

Background: We examined FRET between Kir6.2 and SUR1 domains of KATP channels, in various combinations.
Results: FRET was detected within and between Kir6.2 subunits and between Kir6.2 and split SUR1 N-terminal constructs.
Conclusion: Kir6.2 C termini are centrally located. SUR1 domains can self-associate and are close to Kir6.2 termini in the full complex.
Significance: The results indicate domain architecture of this unique channel complex.
Keywords: ABC Transporter, Fluorescence Resonance Energy Transfer (FRET), Ion Channels, Membrane Proteins, Potassium Channels, Kir6.2, SUR1
Abstract
KATP channels link cell metabolism to excitability in many cells. They are formed as tetramers of Kir6.2 subunits, each associated with a SUR1 subunit. We used mutant GFP-based FRET to assess domain organization in channel complexes. Full-length Kir6.2 subunits were linked to YFP or cyan fluorescent protein (CFP) at N or C termini, and all such constructs, including double-tagged YFP-Kir6.2-CFP (Y6.2C), formed functional KATP channels. In intact COSm6 cells, background emission of YFP excited by 430-nm light was ∼6%, but the Y6.2C construct expressed alone exhibited an apparent FRET efficiency of ∼25%, confirmed by trypsin digestion, with or without SUR1 co-expression. Similar FRET efficiency was detected in mixtures of CFP- and YFP-tagged full-length Kir6.2 subunits and transmembrane domain only constructs, when tagged at the C termini but not at the N termini. The FRET-reported Kir6.2 tetramer domain organization was qualitatively consistent with Kir channel crystal structures: C termini and M2 domains are centrally located relative to N termini and M1 domains, respectively. Additional FRET analyses were performed on cells in which tagged full-length Kir6.2 and tagged SUR1 constructs were co-expressed. These analyses further revealed that 1) NBD1 of SUR1 is closer to the C terminus of Kir6.2 than to the N terminus; 2) the Kir6.2 cytoplasmic domain is not essential for complexation with SUR1; and 3) the N-terminal half of SUR1 can complex with itself in the absence of either the C-terminal half or Kir6.2.
Introduction
ATP-sensitive potassium (KATP) channels couple cellular metabolism to electrical activity in multiple cell types and are formed by a unique combination of four inwardly rectifying potassium channel (Kir6.2) subunits that generate the channel pore, each associated with one sulfonylurea receptor (SUR1)4 subunit to form a higher order octamer (1–3). Extensive studies have indicated that ATP can close the pore-forming Kir6.2 directly, whereas MgADP can bind to the nucleotide-binding domains (NBDs) of SUR1 to activate the channel (4–8).
The crystal structures of prokaryotic and eukaryotic Kir channels reveal a tetrameric arrangement of transmembrane domains, each formed by two α-helices (9, 10). The transmembrane ion permeation pore is constituted by the pore loop, which generates the selectivity filter, whereas the second transmembrane helix lines the inner cavity and generates the gate at the bottom of the inner cavity (11). The N termini of Kir channels consist of a helix (termed the slide helix), which lies parallel to the membrane surface and then extends outwards. The large C termini generate a series of β-sheets that form a cytoplasmic vestibule, which extends the permeation pathway into the cytoplasm (9, 10).
SUR1 is a member of the ATP-binding cassette (ABC) transporter protein family. Structurally, the 17 transmembrane regions of SUR1 can be grouped into three major transmembrane domains (TMD0, transmembrane regions 1–5; TMD1, transmembrane regions 6–11; and TMD2, transmembrane regions 12–17). TMD0 and TMD1 are connected by a long cytosolic loop (L0). TMD1 and TMD2 are connected by a nucleotide-binding domain (NBD1); a second NBD (NBD2) is at the C terminus of TMD2 (4, 7, 8, 12). Low resolution three-dimensional reconstruction of a KATP channel complex suggested a compact architecture, with the central Kir6.2 tetramer intimately surrounded by four SUR1 subunits (13). Crystal structures of isolated NBDs of related ABC proteins indicate that they form dimers (14), and an x-ray scattering study suggested that the NBDs of SUR2A can themselves form tetramers of such dimers, with a maximum dimension of ∼18 nm, reasonably consistent with packing predicted by EM observations (13, 15).
GFP can be fused to KATP channel subunits without compromising channel function; such subunits are visible in vivo within the secretory pathway of transfected cells and form functional KATP channels with characteristic properties (16–19). GFP is a 238-amino acid protein shaped as a tightly packed cylinder 48 Å high and 24 Å in diameter (20). These dimensions should not dramatically change the structure of KATP channels that incorporate GFP pairs, and in the present study, we have developed CFP- and YFP-tagged KATP channel constructs and applied FRET to reveal domain orientation within the KATP channel complex. We and others have previously found that it is possible to generate functional “split” SUR1 constructs in which two “halves” of the protein can be generated at various positions and co-expressed with Kir6.2 to generate functional KATP channels (21–23), enabling us to tag SUR1 with CFP or YFP at positions within the functional complex. Our results indicate that not only full-length Kir6.2, but also the Kir6.2 transmembrane domain alone and SUR1 N-terminal halves (nSUR1, including TMD0-L0-TMD1-NBD1) can all form complexes independent of other subunits. The data also provide a first indication of the location of SUR1 NBDs relative to the Kir6.2 channel.
EXPERIMENTAL PROCEDURES
Constructs and Nomenclature
Multiple Kir6.2 constructs (derived from mouse Kir6.2) and SUR1 constructs (derived from hamster SUR1) were generated, as detailed under “Results.” nSUR1 includes TMD0-L0-TMD1-NBD1 (residues Met-1 to Ala-1000), and cSUR1 includes TMD2-NBD2 (residues Cys-1001 to Lys-1580). The truncation point was previously found to best recapitulate full-length SUR1 function, in terms of KATP channel expression and stimulation by magnesium nucleotides (23). All of the Kir6.2 constructs were expressed in the pCMV6 vector, and all of the SUR1 constructs were in the pECE expression vector.
Cell Transfection and Culture
COSm6 cells were transiently transfected for fluorescence measurements, 86Rb+ efflux measurements, and excised patch clamp recordings. The cells were plated at a density of 0.5–2.5 × 105 cells/well (30-mm 6-well dishes) and cultured in DMEM plus 10 mm glucose, supplemented with FCS (10%). 2 μg of total cDNA/well was transfected into COSm6 cells using FuGENE 6 transfection reagent following standard protocols (Roche Applied Science). The cells were incubated for 24 h for fluorescence measurements or transferred to coverslips for patch clamp recordings after an additional 24–48 h of incubation.
Macroscopic 86Rb+ Flux Experiments
The cells were incubated for 12–24 h in culture medium containing 86RbCl (1 μCi/ml) before measurements. For 86Rb+ efflux measurements, the cells were incubated for 30 min at 25 °C in Ringer's solution: 118 mm NaCl, 25 mm NaHCO3, 10 mm HEPES, 4.7 mm KCl, 2.5 mm CaCl2, 1.2 mm KH2PO4, and 1.2 mm MgSO4, pH 7.4, with or without metabolic inhibitors (2.5 μg/ml oligomycin plus 1 mm 2-deoxy-d-glucose). At selected time points, the solution was aspirated and replaced with fresh solution; after completion of the assay, the cells were lysed with 1% SDS and aspirated. The 86Rb+ in the aspirated solution was counted in a liquid scintillation analyzer (1600TR, Packard).
Electrophysiological Experiments
Patch clamp experiments in the inside-out, excised patch configuration were performed at room temperature, in a perfusion chamber which allowed the solution bathing the exposed (intracellular) surface of the excised patch to be changed in less than 50 ms. Micropipettes were pulled from thin-walled glass (WPI Inc., New Haven, CT) on a horizontal puller (Sutter Instrument Co., Novato, CA). Membrane patches were voltage-clamped with an Axopatch 1B amplifier (Axon Inc., Foster City, CA). The data were filtered at 0.5–3 kHz, and signals were digitized at 22 kHz (Neurocorder; Neurodata, New York, NY) and stored on video tape. Off-line data analysis was performed using ClampFit (Axon Inc.) and Microsoft Excel. The standard bath (intracellular) and pipette (extracellular) solution (Kint) had the following composition: 140 mm KCl, 10 mm K-HEPES, 1 mm K-EGTA, pH 7.3, with additions as described. Wherever possible, the data are presented as the means ± S.E. Microsoft Solver was used to fit data by a least square algorithm.
Fluorescence Measurements
Transiently transfected COSm6 cells were collected from two 3-cm Petri dishes and resuspended in 3 ml of Kint. Fluorescence from intact cells or broken cells (below) was measured in a stirred cuvette in a FluoroMax fluorometer. CFP and YFP were excited at 430 and 480 nm, respectively, and emission scans were performed. Excitation and emission slit widths were 5 nm, and fluorescence data points were acquired in 1-nm wavelength increments. For trypsin treatment, the same amount of cells was mechanically disrupted on ice in a ball-bearing homogenizer. Nuclei and organelles were separated by centrifugation, and the cytoplasmic/microsomal fraction was analyzed for fluorescence as above. Trypsin was added to a final concentration of 0.1 mg/ml, and the cell homogenate was incubated at room temperature. Spectroscopic scans were transferred to Microsoft Excel for analysis. Specific FP-associated fluorescence was obtained by subtraction of non-FP fluorescence from the same volume of nontransfected control cells (see Fig. 3, A and B). In FRET experiments, YFP (525 nm) emission excited by optimal 480-nm excitation was obtained directly from this subtracted spectrum. To obtain YFP (525 nm) emission excited by 430 nm, pure CFP spectrum (obtained from Kir6.2-CFP-only expressing cells under identical experimental conditions) was scaled to provide an optimum least squares fit to the FRET spectrum over the region 450–500 nm and then subtracted from the whole trace (see Fig. 3C). The apparent FRET efficiencies (Eapp) were then calculated as the ratio of YFP (525 nm) emission excited by 430 nm to maximum YFP emission excited by 480 nm (Equation 1),
 |
where F430(525) and F480(525) are the emission of transfected cells at 525 nm, excited at 430 and 480 nm, respectively, F430(525)CFP is the emission of cells transfected with Kir6.2-CFP only.
FIGURE 3.

Calculation of apparent FRET efficiency for CFP/YFP-tagged constructs. A, emission spectra of untransfected cells (dashed lines), 6.2C:6.2Y (1:1) co-transfected cells (thin solid lines), excited at 430 (blue) or 480 nm (yellow), and specific FP-associated fluorescence obtained by subtraction of non-FP fluorescence (thick solid lines). B, specific FP fluorescence of 6.2C (blue) or 6.2Y (yellow) excited at 430 nm (dashed) or 480 nm (solid). 525-nm 6.2Y fluorescence excited by 430 nm (Eapp) was ∼6% of that excited by 480 nm. C, specific FP fluorescence of co-expressed 6.2C:6.2Y (1:1) excited at 430 nm (blue) or 480 nm (yellow), scaled 6.2C fluorescence excited at 430 nm (see text), and subtracted 6.2Y-specific emission. 525 nm 6.2Y fluorescence excited by 430 nm (Eapp) was ∼22% of that excited by 480 nm.
FRET Measurements Using Spectral Imaging and Linear Unmixing
Transiently transfected COSm6 cells were plated on Petri dishes with bottom coverslips (MatTeK Corp.). Image-based FRET examination was performed on a Zeiss LSM510 META NLO multiphoton imaging system. Pure CFP and YFP emission spectra were obtained from COSm6 cells transfected with plasmids carrying only CFP (6.2C/nSUR1/cSUR1) or YFP (6.2/nSUR1Y/cSUR1) fusion tags, respectively. DMEM was replaced by 1× PBS immediately before imaging to reduce background fluorescence. CFP and YFP emission spectra were acquired with beam splitter HFT-KP650, with excitation wavelengths of 820 and 900 nm, respectively. For FRET examination, COSm6 cells were excited by 800 nm, and the CFP emission and sensitized YFP emission caused by FRET were separated by spectral imaging and linear unmixing of the CFP and YFP spectra (emission fingerprinting tool; Zeiss LSM software). Corresponding YFP images were acquired on the same region using 488-nm laser excitation, beam splitter HFT488, and emission filter LP505. All COSm6 cells transfected with different sets of plasmids were imaged under identical conditions.
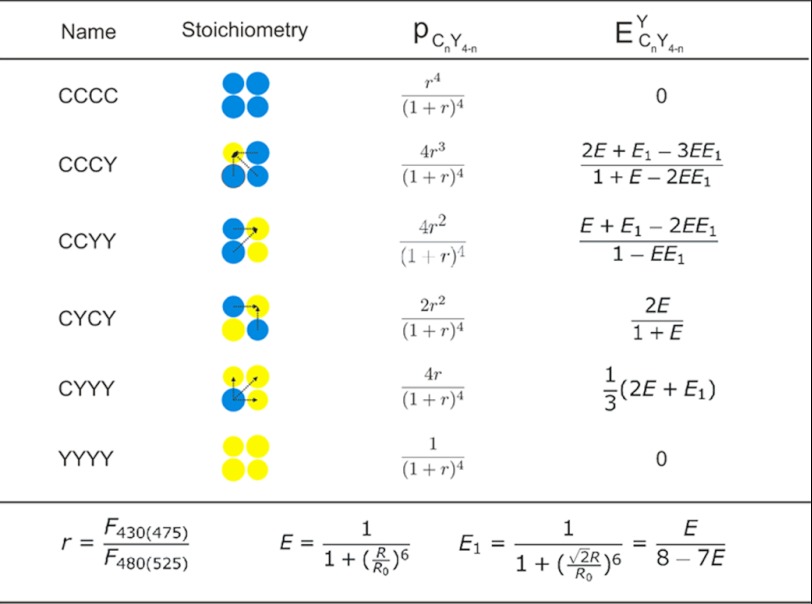
Tetrameric FRET Model
Because Kir6.2 subunits form tetrameric channels, multiple fluorophores may be brought within the FRET distance, and the observed FRET efficiency will differ from that of a single donor-to-acceptor pair. We addressed this issue using a mathematical model developed by Cheng et al. (24) to describe the dependence of FRET on the channel subunit stoichiometry. The model is based on the specific assumptions that 1) CFP/YFP-tagged subunits are all located in tetramers; 2) tagged subunits co-assemble randomly into tetramers according to a binomial distribution; 3) the tetramer is 4-fold symmetric in cross-section; and 4) FRET coupling between the possible pathways within the tetramer is independent and additive.
The first three assumptions are reasonable given that YFP-tagged versus CFP-tagged constructs are identical except for a couple of fluorophore amino acids and that unassembled subunits are likely degraded, although we cannot formally exclude the possibility that differential expression and subunit compositions may be present in different membrane compartments. The fourth assumption is valid only under low excitation light conditions, when the fraction of time that a fluorophore spends in the excited state is low (24). Under our experimental conditions, increasing excitation light intensity resulted in proportional increase in fluorescence emission intensity (not shown), suggesting that this condition is satisfied.
Based on the tetrameric FRET model, the apparent FRET efficiency (Eapp) is the average of the FRET efficiencies from different Kir6.2-C/YFP configurations and can be calculated from the following equation,
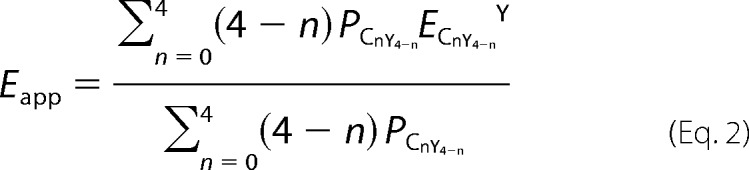 |
with parameters as defined in detail in Table 1. The FRET efficiencies between single CFP and YFP FRET pair (E) fused at adjacent Kir6.2 N or C termini can be determined by fitting Equation 2 to the apparent FRET efficiencies obtained using Equation 1 with varying ratios (r) of CFP/YFP containing subunits. Model fitting in Microsoft Excel was performed using the minimum chi-squared method.
TABLE 1.
Kir6.2-FP fusion protein stoichiometry and tetrameric FRET model
Where r = ratio of CFP to YFP containing subunits in transfected Cosm6 cells, calculated as the ratio of emission at 475 nm (excited at 430 nm, F430(475)) to the emission at 525 nm (excited at 480 nm, F480(525)); CnY4-n is the specific configuration of Kir6.2-CFP/Kir6.2YFP tetramers containing n CFP and 4-n YFP fluorophores; R is the distance between CFP and YFP fused at adjacent subunits of Kir6.2; R0 is the Förster distance of the CFP/YFP FRET pair, which is 30 Å with an orientation factor of 2/3; pCnY4-n is the probability of Kir6.2-C/YFP mixtures forming configurations that contain n CFP and 4-n YFP; ECnY4-nY is the effective FRET efficiencies of Kir6.2-C/Kir6.2YFP tetramers containing n CFP and 4-n YFP, determined by the YFP sensitized emission.
RESULTS
Fluorescent Protein-tagged Fusion Proteins Generate Functional KATP Channels
The tagged constructs used in these experiments are listed graphically in Fig. 1. All full-length Kir6.2 channel constructs were shown to be functional by 86Rb+ flux or patch clamp. Example data only are shown in Fig. 2 for full-length Kir6.2 constructs, tagged at the N terminus (C6.2) or C terminus (6.2C) with CFP, or tagged at each terminus with CFP/YFP pairs (C6.2Y or Y6.2C) and co-expressed with SUR1 in COSm6 cells. 86Rb+ efflux from metabolically inhibited cells (Fig. 2A) indicates that functional KATP channels were generated in each case, as shown previously for Kir6.2 with GFP at the N or C terminus (16, 17, 19). We systematically examined the ATP sensitivity of channels formed from one double fusion construct (Y6.2C) co-expressed with SUR1 using patch clamp measurements on inside-out membrane patches (Fig. 2B). Although channels were still sensitive to ATP, the double fusion channels were ∼30-fold less sensitive to inhibition by ATP than wild type (Fig. 2C). Similar shifts in ATP sensitivity have been reported for similarly GFP-tagged Kir6.2 constructs (1, 3, 16). We have not examined the underlying mechanism in detail, but this effect probably results from a relative stabilization of the open channel (25).
FIGURE 1.

Constructs used in the present work and their corresponding nomenclatures. Full-length and partial Kir6.2 and split SUR1 fusion constructs are shown as linear representations at the left (dashed segments are deletions from the construct), with nomenclatures listed at the right. N′, N termini; C′, C termini; M1, transmembrane helix 1; M2, transmembrane helix 2; SF, selectivity filter; TMD0, transmembrane domain zero; TMD1, transmembrane domain 1; TMD2, transmembrane domain 2; NBD1, nucleotide-binding domain 1; NBD2 , nucleotide-binding domain 2.
FIGURE 2.
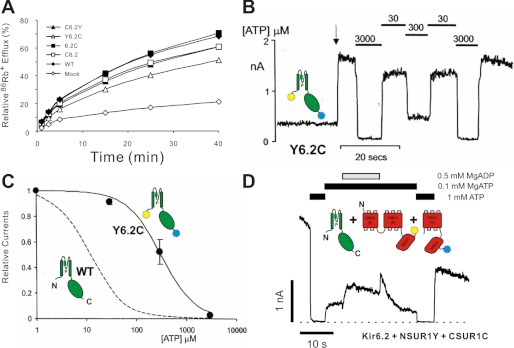
Functional assay of CFP/YFP-tagged KATP channels. A, 86Rb+ efflux from untransfected (mock) COSm6 cells and cells expressing SUR1 and Kir6.2 subunits. The graphs show percentages of 86Rb+ released into the medium as a function of time in the presence of metabolic inhibitors, for one representative experiment. B, representative currents recorded from inside-out membrane patches containing double-tagged Y6.2C co-expressed with SUR1 at −50 mV. The patch was isolated at the arrow and exposed to differing [ATP] as indicated. C, steady-state dependence of membrane current (relative to current in zero ATP) on [ATP] for wild type KATP channels and channels formed from Y6.2C + SUR1 subunits (as indicated). The data points represent the means ± S.E. (n = 3 patches). The fitted lines correspond to least squares fits of the Hill equation (relative current = 100/(1 + ([ATP]/Ki)H), where H = 1.3, and Ki = 12 μm (wild type) and 314 μm (Y6.2C + SUR1). D, representative currents recorded from inside-out membrane patches containing nSUR1Y and cSUR1C co-expressed with Kir6.2 at −50 mV. The patch was exposed to differing [nucleotide] as indicated. In this and the following figures, cartoon representations of different constructs are shown to aid interpretation.
Similarly, we examined the properties of channels formed from FP-tagged “split” SUR1 constructs: nSUR1 tagged with YFP (nSUR1Y) and cSUR1 tagged with CFP (cSUR1C), co-expressed with Kir6.2 (Fig. 2D). Again, 86Rb+ efflux from metabolically inhibited cells (data not shown) revealed that functional KATP channels are generated, and patch clamp measurements on inside-out membrane patches (Fig. 2D) indicate that Kir6.2 + split SUR1 channels are still sensitive both to ATP and, importantly, to MgADP activation, indicating the functional and structural intactness of NBDs in the split SUR1 (23).
Kir6.2 with YFP and CFP Tagged at N or C Termini Exhibits FRET
Doubly tagged YFP-Kir6.2-CFP (Y6.2C) transfectants illuminated with 430-nm light emit spectra with two prominent peaks at 475- and 525-nm maxima, corresponding to CFP and YFP emission peaks (see Fig. 4). The YFP emission of this construct with 430-nm excitation is ∼25% of the emission excited by 480-nm illumination, and this “yellow” emission is not significantly altered in co-expression with SUR1 (data not shown), consistent with earlier reports of FRET with C6.2Y constructs (17, 19). The observed FRET in expressed Y6.2C subunits could result from inter- or intrasubunit CFP-YFP interactions, because the full-length construct can assemble into a tetramer and form functional channels (Fig. 2). A series of control emission scans of Kir6.2 or isolated transmembrane domains and cytoplasmic domains fused only to YFP (see Fig. 4A), without co-expression with SUR1, showed that the intensity of YFP emission excited by 430-nm light does not exceed ∼6% of the YFP emission induced by 480 nm in each case (see Fig. 4A). This indicates that the major component of the yellow emission in the double-tagged constructs is due to FRET. To further demonstrate the FRET nature of the yellow peak in Y6.2C excited with 430-nm light, we disrupted the YFP to CFP linkage by trypsin treatment in the full-length Y6.2C construct (see Fig. 4B). Trypsin (0.1 mg/ml) is known to disrupt the functional coupling of Kir6.2 to SUR1, presumably by cleavage of KATP cytoplasmic domains (26, 27). However, GFP is resistant to trypsin at this concentration (28), and similarly, yellow emission of single YFP-tagged constructs was insensitive to short (<30 min) exposure (data not shown). In the double-tagged Y6.2C construct, the yellow peak decreased significantly, and the cyan peak increased, after trypsin cleavage (see Fig. 4B), even though the yellow emission with 480-nm excitation was unaffected by trypsin treatment (not shown).
FIGURE 4.

Kir6.2 constructs with YFP and CFP tagged at N and C termini exhibit FRET. A, apparent FRET efficiencies of singly YFP-tagged and CFP/YFP double-tagged Kir6.2 constructs. These constructs were expressed in COSm6 cells, and apparent FRET efficiencies were measured as the ratio of YFP emission (525 nm) excited by 430 nm to maximum YFP emissions (525 nm) excited by 480-nm light. All of the data are presented as the means ± S.E. with n = 6–10. B, emission spectra (Ex = 430 nm) of COSm6 cell extracts expressing Y6.2C fusion constructs before and after 0.1 mg/ml trypsin digestion. The increase in donor (CFP) emission and the decrease in acceptor (YFP) emission are marked by dashed arrows.
As discussed below, these experiments demonstrate the true FRET nature of the 430-nm excited yellow emission in the double-tagged constructs. Specific disruption of the FRET signal by trypsin should ideally provide a means of quantifying the FRET efficiency. However, it is not clear that in Fig. 4B, for instance, the disruption of FRET by trypsin is complete, because there is still a small yellow component when trypsinized spectra are compared with pure CFP emission spectra (see Fig. 4B). Furthermore, it is difficult to control the trypsin action, because prolonged exposure (>30 min) does cause gradual loss of the fluorophore emission (not shown). In the following experiments, as a practical means of quantifying the FRET signal, we therefore routinely subtracted a pure 430-nm excited cyan emission spectrum from the FRET construct emission spectrum, as shown in Fig. 3. The residual 525-nm emission excited at 430 nm is then expressed as a fraction of the 525-nm emission when excited at 480 nm. In this way, apparent FRET signals of less than 6% are dismissed as being caused by YFP emission excited directly by 430-nm light, as in Figs. 3B and 4A, and signals above this level are considered to be due to true FRET.
Certain Combinations of Co-expressed CFP- and YFP-tagged Kir6.2 Subunits Exhibit FRET
To examine energy transfer between subunits, we tested four combinations of co-expressed Kir6.2 subunits tagged with either CFP or YFP at either N or C termini. In crystal structures of eukaryotic Kir2.2 and Kir3.2 (10, 29, 30), the N termini are located at the outer surfaces of the channel, beneath the membrane, whereas the C termini form short α-helices projecting straight down into the cytoplasm. When separate subunits are tagged at the same location, variable numbers of donors (CFP) or acceptors (YFP) will be incorporated into any given Kir6.2 tetramer, and therefore multiple energy transfer pathways will exist in the complex. We assumed a FRET model based on fluorescence energy transfer rate theory with the assumption that CFP and YFP containing Kir6.2 subunits were randomly assembled into tetramers (Table 1) (24). Apparent FRET efficiencies for varying ratios of CFP- and YFP-tagged constructs were well fit with the model (Fig. 5, A and B), allowing FRET efficiency at a 1:1 fluorophore ratio to be reliably estimated. The results indicate that N-terminally CFP/YFP-tagged Kir6.2 (C6.2/Y6.2) exhibit no FRET, which may be due to their relative remoteness from one another or unfavorable orientation (Fig. 5, A and C). In contrast, tetramers generated from subunits tagged with CFP/YFP fused at C termini revealed marked FRET signals with FRET efficiencies of ∼25% (Fig. 5C).
FIGURE 5.

Inter-Kir6.2 subunit FRET efficiencies. A and B, FRET efficiency between CFP and YFP fused at N- or C-terminal of full-length Kir6.2 or transmembrane domains. Apparent FRET efficiencies obtained with different measured CFP/YFP ratios were fitted with equations derived from tetrameric FRET model based on FRET rate theory (see text); FRET efficiency between 6.2C/6.2Y, 6.2tmC/6.2tmY, and C6.2/Y6.2 (24.5, 28.9, and 0.3%), indicate close proximity of C termini, but not N termini; C6.2/6.2Y, but not Y6.2/6.2C indicates proximity of C and N termini, but with clear anisotropy; only C-terminal CFP can transfer energy to N-terminal YFP. C, FRET efficiencies of CFP/YFP tags at N or C termini of Kir6.2 subunits, calculated using the tetrameric FRET model described by Cheng et al. (24). D, FRET efficiencies of CFP/YFP fused at N or C termini of truncated Kir6.2 transmembrane domain.
Transmembrane pore-only cation channels, in which cytoplasmic and other domains have been removed, have been generated, and have been shown to be functional for other cation channels (31, 32), although no pore-only Kir channels have been reported. Truncated Kir6.2 constructs with only transmembrane domains present (6.2tm) are nonfunctional (data not shown), but by CFP/YFP tagging these constructs, we can show that they are expressed and form complexes with apparent FRET efficiencies for combinations of N- and C-terminally tagged CFP/YFP that are very well fitted with the tetrameric FRET model (Fig. 5, A and D), indicating that these truncated subunits still assemble as tetramers. Moreover, FRET signals generated by such constructs show a qualitative pattern almost identical to that obtained with the full-length constructs (Fig. 5, compare C with D), with strong FRET between C-terminally tagged subunits, but no FRET between N-terminally tagged constructs. Interestingly, FRET between the N and C termini of both full-length Kir6.2 and 6.2tm constructs was one-directional, i.e., FRET signals were only detected in proteins with donor CFP at N termini and acceptor YFP at C termini (Fig. 5, C and D). This lack of reversibility may imply anisotropy of the fluorophores, suggesting restriction of the N-terminally tagged fluorophore mobility within the Kir6.2-SUR1 complex.
Domain Organization of the KATP-SUR1 Complex
We and others have previously demonstrated that splitting of SUR1 at the C termini of NBD1 does not abolish function (21–23) and that channel complexes formed by split SUR1 and Kir6.2 can still be activated by Mg-ADP (Fig. 2D). As with Kir6.2 constructs, YFP-only tagged nSUR1 (nSUR1Y) constructs again gave apparent FRET efficiency of ∼6% (Fig. 6). To examine SUR1 domain assembly, we co-expressed nSUR1Y + cSUR1C or nSUR1C + cSUR1Y constructs together with Kir6.2. Co-expression of nSUR1/cSUR1 FRET constructs together with untagged Kir6.2 exhibited ∼15% apparent FRET (Fig. 6), consistent with NBD1 and NBD2 forming heterodimers in vivo (15, 33).
FIGURE 6.

Apparent FRET efficiencies between tagged Kir6.2 and SUR1subunits. Apparent FRET efficiency between nSUR1Y and different tagged and untagged Kir6.2 subunits (top), between nSUR1C and nSUR1Y with additional untagged subunits (center), between Kir6.2C and full-length SUR1Y (below, from Ref. 40), and between nSUR1Y and cSUR1C (bottom). CFP- and YFP-tagged DNA constructs were expressed at 1:1 ratio in each case. All of the data are presented as the means ± S.E. with n = 3–5, except for nSUR1Y/cSUR1C mixtures (average of n = 2).
Both N- and C-terminally CFP-tagged Kir6.2 (6.2C) exhibited significant FRET when co-expressed with nSUR1Y, with higher apparent FRET efficiency for C-terminally tagged than for N-terminally tagged C6.2, which suggests that the C termini of Kir6.2 are relatively closer to NBD1 than the N termini (Fig. 6). FRET signals were also detected between nSUR1Y and C-terminally CFP-tagged Kir6.2 transmembrane domain (6.2tmC), which implies that the cytoplasmic C terminus of Kir6.2 is not necessary for Kir6.2-SUR1 complex formation (Fig. 6). Finally, significant FRET between nSUR1C + nSUR1Y indicates that nSUR1, which includes only TMD0-L0-TMD1-NBD1, can form a complex with itself, in the presence or absence of either Kir6.2 or cSUR1 (see Fig. 8).
FIGURE 8.
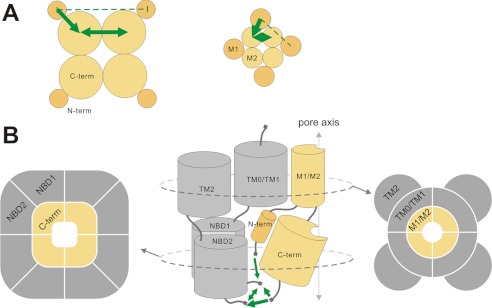
Domain organization in the KATP channel complex as reported by FRET. Domain organization in full-length Kir6.2 (left) and in transmembrane domains (M1 and M2) of Kir6.2 (right) (A) and the full KATP channel complex (B), viewed in plane of the membrane (center) or in cross-sections within the membrane (right) or within the cytoplasmic domains (left). The thickness and direction of the green arrows indicates relative FRET intensities between domains (see text). The dashed line indicates no FRET.
A potential concern for the present studies is that even though specific constructs might associate normally, differential subcellular location of expressed constructs may lead to aberrantly low or no FRET signals. This could be particularly problematical for co-expression of the dissimilar SUR1 and Kir6.2 constructs. To assess this point, we have also examined FRET between tagged Kir6.2/SUR1 constructs by cellular imaging (Fig. 7). We have utilized the emission fingerprinting imaging tool of the Zeiss LSM510 META NLO multiphoton imaging system, which can perform spectral imaging and linear unmixing simultaneously, thereby providing sensitized YFP emissions caused by FRET. Fig. 7A shows measured emission spectra from COSm6 cells transfected with only CFP (6.2C) or YFP (nSUR1Y) constructs; these spectra are quite similar to these from cuvette-based measurements (Fig. 3).
FIGURE 7.

Construct localization and FRET signals in living cells. A, CFP and YFP emission spectra acquired for fingerprinting imaging were obtained from COSm6 cells transfected with constructs containing only CFP or YFP fusion tags. B, FRET examination on living cells using fingerprinting imaging. COSm6 cells transfected with different plasmid sets were excited by 800 nm, and emission spectra from 441 to 602 nm were imaged pixel by pixel with a beam splitter HFT-KP650. The fluorescence emission from CFP (marked as CFP) and sensitized emission from YFP caused by FRET (marked as FRET) were separated by linearly unmixing using standard CFP and YFP spectra obtained in A. The YFP emission (marked as YFP) was imaged separately using a 488-nm laser. The scale bars in the middle panels are 20 μm. All of the cells were imaged and displayed under identical configurations.
In cells that express plasmid sets with either CFP or YFP fusion tags only, no sensitized YFP emissions were detected when they were excited by 800-nm laser, which indicates no leak through to the FRET channel under such excitation and imaging configurations (Fig. 7B, middle panels). In contrast, significant FRET signals were detected between CFP fused at N or C termini of Kir6.2 and YFP fused at C termini of NBD1 of SUR1. Moreover, FRET signals between 6.2C and nSUR1Y are visibly stronger than between C6.2 and nSUR1Y, consistent with our cuvette-based FRET measurements (Fig. 6). It is apparent from the raw images that although both CFP and YFP constructs show both membrane and intracellular distributions, both subunits are qualitatively co-localized and there are no obvious differences between FRET signals arising from intracellular membranes and plasma membranes, suggesting that differential subcellular localization of specific constructs is not a significant confounding issue in the cuvette-based analyses.
DISCUSSION
KATP Domain Organization
KATP channels are large eukaryotic membrane protein complexes with molecular masses close to 1 MDa, making them difficult targets for high resolution crystal structures. In 2005, a low resolution EM reconstruction provided an envelope architecture of functional Kir6.2-SUR1 complexes, but the domain organization within the complex is not determined at this low resolution (13). A more recent x-ray scattering study suggested a dimeric tetramer of NBD domains of SUR2A (15). However, without Kir6 subunits, the implications for understanding structural mechanisms of the KATP channel complex are unclear. FRET is a powerful tool for the resolution of static and dynamic structures of protein molecules. Theoretically, it can provide absolute structural constraints if relevant parameters, particularly the orientation factor of the fluorophores (κ), are precisely determined (34). We probed the domain organization of the KATP channel complex by measuring FRET between CFP/YFP fused at different termini of Kir6.2 and SUR1 subunits. For most constructs, the fluorophores were linked to Kir6.2 or SUR1 subunits at N or C termini. Assuming a κ value of ⅔ (i.e., freely mobile fluorophores) for the C termini of Kir6.2, the predicted interfluorophore distance is ∼35 Å, close to the predicted distance of 37 Å, based on the crystal structure of chicken Kir2.2 (10). However, the large size of the fluorophores, as well as unknown spatial orientation of the donor and acceptor dipoles, precludes any serious attempt to calculate absolute physical distances between fluorophores. Instead, the simple, and reasonable, assumption that bigger FRET signals reflect shorter distances between the fused fluorophores may allow the FRET efficiencies measured here to be used to indicate relative proximity of positions labeled with CFP/YFP.
Domain Orientation of KATP Channels in Intact Cells
Crystal structures of eukaryotic Kir channels show that the core of the cytoplasmic domain is formed by the C termini, which line an extension of the pore, with the N termini extending to the outer edges of the protein (10, 29, 30). CFP/YFP-tagged full-length Kir6.2 constructs generated fully functional channels (Fig. 2), indicating that the FP tagging does not disrupt the channel organization significantly, nor does it prevent subunit-subunit association. We initially validated our approach by demonstrating large, trypsin-sensitive, FRET signals with double-tagged full-length Kir6.2. Estimation of fluorophore proximity by FRET intensity provides certain constraints on domain orientation in the Kir6.2 channel tetramer and leads us to the cartoon model of domain orientation shown in Fig. 8. Strong FRET signals between Kir6.2 C termini are consistent with the C-terminal domains being centrally located, lining the extended cytoplasmic pore down the central axis, as is indeed observed in Kir channel crystal structures, and as previously shown for similar CFP/YFP-tagged Kir3 channel complexes (35, 36). Reuveny and co-workers (35, 36) detected changes of FRET associated with G-protein gating, although we did not find any measurable changes in FRET signals for tagged constructs, in response to either diazoxide- or sulfonylurea-dependent opening or closing, nor in broken cells in the presence or absence of ATP (data not shown). Available crystal structures of Kir and KirBac proteins suggest that the tetrameric cytoplasmic domain structure is unlikely to change dramatically during gating (11, 29, 30, 38). Using small molecule FRET probes on purified KirBac1.1 proteins reconstituted into liposomes to assess the gating associated conformational changes of KirBac1.1 during PIP2 gating, we have shown directly that secondary structures do undergo some motions, but they are indeed very small (41), and it seems highly unlikely that such changes will lead to measurable motions of large terminally attached FP domains.
FRET observations on tagged transmembrane domain-only constructs demonstrate essentially identical patterns of FRET (Fig. 5), implying a maintained domain organization in the truncated constructs, which is consistent with the idea that Kir tetramerization is primarily driven by the transmembrane regions rather than cytoplasmic domains (37). We detect no FRET signal between the N termini of full-length Kir6.2, which suggests that the N termini of Kir6.2 subunits may be relatively far from each other. In this regard, it is striking that we do observe FRET between the Kir6.2 N termini and nSUR1 (Fig. 6), which implies that the Kir6.2 N termini must reside close enough to NBD1, which itself is likely held in a NBD1-NBD2 heterodimer, reflected in significant FRET between tagged nSUR1 and cSUR1 constructs (Fig. 6). Based on these FRET measurements, we therefore propose that the NBD1 (and by extension, the NBD1-NBD2 dimer) is located relatively close to the N termini and even closer to the C termini of Kir6.2 subunits (Fig. 8B).
SURs have 17 transmembrane regions, consisting of two six-TM ABC transporter protein domains and an additional five-TM domain (TMD0) that is only present in a few ABC transporter subfamilies (7, 12). So far, the only high resolution structural templates for SURs are from their distant bacterial ABC transporter relatives, such as Sav1866, which function as homodimers (39). How the different structural domains of SUR are arranged with respect to each other and to Kir subunits to generate a coordinated functional complex is not known. Based on the previous reports of Mikhailov et al. (13) and Park and Terzic (15), together with our FRET observations, we propose a structural model of the Kir6-SUR complex as in Fig. 8B. The key features are: 1) the N-terminal domains of SUR1 is located closer to the central axis than the C-terminal domains, such that they are close enough to show FRET interactions with each other, as well as with the C-terminal domains; and 2) the N termini of Kir6.2 subunits interact with the SUR1 N-terminal domains, as well as with Kir6.2 C termini. Such an intimate arrangement could explain why NBD1, but not NBD2, is necessary for stabilizing the KATP channel in active conformation (23) and is consistent with the proposed model of Mikhailov et al. (13), based on low resolution electron microscopic analysis.
Acknowledgments
We are grateful to the Washington University Diabetes Research and Training Center for continued provision of molecular biology reagents and to Dr. John Cooper for the use of the fluorometer.
This work was supported, in whole or in part, by National Institutes of Health Grant HL45742 (to C. G. N.). This work was also supported by American Heart Association Postdoctoral Fellowship 10POST4280056 (to S. W.) and National Institutes of Health Neuroscience Blueprint Center Core Grant P30 NS057105 (to Washington University).
- SUR
- sulfonylurea receptor
- CFP
- cyan fluorescent protein
- NBD
- nucleotide-binding domain
- ABC
- ATP-binding cassette
- TM
- transmembrane
- TMD
- TM domain
- FP
- fluorescent protein.
REFERENCES
- 1. Clement J. P., 4th, Kunjilwar K., Gonzalez G., Schwanstecher M., Panten U., Aguilar-Bryan L., Bryan J. (1997) Association and stoichiometry of KATP channel subunits. Neuron 18, 827–838 [DOI] [PubMed] [Google Scholar]
- 2. Inagaki N., Gonoi T., Seino S. (1997) Subunit stoichiometry of the pancreatic β-cell ATP-sensitive K+ channel. FEBS Lett. 409, 232–236 [DOI] [PubMed] [Google Scholar]
- 3. Shyng S., Nichols C. G. (1997) Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 110, 655–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Babenko A. P., Aguilar-Bryan L., Bryan J. (1998) A view of sur/KIR6.X, KATP channels. Annu. Rev. Physiol. 60, 667–687 [DOI] [PubMed] [Google Scholar]
- 5. Tucker S. J., Gribble F. M., Proks P., Trapp S., Ryder T. J., Haug T., Reimann F., Ashcroft F. M. (1998) Molecular determinants of KATP channel inhibition by ATP. EMBO J. 17, 3290–3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matsuoka T., Matsushita K., Katayama Y., Fujita A., Inageda K., Tanemoto M., Inanobe A., Yamashita S., Matsuzawa Y., Kurachi Y. (2000) C-terminal tails of sulfonylurea receptors control ADP-induced activation and diazoxide modulation of ATP-sensitive K+ channels. Circ. Res. 87, 873–880 [DOI] [PubMed] [Google Scholar]
- 7. Nichols C. G. (2006) KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476 [DOI] [PubMed] [Google Scholar]
- 8. Hibino H., Inanobe A., Furutani K., Murakami S., Findlay I., Kurachi Y. (2010) Inwardly rectifying potassium channels. Their structure, function, and physiological roles. Physiol. Rev. 90, 291–366 [DOI] [PubMed] [Google Scholar]
- 9. Kuo A., Gulbis J. M., Antcliff J. F., Rahman T., Lowe E. D., Zimmer J., Cuthbertson J., Ashcroft F. M., Ezaki T., Doyle D. A. (2003) Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300, 1922–1926 [DOI] [PubMed] [Google Scholar]
- 10. Tao X., Avalos J. L., Chen J., MacKinnon R. (2009) Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science 326, 1668–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bavro V. N., De Zorzi R., Schmidt M. R., Muniz J. R., Zubcevic L., Sansom M. S., Vénien-Bryan C., Tucker S. J. (2012) Structure of a KirBac potassium channel with an open bundle crossing indicates a mechanism of channel gating. Nat. Struct. Mol. Biol. 19, 158–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aguilar-Bryan L., Nichols C. G., Wechsler S. W., Clement J. P., 4th, Boyd A. E., 3rd, González G., Herrera-Sosa H., Nguy K., Bryan J., Nelson D. A. (1995) Cloning of the beta cell high-affinity sulfonylurea receptor. A regulator of insulin secretion. Science 268, 423–426 [DOI] [PubMed] [Google Scholar]
- 13. Mikhailov M. V., Campbell J. D., de Wet H., Shimomura K., Zadek B., Collins R. F., Sansom M. S., Ford R. C., Ashcroft F. M. (2005) 3-D structural and functional characterization of the purified KATP channel complex Kir6.2-SUR1. EMBO J. 24, 4166–4175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hollenstein K., Dawson R. J., Locher K. P. (2007) Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 17, 412–418 [DOI] [PubMed] [Google Scholar]
- 15. Park S., Terzic A. (2010) Quaternary structure of KATP channel SUR2A nucleotide binding domains resolved by synchrotron radiation x-ray scattering. J. Struct. Biol. 169, 243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Makhina E. N., Nichols C. G. (1998) Independent trafficking of KATP channel subunits to the plasma membrane. J. Biol. Chem. 273, 3369–3374 [DOI] [PubMed] [Google Scholar]
- 17. Tsuboi T., Lippiat J. D., Ashcroft F. M., Rutter G. A. (2004) ATP-dependent interaction of the cytosolic domains of the inwardly rectifying K+ channel Kir6.2 revealed by fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. U.S.A. 101, 76–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. John S. A., Monck J. R., Weiss J. N., Ribalet B. (1998) The sulphonylurea receptor SUR1 regulates ATP-sensitive mouse Kir6.2 K+ channels linked to the green fluorescent protein in human embryonic kidney cells (HEK 293). J. Physiol. 510, 333–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lippiat J. D., Albinson S. L., Ashcroft F. M. (2002) Interaction of the cytosolic domains of the Kir6.2 subunit of the KATP channel is modulated by sulfonylureas. Diabetes 51, (Suppl. 3) S377–S380 [DOI] [PubMed] [Google Scholar]
- 20. Ormö M., Cubitt A. B., Kallio K., Gross L. A., Tsien R. Y., Remington S. J. (1996) Crystal structure of the Aequorea victoria green fluorescent protein. Science 273, 1392–1395 [DOI] [PubMed] [Google Scholar]
- 21. Babenko A. P., Bryan J. (2003) Sur domains that associate with and gate KATP pores define a novel gatekeeper. J. Biol. Chem. 278, 41577–41580 [DOI] [PubMed] [Google Scholar]
- 22. Chan K. W., Zhang H., Logothetis D. E. (2003) N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits. EMBO J. 22, 3833–3843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Masia R., Caputa G., Nichols C. G. (2007) Regulation of KATP channel expression and activity by the SUR1 nucleotide binding fold 1. Channels 1, 315–323 [DOI] [PubMed] [Google Scholar]
- 24. Cheng W., Yang F., Takanishi C. L., Zheng J. (2007) Thermosensitive TRPV channel subunits coassemble into heteromeric channels with intermediate conductance and gating properties. J. Gen. Physiol. 129, 191–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Enkvetchakul D., Nichols C. G. (2003) Gating mechanism of KATP channels. Function fits form. J. Gen. Physiol. 122, 471–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nichols C. G., Lopatin A. N. (1993) Trypsin and α-chymotrypsin treatment abolishes glibenclamide sensitivity of KATP channels in rat ventricular myocytes. Pflugers Arch. 422, 617–619 [DOI] [PubMed] [Google Scholar]
- 27. Proks P., Ashcroft F. M. (1993) Modification of K-ATP channels in pancreatic beta-cells by trypsin. Pflugers Arch. 424, 63–72 [DOI] [PubMed] [Google Scholar]
- 28. Mitra R. D., Silva C. M., Youvan D. C. (1996) Fluorescence resonance energy transfer between blue-emitting and red-shifted excitation derivatives of the green fluorescent protein. Gene 173, 13–17 [DOI] [PubMed] [Google Scholar]
- 29. Hansen S. B., Tao X., MacKinnon R. (2011) Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Whorton M. R., MacKinnon R. (2011) Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147, 199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen Z., Alcayaga C., Suarez-Isla B. A., O'Rourke B., Tomaselli G., Marban E. (2002) A “minimal” sodium channel construct consisting of ligated S5-P-S6 segments forms a toxin-activatable ionophore. J. Biol. Chem. 277, 24653–24658 [DOI] [PubMed] [Google Scholar]
- 32. McCusker E. C., D'Avanzo N., Nichols C. G., Wallace B. A. (2011) Simplified bacterial “pore” channel provides insight into the assembly, stability, and structure of sodium channels. J. Biol. Chem. 286, 16386–16391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Masia R., Nichols C. G. (2008) Functional clustering of mutations in the dimer interface of the nucleotide binding folds of the sulfonylurea receptor. J. Biol. Chem. 283, 30322–30329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd ed., Springer, New York [Google Scholar]
- 35. Riven I., Iwanir S., Reuveny E. (2006) GIRK channel activation involves a local rearrangement of a preformed G protein channel complex. Neuron 51, 561–573 [DOI] [PubMed] [Google Scholar]
- 36. Riven I., Kalmanzon E., Segev L., Reuveny E. (2003) Conformational rearrangements associated with the gating of the G protein-coupled potassium channel revealed by FRET microscopy. Neuron 38, 225–235 [DOI] [PubMed] [Google Scholar]
- 37. Wang S., Alimi Y., Tong A., Nichols C. G., Enkvetchakul D. (2009) Differential roles of blocking ions in KirBac1.1 tetramer stability. J. Biol. Chem. 284, 2854–2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Clarke O. B., Caputo A. T., Hill A. P., Vandenberg J. I., Smith B. J., Gulbis J. M. (2010) Domain reorientation and rotation of an intracellular assembly regulate conduction in Kir potassium channels. Cell 141, 1018–1029 [DOI] [PubMed] [Google Scholar]
- 39. Dawson R. J., Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 40. Sala-Rabanal M., Wang S., Nichols C. G. (2012) On potential interactions between non-selective cation channel TRPM4 and sulfonylurea receptor SUR1. J. Biol. Chem. 287, 8746–8756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang S., Lee S. J., Heyman S., Enkvetchakul D., Nichols C. G. (2012) Structural rearrangements underlying ligand-gating in Kir channels. Nat. Commun. 3, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]