. 1B).
The absence of an effect in the case of superoxide dismutase, which facilitates the rapid dismutation of O2˙̄ into H2O2 and O2, agrees with most previous work (25), although at least one conflicting report exists (26). However, both effects are very small compared with the effect of AHSP binding to αO2.
We also examined H2O2 and O2˙̄ production from AHSP·αO2 complexes during autoxidation. We recorded spectral changes during the co-oxidation of epinephrine to adrenochrome in the same buffer and at the same temperature. We recorded changes in absorbance at 475 nm in the presence and absence of catalase and superoxide dismutase (25). This process has previously been used to assay for H2O2 and O2˙̄ production during HbA autoxidation (25). Representative data from these studies are shown in Fig. 1C. Both enzymes showed additive effects on the diminution of the observed rate of co-oxidation of epinephrine. Similar effects of catalase and superoxide dismutase are observed for the autoxidation of native HbA (25).
Ferryl Heme Species
In addition to the oxidation of ferrous heme to the ferric heme state, iron atoms within HbA can undergo further oxidation to the ferryl heme state in the presence of reactive oxygen species (6, 7). This process has been associated with radical formation both in vitro and in vivo (36, 37), and significant work on HbA shows that ferryl heme formation occurs following oxidation of both ferrous and ferric protein by H2O2 (6, 7, 36). Recently, unique tyrosine-mediated inter-subunit electron transfer pathways have been proposed for intact HbA (20). Because autoxidation of oxy-Hb produces O2˙̄, which spontaneously dismutates to form H2O2, we sought to characterize the behavior of both subunits in isolation with respect to H2O2 reactivity.
HbA oxidation to the ferryl heme state can be induced in vitro by subjecting the protein to moderate excesses of H2O2 (36). Ferryl heme formation causes the emergence of two broad optical absorbance peaks with maxima at ∼545 and 585 nm (36). We first mixed ferrous oxygenated HbA, α, β, and AHSP·α-subunit complexes with H2O2 using conditions similar to those reported by Tomoda et al. (38). These reactions were spectrally monitored for up to 2 min in a benchtop optical absorbance spectrophotometer. In agreement with the previous report (38), we found that isolated ferrous αO2- and βO2-subunits form hemichromes and precipitate on this time scale. In contrast, we observed that aquo- or hydroxy-(met) α- and β-subunits form relatively stable, transient ferryl heme complexes, which are similar to those observed for tetrameric met-HbA.
As shown in Fig. 2A, mixing 30 μm ferric β-subunits with 3 mm H2O2 (post-mixing concentrations) results in the emergence of broad absorbance bands at ∼547 and 585 nm, which are indicative of formation of a Fe(IV) = O complex. The reaction of H2O2 with ferric α-subunits under equivalent conditions results in similar spectral transitions, with the emergence of ferryl-hemichrome-like spectra with absorbance peaks at ∼543 and ∼582 nm (Fig. 2B). The red lines in Fig. 2 represent spectra obtained for the reaction of intact ferric HbA with H2O2 using identical experimental conditions.
FIGURE 2.

Ferryl complex formation after reaction with H2O2. A, β. B, α. C, AHSP·α. Spectra were recorded approximately every 2 s for 20 s following rapid mixing of 30 μm ferric subunits with 3 mm H2O2 (post-mixing concentrations) in 10 mm potassium phosphate buffer, pH 7.0, at 8 °C. Subunits were used within minutes of oxidation to the ferric state. Red lines represent end point spectra of HbA derived from experiments using the same time scales and equivalent concentrations of heme and H2O2. Arrows depict the direction of change post-mixing.
Fig. 2C shows spectra for the reaction of AHSP·(met)-α-subunit complexes with H2O2 using identical experimental conditions. We observed no absorbance changes between 3 ms and 20 s post-mixing. These data suggest that AHSP completely inhibits ferryl heme formation on these time scales (Fig. 2C), even in the presence of large excesses of H2O2.
On longer time scales between 20 s and 2 min, isolated ferric subunits precipitated following exposure to H2O2, as evidenced by marked increases in solution turbidity (not shown). In agreement with other reports (13, 15), however, we find that AHSP binding to α-subunits inhibits this H2O2-induced precipitation. We detect no spectral evidence for ferryl heme intermediates under any conditions examined.
Because the absorbance spectra of hemichrome and ferryl heme species are similar (compare red spectra in Fig. 2, A and B, with the black spectrum in C), we used sodium sulfide (Na2S) to further characterize the extent of ferryl heme formation in isolated met-subunits and AHSP·(met)-α-subunit complexes. This reagent has previously been shown to react with ferryl heme groups in HbA and myoglobin to produce quantifiable derivatives that give an absorbance peak at ∼620 nm (27). The iron in the ferryl heme is reduced to the ferrous state and the sulfur is incorporated onto the porphyrin (27). This reaction depends both on the kinetics of ferryl heme formation and its rate of reaction with Na2S, and has widely been used to derivatize ferryl Hb in vitro and in biological samples (27, 39).
We added 90 μm H2O2 to 60 μm ferric α-subunits, β-subunits, HbA, and AHSP·α-subunit complexes, incubated the samples at room temperature for 60–120 s (see the legend of Fig. 3 for the precise conditions), and recorded spectra before and after this incubation. After recording spectra at the completion of each incubation, 2 mm Na2S was rapidly added to each sample, and again spectra were recorded. Representative spectra of the absorbance peaks at ∼620 nm are shown in Fig. 3, A, B, and D. These data indicate that H2O2-treated met-HbA and met-β form sulfheme species quite readily (Fig. 3). However, α-subunit sulfheme formation is more limited, due in part to a slower rate of ferryl heme formation and a greater rate of precipitation (Fig. 3D). In contrast, the AHSP·(met)-α-subunit complexes do not form any detectable sulfheme species, verifying the spectral measurements in Fig. 2 (see Fig. 3C). Using a previously published extinction coefficient (27), the calculated sulfheme concentrations in our HbA, α, β, and AHSP·α samples were 26.6 ± 0.4, 9.5 ± 0.4, 29.4 ± 0.7, and 0 μm, respectively, of a total of 60 μm heme in each sample.
FIGURE 3.

Sulfheme formation after the derivatization of ferryl heme with Na2S. A, HbA. B, β. C, AHSP·α. D, α. Following the incubation of 60 μm ferric proteins with 90 μm H2O2 in 10 mm potassium phosphate buffer, pH 7.0, at 22 °C, 2 mm Na2S was added and spectra were immediately recorded. Na2S was added 70 s following H2O2 addition in the case of α and AHSP·α, 90 s in the case of β, and 120 s in the case of HbA. Various other incubation times were also assayed (not shown). However, simultaneous oxidation and precipitation of ferric subunits creates a limited time in which ferryl heme intermediates can be detected. The peak at ∼620 nm is due to ferrous sulfhemoglobin formation (27). Each panel contains an overlay of ferric (black), ferryl (red), and sulfheme (blue) Hb spectra.
To further investigate our findings, we altered the evolutionarily conserved AHSP proline 30 in recombinant AHSP to generate AHSP(P30A) and AHSP(P30W) mutant proteins (17, 40). Although these mutations do not detectably perturb erythropoiesis in mouse models, AHSP(P30W) has been shown to bind oxygenated α-subunits with a 30-fold increased affinity in vitro, and both variants cause decreased rates of autoxidation and increased rates of hemin loss relative to wild-type AHSP (17, 40). In conducting experiments analogous to those presented in Fig. 3, we found that these variants exhibit the same behavior as wild-type AHSP (data not shown). This suggests that once the hemichrome is formed, the reaction with H2O2 is strongly inhibited through a mechanism that is independent of the specific residue at position 30.
Alkaline conditions have previously been reported to stabilize the ferryl heme oxidation state (28, 41). We therefore examined the H2O2 reactions and sulfide addition at various pH values using isolated ferric α- and β-subunits and AHSP·α-subunit complexes, using previously reported experimental conditions (28). We were able to detect large amounts of the ferryl heme state in β, small amounts in α, but none in the AHSP·α-subunit complex at any pH (Fig. 4).
FIGURE 4.

pH dependence of sulfheme formation. A, α-subunit. B, β-subunit. C, AHSP·α-subunit complex. The reactions of Fig. 3 were repeated at pH values 5–8 at 23 °C. Buffers used include 200 mm potassium phosphate (pH 7.0 and 8.0 at 23 °C) and 200 mm sodium acetate (pH 5.0 and 6.0 at 23 °C) (27). Spectra were each offset by a constant absorbance value to aid in visualization.
Midpoint Redox Potential
The redox potentials of isolated HbA subunits have previously been investigated by two groups using potentiometric methods (42, 43). Neither group reported the effects of AHSP binding to α-subunits on midpoint potential. Our previous measurements showed that AHSP binds met-α with a much higher affinity than αCO, αO2, and deoxy-α (17, 44). This result predicts that AHSP binding should markedly decrease the reduction potential of isolated α-subunits, provided the binding equilibrium is significantly dependent on the oxidation state of heme.
To verify this prediction, we measured the reduction potential of α-subunits in the presence and absence of AHSP under anaerobic conditions using a spectroelectrochemical method. As shown in Fig. 5, the Nernst plots for isolated α-subunits and AHSP·α-subunit complexes are linear and show evidence of a simple, noncooperative, single electron reduction process. Based on these data, we calculate an E½o of +40 (versus normal hydrogen electrode) for free α-subunits, which agrees with the reports of both Abraham and Taylor (42) and Banerjee and Cassoly (43). Binding to AHSP lowers the midpoint potential to −78 mV. These values are listed in Table 1, along with the reduction potentials we determined for HbA and Hp·Hb complexes. Like AHSP, Hp binding also lowers the reduction potential of HbA, but to a lesser extent, from +120 to +54 mV (24). Interestingly, the redox process involving AHSP·α-subunit complexes was completely reversible as observed by reversing the polarity of the working electrode at the end of each spectroelectrochemical experiment. This result indicates that reduction does not dissociate the AHSP·α complex at the micromolar protein concentrations used in the measurements, as predicted from previous measurements of the KD for binding of the ferric (0.0006 μm) and ferrous (0.017 μm) forms of α to AHSP (17).
FIGURE 5.

Redox potential of AHSP·α-subunit complex. A, spectral changes in AHSP·α-subunit complexes following exposure to increasingly negative electrical potentials. The initial AHSP·(met)-α-subunit band at 413 nm shows a progressively decreasing intensity at increasingly negative potentials. These data also show a concomitant increase in absorbance at 430 nm. These transitions reflect a conversion to the ferrous oxidation state from the ferric state. AHSP·(met)-α-subunit complex spectra exhibit two hemichrome peaks at 535 and 565 nm, and the spectra of reduced complexes exhibit maxima at ∼558 nm. B, Nernst plots for AHSP·α (open square) and isolated α-subunits (open circle). The dots represent actual data points and the smooth lines are the best fit to the data sets. Data points were obtained using different absorbance data at the Soret regions at all applied potentials for different species. All spectroelectrochemical experiments were done in 1 m glycine at pH 6.0 at 8 °C, using heme concentrations of 80 μm for free α-subunits and AHSP·α-subunit complexes.
TABLE 1.
Midpoint potentials of the indicated samples
Measurements were made using 1 m glycine buffer, pH 6.0, at 8 °C using a 0.6-mm path length cell. Protein concentrations were 80 μm in heme equivalents. Values for HbA and αβ·Hp complexes were measured using 200 mm potassium phosphate buffer, pH 7.0, at 22 °C.
a From Banerjee et al. (24).
E½o values measured for isolated β-subunits were variable and differed substantially from those reported by other groups (42, 43). Although the reason for this is unclear, we found that irreversible β-subunit denaturation during our experiments prevented reliable measurement. Also, β-subunits readily self-associate into homotetramers (45). Neither our experiments nor those of Abraham and Taylor (42) or Banerjee and Cassoly (43) adequately controlled for this phenomenon.
Radical Formation
We also investigated the effects of AHSP on protein radical formation using low temperature EPR spectroscopy. We prepared ferric α-subunits, β-subunits, HbA, AHSP·α-subunits, and Hb·Hp complexes using the methods described under “Experimental Procedures,” and recorded spectra before and following the addition of either 0.1 m sodium fluoride or a 1.5-fold molar excesses of H2O2.
Fig. 6A contains spectra recorded in the presence or absence of sodium fluoride, which normally converts aquo-met or hydroxy-met samples into completely high spin forms. All three ferric resting samples showed EPR spectra indicative of a mixture of high and low spin ferric iron. The g values of the low spin α-subunits (2.792, 2.248, and 1.687) and AHSP·(met)-α-subunit complexes (2.932, 2.247, and 1.737) indicate that both species have a histidine imidazole distal ligand according to modified Truth-Diagram analysis (46), and the g values for the low spin β (2.752, 2.253, and 1.830) indicate a hydroxide ligand (Fig. 6A). The amount of low spin ferric iron in all three samples is substantial and should not be underestimated by directly comparing the apparent amplitudes of the high and low spin heme signals. The extent of low spin character is revealed by comparing the heights of the g = 6 signals of the met samples with those in the EPR spectrum of the corresponding fluoride derivatives. Fluoride binding converts all Fe3+ atoms into the high spin state with the g‖ component at g = ∼2 showing a doublet due to the nuclear hyperfine of the fluorine (Fig. 6A) and a maximal intrinsic g = ∼6 signal, which can be used to estimate the total amount of Fe3+ in each sample by converting it all to high spin. The intensity of the observed g = 6 signals for the untreated samples can then be compared with that for the met·fluoride complex to estimate the amount of high spin iron. Thus, the high spin heme signals of α- and β-subunits are ∼22 and 21% of the total heme iron, respectively, assuming that the height of the derivative signals remain proportional to the concentration of high spin heme detected at g = 6 in each case.
Fluoride failed to convert the low spin ferric heme iron in the AHSP·(met)-α-subunit complex to high spin, indicating that fluoride cannot access the iron once AHSP is bound due to its strong stabilization of bis-His coordination. We estimated the amount of high spin heme in AHSP·α-subunit complexes using the amplitude of the EPR g = 6 signal for the fluoride complexes of the α- and β-subunits. As shown in Table 2, the percentage of high spin heme is ≤10% in the AHSP·(met)-α-subunit complex, much less than that of the α- and β-subunits, and in sharp contrast to that of met-HbA, which is as high as 76.4%. This much reduced level of high spin heme in AHSP·(met)-α-subunit complex is clearly due to bis-His coordination. Estimates of the amounts of high and low spin ferric α-subunit, β-subunit, HbA, AHSP·α-subunit complex, and Hp·αβ are summarized in Table 2.
Fig. 6B shows spectra recorded from samples incubated with H2O2 for 10 s prior to freezing. β-Subunits treated with H2O2 show a significant amount of protein-based free radical signal at g = 2.0 under these conditions. The H2O2-reacted met-α sample shows less signal, and the AHSP·(met)-α sample shows almost no detectable free radical peak g = 2.0.
EPR spectra of the radical species recorded in the 100 gauss range are presented in Fig. 7. The radical generated in the α-subunit is centered at g = 2.0038 with a symmetric line shape and an overall width of 20 gauss. The radical signal found for β-subunits, although with a similar center at g = 2.0039, is asymmetric with an additional low field component at 2.033 and clear hyperfine structures in the major signal, with an overall width of 24.5 gauss. The half-saturation power is also different, 2.4 versus 10.4 milliwatts for the α- and β-subunits, respectively, indicating the location and structure of the protein radical found in the α- and β-subunits are likely different.
The percentages of radical yields are shown in Table 2, together with similar data obtained for the Hp·Hb complex for comparison under equivalent experimental conditions. The reaction of H2O2 with Hp-met-Hb dimers shows a level of radical signal, which is comparable with those observed for isolated met-β or met-HbA, consistent with a recent observation (47), and is 50-fold greater than that seen for the AHSP·(met)-α complex.
DISCUSSION
The reaction between H2O2 and ferrous Hb is a two-electron process that results in the formation of ferryl heme iron (6). If H2O2 reacts with ferric Hb, a radical species is formed in addition to the oxoferryl species (Eqs. 1 and 2) (6). Both the ferryl heme and its associated protein radical can induce a wide variety of oxidative reactions that affect nearby molecules due to their high midpoint redox potentials, ∼1 V (48). Specific chemical modifications to free Hb have been observed if proteins are challenged with excess H2O2 in vitro. First, the heme vinyl groups may become modified and covalently linked to the protein (48). Second, extensive globin chain cross-links and irreversible modifications of several amino acids, primarily in β-subunits, have been observed (49). Studies have also revealed the presence of oxidative changes to the surrounding tissues following exposure to extracellular Hb, both in animals infused with these proteins and when Hb is released from human red cells leading to kidney or brain injury (8, 50, 51). It is not surprising that several pathways exist in mammals to control these events. Two key globin-binding proteins that appear to inhibit oxidative damage are: (a) AHSP, which provides protection against oxidative damage to α-subunits and surrounding proteins during erythropoiesis (12–14) and (b) Hp and the CD163 receptor on macrophages, which coordinate Hb dimer clearance during hemolysis (50). Our results indicate that AHSP and Hp exert these functions through distinct mechanisms.
Preferential Binding of AHSP to Met-α
Current evidence suggests that AHSP protects free α-subunits from oxidative degradation by preferentially binding to the ferric form and rapidly inducing structural changes that generate a stable hexacoordinated species (12, 13, 15, 17). Our spectroelectrochemical experiments confirm that AHSP binds preferentially to met-α-subunits. The midpoint reduction potential of free α-subunits decreases from +40 to −78 mV when the subunit is bound to AHSP (Fig. 5B, Table 1). These values can be used to independently estimate the ratio of the equilibrium dissociation constants for AHSP binding to reduced deoxy-α and met-α-subunits (i.e. KD,red/KD,ox). A complete derivation of the effect of AHSP binding on the reduction potential of α-subunits is given in the Supplemental Scheme 1 in Supplemental Derivation. Using this method and the parameters in Table 1, the computed ratio is ∼130, which is almost identical to the ratio computed from the bimolecular association and unimolecular dissociation rate constants (i.e. 100) reported by Mollan et al. (17).
Interestingly, under comparable experimental conditions, the midpoint potential of HbA was found to decrease from +120 mV in the tetrameric form to +54 mV in the Hp-bound αβ-dimeric form (Table 1) (24). Thus, both Hb-binding proteins facilitate oxidation, AHSP by inducing hemichrome formation and Hp by promoting dissociation into more easily oxidized dimers. These two proteins also deal with peroxide-induced radical chemistry in markedly different ways (see below).
Inhibition of H2O2 Reactions by AHSP
Early structural work on AHSP·α-subunit complexes led to the proposal that the bis-histidyl conformation strongly inhibits ferric-ferryl redox cycling following exposure to H2O2 (15, 52). It was estimated that at least 33% of the heme in ferrous αΟ2·AHSP complexes converts to the ferryl heme form following H2O2 exposure, whereas less than 10% of the heme in ferric met-α·AHSP complexes converts to the ferryl heme form following the same exposure (15). A more recent report indicates that H2O2-induced covalent modification of α-subunits can be prevented by AHSP binding, and that two exposed α-subunit Tyr residues (Tyr-24 and Tyr-42) are unable to take part in electron transfer and ferryl heme protein radical formation (Fig. 8) (18).
FIGURE 8.

Model structure of ferrous α-subunit, β-subunit, and ferric AHSP·α-subunit complex and oxidation reactions at the heme vicinity. A, α-subunit. B, β-subunit. C, AHSP·α-subunit complex. D, oxidation scheme. AHSP, α-subunit, and β-subunit structures are shown as ribbons in cyan, silver, and gold, respectively. Heme or heme groups are shown using pink stick structures, with αTyr-42 and βPhe-41 sticks shown in green and blue, respectively. Distal and proximal histidines are also shown using stick structures. Corey-Pauling-Koltun coloring is otherwise used throughout. Structure images were created using the PyMOL Molecular Graphics System and PDB files 1Z8U and 1HHO (15, 58).
In this work, we focused on ferric-ferryl redox transitions within the AHSP·α complex. We confirmed that AHSP·α autoxidizes ∼10-fold more rapidly than isolated α-subunits (13, 16), and found that this process both produces and consumes H2O2. We also found that in the presence of molar excesses of H2O2, the ferric forms of HbA, α, and β can react to form ferryl heme intermediates, which can be detected using Na2S. By contrast, AHSP·(met)-α-subunit complexes show no evidence of H2O2-induced ferryl heme intermediates or species (Figs. 2 and 3). This lack of peroxidase activity is likely a consequence of low accessibility of H2O2 to the heme iron once AHSP is bound and induces bis-His coordination (Fig. 8C). Internal hexacoordination also explains the lack of conversion from low spin to high spin Fe3+ by addition of excess fluoride to the met-α·AHSP complex (Fig. 6A). These findings are confirmed in our experiments using buffers with pH values varying from 5.0 to 8.0 (Fig. 4). Fig. 8D outlines a scheme of AHSP function based on these findings.
Differences between Met-α, Met-β, and AHSP·(met)-α-Subunits
Although our data show that isolated α-subunits behave markedly differently than isolated β-subunits with respect to ferryl heme and protein radical formation, it is clear that more work is needed to determine the underlying mechanistic differences, including more directed measurements of the rates of formation and decay of other intermediates and site-directed mutagenesis studies designed to determine the structural origin of the radical signals in Fig. 7.
For example, the smaller radical and ferryl heme signals for α-subunits could be due to stronger water coordination to Fe(III), which inhibits the initial reaction with H2O2 (53). There is evidence that the distal histidine in α-subunits stabilizes bound ligands and distal pocket water to a greater extent than in β-subunits because of a more rigid conformation and closer proximity of the imidazole side chain to the coordinated O atom (Fig. 8, A and B) (54, 55).
In addition, Reeder et al. (20) have shown that ferryl-α-subunits both autoreduce or react with added reducing agents ∼10 times more rapidly than ferryl-β-subunits, and that these differences are due in part to facilitation of electron transfer by the solvent-exposed Tyr-42 side chain at the CD corner of the α- but not β-subunits (Fig. 8). However, the extent of αTyr-42 participation in the peroxidase reaction is not fully defined and studies with isolated mutant subunits remain to be done.
By contrast, the mechanistic cause of the lack of reactivity of the AHSP·(met)-α complex with H2O2 and formation of ferryl heme species is much clearer. A dramatic conformational change in the CD corner of α-globin occurs on binding to AHSP and autoxidation, which results in hemichrome formation with both His-58(E7) and His-87(F8) coordinated to the Fe(III) atom (Fig. 8C). This hexacoordination appears to completely inhibit the reaction of AHSP·(met)-α with H2O2 to form the initial intermediates observed in free isolated subunits.
Summary and Physiological Relevance
Our study provides three biochemical findings of relevance to erythroid physiology. First, met-β-subunits are more prone to the H2O2-induced formation of ferryl heme species and protein-based radicals than met-α-subunits. This may explain why α- and β-thalassemic erythroid cells exhibit distinct membrane abnormalities and oxidative modifications (56, 57). It also suggests that Hb-based O2 therapeutics should be designed with more attention directed at mitigating oxidative reactions of β-subunits. Second, AHSP binding renders met-αHb nearly inert to oxidative degradation by H2O2, with no ferryl heme species or protein-based radicals detected by either optical absorbance or EPR. Third, AHSP binding dramatically lowers the redox potential of α to a much more negative value, and thermodynamically favors the ferric over ferrous iron.
Although AHSP and Hp both protect against Hb-mediated oxidative damage, the biochemical mechanisms are distinct. AHSP stabilizes nascent α-subunits by inducing the formation of a stable hemichrome that inhibits interactions with H2O2 and other ligands. By contrast, Hp-bound αβ dimers retain their pseudoperoxidase activity, although the resulting ferryl heme species appear to be less reactive due in part to the stabilization of a protein-based radical on βTyr-145 (47).
Supplementary Material
Acknowledgments
We acknowledge the generous assistance of Antony Mathews, Francine Wood, and Eileen Singleton for their help with HbA subunit isolation.
This work was supported, in whole or in part, by National Institutes of Health Grants HL110900 (to A. I. A.), HL095821 (to A.-L. T.), HL47020 (to J. S. O.), HL110900 (to J. S. O.), GM35649 (to J. S. O.), DK61692 (to M. J. W.), HL087427 (to M. J. W.), and GM008362 (to T. L. M.), Welch Grant C-0612 (to J. S. O.), National Science Foundation Grant CHE 0809466 (to A. L. C.), and the United States Food and Drug Administration (MODSCI 2011) (to A. I. A.).

This article contains a Supplemental Derivation.
- AHSP
- α-hemoglobin stabilizing protein
- ferrous
- Fe2+
- ferric
- Fe3+
- ferryl
- Fe4+
- hemichrome
- endogenous hexacoordination of iron within HbA (bis-histidyl)
- Hb
- hemoglobin
- HbA
- wild-type adult human Hb
- heme
- ferroprotoporphyrin IX
- Hp
- haptoglobin
- met
- ferric iron oxidation
- O2˙̄
- superoxide radical.
REFERENCES
- 1. Jarolim P., Lahav M., Liu S. C., Palek J. (1990) Effect of hemoglobin oxidation products on the stability of red cell membrane skeletons and the associations of skeletal proteins. Correlation with a release of hemin. Blood 76, 2125–2131 [PubMed] [Google Scholar]
- 2. Flynn T. P., Allen D. W., Johnson G. J., White J. G. (1983) Oxidant damage of the lipids and proteins of the erythrocyte membranes in unstable hemoglobin disease. Evidence for the role of lipid peroxidation. J. Clin. Invest. 71, 1215–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tsantes A. E., Bonovas S., Travlou A., Sitaras N. M. (2006) Redox imbalance, macrocytosis, and RBC homeostasis. Antioxid. Redox Signal. 8, 1205–1216 [DOI] [PubMed] [Google Scholar]
- 4. Reeder B. J., Sharpe M. A., Kay A. D., Kerr M., Moore K., Wilson M. T. (2002) Toxicity of myoglobin and haemoglobin. Oxidative stress in patients with rhabdomyolysis and subarachnoid haemorrhage. Biochem. Soc. Trans. 30, 745–748 [DOI] [PubMed] [Google Scholar]
- 5. Bunn H., Forget B. (1986) Hemoglobin: Molecular, Genetic, and Clinical Aspects, WB Saunders Co., Philadelphia, PA [Google Scholar]
- 6. Rifkind J. M., Ramasamy S., Manoharan P. T., Nagababu E., Mohanty J. G. (2004) Redox reactions of hemoglobin. Antioxid. Redox Signal. 6, 657–666 [DOI] [PubMed] [Google Scholar]
- 7. Reeder B. J., Svistunenko D. A., Cooper C. E., Wilson M. T. (2004) The radical and redox chemistry of myoglobin and hemoglobin. From in vitro studies to human pathology. Antioxid. Redox Signal. 6, 954–966 [DOI] [PubMed] [Google Scholar]
- 8. Baek J. H., D'Agnillo F., Vallelian F., Pereira C. P., Williams M. C., Jia Y., Schaer D. J., Buehler P. W. (2012) Hemoglobin-driven pathophysiology is an in vivo consequence of the red blood cell storage lesion that can be attenuated in guinea pigs by haptoglobin therapy. J. Clin. Invest. 122, 1444–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mollan T. L., Yu X., Weiss M. J., Olson J. S. (2010) The role of α-hemoglobin stabilizing protein in redox chemistry, denaturation, and hemoglobin assembly. Antioxid. Redox Signal. 12, 219–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weiss M. J., dos Santos C. O. (2009) Chaperoning erythropoiesis. Blood 113, 2136–2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weiss M. J., Zhou S., Feng L., Gell D. A., Mackay J. P., Shi Y., Gow A. J. (2005) Role of α-hemoglobin-stabilizing protein in normal erythropoiesis and β-thalassemia. Ann. N.Y. Acad. Sci. 1054, 103–117 [DOI] [PubMed] [Google Scholar]
- 12. Gell D., Kong Y., Eaton S. A., Weiss M. J., Mackay J. P. (2002) Biophysical characterization of the α-globin binding protein α-hemoglobin stabilizing protein. J. Biol. Chem. 277, 40602–40609 [DOI] [PubMed] [Google Scholar]
- 13. Kihm A. J., Kong Y., Hong W., Russell J. E., Rouda S., Adachi K., Simon M. C., Blobel G. A., Weiss M. J. (2002) An abundant erythroid protein that stabilizes free α-haemoglobin. Nature 417, 758–763 [DOI] [PubMed] [Google Scholar]
- 14. Kong Y., Zhou S., Kihm A. J., Katein A. M., Yu X., Gell D. A., Mackay J. P., Adachi K., Foster-Brown L., Louden C. S., Gow A. J., Weiss M. J. (2004) Loss of α-hemoglobin-stabilizing protein impairs erythropoiesis and exacerbates β-thalassemia. J. Clin. Invest. 114, 1457–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Feng L., Zhou S., Gu L., Gell D. A., Mackay J. P., Weiss M. J., Gow A. J., Shi Y. (2005) Structure of oxidized α-haemoglobin bound to AHSP reveals a protective mechanism for haem. Nature 435, 697–701 [DOI] [PubMed] [Google Scholar]
- 16. Zhou S., Olson J. S., Fabian M., Weiss M. J., Gow A. J. (2006) Biochemical fates of α-hemoglobin bound to α-hemoglobin stabilizing protein. J. Biol. Chem. 281, 32611–32618 [DOI] [PubMed] [Google Scholar]
- 17. Mollan T. L., Khandros E., Weiss M. J., Olson J. S. (2012) The kinetics of α-globin binding to α-hemoglobin stabilizing protein (AHSP) indicate preferential stabilization of a hemichrome folding intermediate. J. Biol. Chem. 287, 11338–11350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hamdane D., Vasseur-Godbillon C., Baudin-Creuza V., Hoa G. H., Marden M. C. (2007) Reversible hexacoordination of α-hemoglobin stabilizing protein. α-Hemoglobin versus pressure. Evidence for protection of the α chains by their chaperone. J. Biol. Chem. 282, 6398–6404 [DOI] [PubMed] [Google Scholar]
- 19. Gray H. B., Winkler J. R. (2010) Electron flow through metalloproteins. Biochim. Biophys. Acta 1797, 1563–1572 [DOI] [PubMed] [Google Scholar]
- 20. Reeder B. J., Grey M., Silaghi-Dumitrescu R.L., Svistunenko D. A., Bülow L., Cooper C. E., Wilson M. T. (2008) Tyrosine residues as redox cofactors in human hemoglobin. J. Biol. Chem. 283, 30780–30787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wiedermann B. L., Olson J. S. (1975) Acceleration of tetramer formation by the binding of inositol hexaphosphate to hemoglobin dimers. J. Biol. Chem. 250, 5273–5275 [PubMed] [Google Scholar]
- 22. Geraci G., Parkhurst L. J., Gibson Q. H. (1969) Preparation and properties of α and β chains from human hemoglobin. J. Biol. Chem. 244, 4664–4667 [PubMed] [Google Scholar]
- 23. Banerjee R., Alpert Y., Leterrier F., Williams R. J. (1969) Visible absorption and electron spin resonance spectra of the isolated chains of human hemoglobin. Discussion of chain-mediated heme-heme interaction. Biochemistry 8, 2862–2867 [DOI] [PubMed] [Google Scholar]
- 24. Banerjee S., Jia Y., Siburt C. J., Abraham B., Wood F., Bonaventura C., Henkens R., Crumbliss A. L., Alayash A. I. (2012) Haptoglobin alters oxygenation and oxidation of hemoglobin and decreases propagation of peroxide-induced oxidative reactions. Free Radic. Biol. Med. 53, 1317–1326 [DOI] [PubMed] [Google Scholar]
- 25. Misra H. P., Fridovich I. (1972) The generation of superoxide radical during the autoxidation of hemoglobin. J. Biol. Chem. 247, 6960–6962 [PubMed] [Google Scholar]
- 26. Watkins J. A., Kawanishi S., Caughey W. S. (1985) Autooxidation reactions of hemoglobin A free from other red cell components. A minimal mechanism. Biochem. Biophys. Res. Commun. 132, 742–748 [DOI] [PubMed] [Google Scholar]
- 27. Berzofsky J. A., Peisach J., Blumberg W. E. (1971) Sulfheme proteins. I. optical and magnetic properties of sulfmyoglobin and its derivatives. J. Biol. Chem. 246, 3367–3377 [PubMed] [Google Scholar]
- 28. Silaghi-Dumitrescu R., Reeder B. J., Nicholls P., Cooper C. E., Wilson M. T. (2007) Ferryl haem protonation gates peroxidatic reactivity in globins. Biochem. J. 403, 391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taboy C. H., Bonaventura C., Crumbliss A. L. (1999) Spectroelectrochemistry of heme proteins: effects of active-site heterogeneity on Nernst plots. Bioelectrochem. Bioenerg. 48, 79–86 [DOI] [PubMed] [Google Scholar]
- 30. Faulkner K. M., Bonaventura C., Crumbliss A. L. (1995) A spectroelectrochemical method for differentiation of steric and electronic effects in hemoglobins and myoglobins. J. Biol. Chem. 270, 13604–13612 [DOI] [PubMed] [Google Scholar]
- 31. Kaim W., Klein A. (2008) Spectroelectrochemistry. RCS Publishing, Cambridge, UK [Google Scholar]
- 32. Kemmer G., Keller S. (2010) Nonlinear least-squares data fitting in Excel spreadsheets. Nat. Protoc. 5, 267–281 [DOI] [PubMed] [Google Scholar]
- 33. Brantley R. E., Jr., Smerdon S. J., Wilkinson A. J., Singleton E. W., Olson J. S. (1993) The mechanism of autooxidation of myoglobin. J. Biol. Chem. 268, 6995–7010 [PubMed] [Google Scholar]
- 34. Tsuruga M., Matsuoka A., Hachimori A., Sugawara Y., Shikama K. (1998) The molecular mechanism of autoxidation for human oxyhemoglobin. Tilting of the distal histidine causes nonequivalent oxidation in the β chain. J. Biol. Chem. 273, 8607–8615 [DOI] [PubMed] [Google Scholar]
- 35. Vasseur-Godbillon C., Hamdane D., Marden M. C., Baudin-Creuza V. (2006) High-yield expression in Escherichia coli of soluble human α-hemoglobin complexed with its molecular chaperone. Protein Eng. Des. Sel. 19, 91–97 [DOI] [PubMed] [Google Scholar]
- 36. Giulivi C., Davies K. J. (1994) Hydrogen peroxide-mediated ferrylhemoglobin generation in vitro and in red blood cells. Methods Enzymol. 231, 490–496 [DOI] [PubMed] [Google Scholar]
- 37. Svistunenko D. A., Patel R. P., Voloshchenko S. V., Wilson M. T. (1997) The globin-based free radical of ferryl hemoglobin is detected in normal human blood. J. Biol. Chem. 272, 7114–7121 [DOI] [PubMed] [Google Scholar]
- 38. Tomoda A., Sugimoto K., Suhara M., Takeshita M., Yoneyama Y. (1978) Haemichrome formation from haemoglobin subunits by hydrogen peroxide. Biochem. J. 171, 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maiorino M., Ursini F., Cadenas E. (1994) Reactivity of metmyoglobin towards phospholipid hydroperoxides. Free Radic. Biol. Med. 16, 661–667 [DOI] [PubMed] [Google Scholar]
- 40. Khandros E., Mollan T. L., Yu X., Wang X., Yao Y., D'Souza J., Gell D. A., Olson J. S., Weiss M. J. (2012) Insights into hemoglobin assembly through in vivo mutagenesis of α-hemoglobin stabilizing protein. J. Biol. Chem. 287, 11325–11337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reeder B. J., Wilson M. T. (2001) The effects of pH on the mechanism of hydrogen peroxide and lipid hydroperoxide consumption by myoglobin. A role for the protonated ferryl species. Free Radic. Biol. Med. 30, 1311–1318 [DOI] [PubMed] [Google Scholar]
- 42. Abraham E. C., Taylor J. F. (1975) Oxidation-reduction potentials of human fetal hemoglobin and γ chains. Effects of blocking sulfhydryl groups. J. Biol. Chem. 250, 3929–3935 [PubMed] [Google Scholar]
- 43. Banerjee R., Cassoly R. (1969) Preparation and properties of the isolated alpha and beta chains of human hemoglobin in the ferri state. Investigation of oxidation-reduction equilibria. J. Mol. Biol. 42, 337–349 [DOI] [PubMed] [Google Scholar]
- 44. Gell D. A., Feng L., Zhou S., Jeffrey P. D., Bendak K., Gow A., Weiss M. J., Shi Y., Mackay J. P. (2009) A cis-proline in α-hemoglobin stabilizing protein directs the structural reorganization of α-hemoglobin. J. Biol. Chem. 284, 29462–29469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Valdes R., Jr., Ackers G. K. (1978) Self-association of hemoglobin beta chains is linked to oxygenation. Proc. Natl. Acad. Sci. U.S.A. 75, 311–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsai A.-L., Berka V., Chen P. F., Palmer G. (1996) Characterization of endothelial nitric-oxide synthase and its reaction with ligand by electron paramagnetic resonance spectroscopy. J. Biol. Chem. 271, 32563–32571 [DOI] [PubMed] [Google Scholar]
- 47. Cooper C., Schaer D., Buehler P. W., Wilson M., Reeder B. J., Silkstone G., Svistunenko D., Bulow L., Alayash A. I. (2012) Haptoglobin binding stabilizes hemoglobin ferryl iron and the globin radical on tyrosine β-145. Antioxid. Redox Signal., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reeder B. J. (2010) The redox activity of hemoglobins. From physiologic functions to pathologic mechanisms. Antioxid. Redox Signal. 13, 1087–1123 [DOI] [PubMed] [Google Scholar]
- 49. Vollaard N. B., Reeder B. J., Shearman J. P., Menu P., Wilson M. T., Cooper C. E. (2005) A new sensitive assay reveals that hemoglobin is oxidatively modified in vivo. Free Radic. Biol. Med. 39, 1216–1228 [DOI] [PubMed] [Google Scholar]
- 50. Buehler P. W., Abraham B., Vallelian F., Linnemayr C., Pereira C. P., Cipollo J. F., Jia Y., Mikolajczyk M., Boretti F. S., Schoedon G., Alayash A. I., Schaer D. J. (2009) Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification. Blood 113, 2578–2586 [DOI] [PubMed] [Google Scholar]
- 51. Boretti F. S., Buehler P. W., D'Agnillo F., Kluge K., Glaus T., Butt O. I., Jia Y., Goede J., Pereira C. P., Maggiorini M., Schoedon G., Alayash A. I., Schaer D. J. (2009) Sequestration of extracellular hemoglobin within a haptoglobin complex decreases its hypertensive and oxidative effects in dogs and guinea pigs. J. Clin. Invest. 119, 2271–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Feng L., Gell D. A., Zhou S., Gu L., Kong Y., Li J., Hu M., Yan N., Lee C., Rich A. M., Armstrong R. S., Lay P. A., Gow A. J., Weiss M. J., Mackay J. P., Shi Y. (2004) Molecular mechanism of AHSP-mediated stabilization of α-hemoglobin. Cell 119, 629–640 [DOI] [PubMed] [Google Scholar]
- 53. Brittain T., Baker A. R., Butler C. S., Little R. H., Lowe D. J., Greenwood C., Watmough N. J. (1997) Reaction of variant sperm-whale myoglobins with hydrogen peroxide. The effects of mutating a histidine residue in the haem distal pocket. Biochem. J. 326, 109–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Birukou I., Schweers R. L., Olson J. S. (2010) Distal histidine stabilizes bound O2 and acts as a gate for ligand entry in both subunits of adult human hemoglobin. J. Biol. Chem. 285, 8840–8854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Park S. Y., Yokoyama T., Shibayama N., Shiro Y., Tame J. R. (2006) 1.25-Å Resolution crystal structures of human haemoglobin in the oxy, deoxy, and carbonmonoxy forms. J. Mol. Biol. 360, 690–701 [DOI] [PubMed] [Google Scholar]
- 56. Schrier S. L., Rachmilewitz E., Mohandas N. (1989) Cellular and membrane properties of α- and β-thalassemic erythrocytes are different. Implication for differences in clinical manifestations. Blood 74, 2194–2202 [PubMed] [Google Scholar]
- 57. Nasimuzzaman M., Khandros E., Wang X., Kong Y., Zhao H., Weiss D., Rivella S., Weiss M. J., Persons D. A. (2010) Analysis of α-hemoglobin stabilizing protein overexpression in murine β-thalassemia. Am. J. Hematol. 85, 820–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shaanan B. (1983) Structure of human oxyhaemoglobin at 2.1-Å resolution. J. Mol. Biol. 171, 31–59 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.