

Abstract
Aims
Recent evidence suggests that both Ccr7 and its ligands, Ccl19 and Ccl21, are present in mouse and human atherosclerotic plaques; however, the role of Ccr7 in atherogenesis is still controversial. Here, we addressed this question by using the classic apolipoprotein E-deficient (ApoE−/−) mouse model of atherosclerosis.
Methods and results
Ccr7−/−ApoE−/− double knockout mice and Ccr7+/+ApoE−/− littermates were generated and maintained on a high-fat Western diet for 8 weeks to induce atherosclerosis. Ccr7−/−ApoE−/− mice showed an ∼80% increase in atherosclerotic lesion size in the whole aorta and a two-fold increase in the aortic root compared with Ccr7+/+ApoE−/− mice. Ccr7−/−ApoE−/− mice had increased T cells in the blood, bone marrow, and spleen, as well as in atherosclerotic lesions. Competitive repopulation experiments revealed that T cells from Ccr7−/−ApoE−/− mice migrated poorly into lymph nodes but better into mouse aortas compared with T cells from Ccr7+/+ApoE−/− mice. Transplantation of the bone marrow from Ccr7−/−ApoE−/− mice into lethally irradiated Ccr7+/+ApoE−/− mice resulted in ∼60% more atherosclerotic lesions compared with Ccr7+/+ApoE−/− donor bone marrow, suggesting that exacerbation was mediated by a Ccr7+ bone marrow-derived cell(s). Furthermore, in Ccr7−/−ApoE−/− mice the serum level of IL-12 was markedly increased, whereas the level of transforming growth factor beta (TGF-β) was significantly decreased, suggesting an imbalance of T cell responses in these mice.
Conclusion
Our data suggest that genetic deletion of Ccr7 exacerbates atherosclerosis by increasing T cell accumulation in atherosclerotic lesions.
Keywords: Atherosclerosis, Inflammation, Leucocytes, Aorta
1. Introduction
Atherosclerosis is the major cause of cardiovascular diseases and is the number one cause of mortality worldwide.1 Currently, atherosclerosis is considered as a chronic inflammatory disease, in which both innate and adaptive immunity are involved.2 The early stage of atherogenesis is characterized by endothelial injury, platelet adhesion/aggregation, and monocyte recruitment to the vessel wall.1,2 Monocytes differentiate into macrophages and become foam cells after digestion of oxidized low-density lipoprotein, secreting reactive oxygen species, lipid mediators, and various inflammatory cytokines, which attract more monocytes, as well as T cells and B cells into atherosclerotic lesions.3 Eventually, the normal blood flow is blocked by plaques formed in the intima and the rupture of unstable plaques may result in angina, myocardial infarction, stroke, and peripheral vascular disease.1,3
Recent studies suggested that the recruitment of inflammatory cells into the vessel wall is dependent on chemokines and their corresponding G protein-coupled receptors.4 For example, the migration of monocytes, neutrophils, and Th1 cells into atherosclerotic plaque is mainly mediated by the chemokine receptor subtypes Ccr2/Ccr5/Ccr6/Cx3cr1, Cxcr2/Cxcr4, and Ccr5/Cxcr3, respectively.4,5 Ccr7 is a chemokine receptor expressed on dendritic cells (DCs), naïve T and B cells, central memory T cells, regulatory T cells (Tregs), and neutrophils.6,7 Both Ccr7 and its two ligands, Ccl19 and Ccl21, have been identified in mouse and human atherosclerotic lesions,8 indicating that Ccr7 may be involved in atherosclerosis development. However, the role of Ccr7 in atherogenesis is still controversial since Ccr7 was originally suggested to be atheroprotective in a regression mouse model,9 but later studies in other mouse models gave contradictory results.10,11 Thus, additional work is needed to clarify the role of Ccr7 in atherogenesis in mouse models and in humans.
In the current study, we deleted Ccr7 in ApoE−/− mice and examined its effect on atherosclerosis in both male and female mice. Our study reveals that Ccr7 deficiency on a bone marrow-derived cell in ApoE−/− mice significantly increased atherosclerosis and promoted accumulation of T cells in atherosclerotic plaques.
2. Methods
2.1. Animals
Ccr7−/−ApoE−/− mice were obtained by crossing ApoE−/− mice and Ccr7−/− mice, which were both on a C57BL/6J background and purchased from Jackson Laboratories (Bar Harbor, ME, USA). Littermate female and male mice were fed a normal chow diet for 6 weeks and then a high-fat Western diet (TD.88137; Harlan Teklad, Madison, WI, USA) for another 8 weeks. Female mice sacrificed at 14 weeks of age were subjected to all the analyses detailed below unless specified. All mice were kept in pathogen-free conditions and all experiments were approved by the Animal Care and Use Committee of the NIAID, NIH.
2.2. Atherosclerotic lesion analysis
The atherosclerotic lesion size in whole aortas and aortic roots were analysed as previously described.5 Mice were anaesthetized with ketamine/xylazine cocktail through intraperitoneal injection (ketamine, 60 mg/kg; xylazine, 8 mg/kg) and the adequacy of anaesthesia was continually monitored by assessing reflexes and respiration, and the death was confirmed by cervical dislocation. After perfusion, mouse aortas and hearts were collected, with aortas stained by Sudan IV. Mouse hearts were snap frozen in OCT and the blocks were cut at 100 μm increments until the valves appeared. Then the sections were cut at 10 μm thickness and six consecutive sections (50 μm apart, with three leaflets of the aortic valve) were stained with Oil Red O and counterstained with haematoxylin (Histoserv, Inc., Germantown, MD, USA). Images of the entire aorta and aortic root were analysed by Image J (NIH) and IVision software (Biovision, Inc., Exton, PA, USA), respectively.
2.3. Lipid analysis
Mouse serum samples were analysed either by the EnzyChrom Kit (BioAssay Systems, Hayward, CA, USA) for total cholesterol, HDL, and LDL/VLDL levels or by the Stanbio Triglyceride LiquiColor assay (Stanbio Lab., Boerne, TX, USA) for triglyceride levels.
2.4. Immunohistochemistry
Frozen aortic root sections were stained with either Alexa Fluor 488 anti-mouse CD3 (BioLegend, San Diego, CA, USA; Cat#: 100210) or Alexa Fluor 488 anti-mouse CD11c (BioLegend, Cat#: 117311) for T cell and DC quantification. For macrophages, the sections were first stained with the primary antibody, rat anti mouse macrophages/monocytes clone MOMA-2 (MCA519G; Serotec, Raleigh, NC, USA), and then with the secondary antibody, goat anti-rat Alexa Fluor 488 (Molecular Probes, Carlsbad, CA, USA). Masson's trichrome staining was used for collagen quantification (Histoserv, Inc., Germantown, MD, USA). Images were captured by a Zeiss microscope (Jena, Germany) and analysed by the IVision software (Biovision, Inc., Exton, PA, USA).
2.5. Bone marrow transplantation
5 × 106 bone marrow cells from either Ccr7−/−ApoE−/− mice or Ccr7+/+ApoE−/− mice were transplanted to Ccr7+/+ApoE−/− mice that have been irradiated with a dose of 900 rads (all the mice are female, 8 weeks old, on Chow diet). Bone marrow cells were obtained by flushing the femurs and tibias of female donor mice, as previously described.5 The donor mice were euthanized by cervical dislocation and the collected bone marrow cells were transplanted into recipient mice by tail vein injection. Mice were maintained on a chow diet for 7 weeks to recover after bone marrow transplantation. Then they were switched to a Western diet for another 8 weeks, followed by the analysis of atherosclerotic lesions in the whole aorta.
2.6. Cell isolation and flow cytometry
Primary leucocytes were harvested from mouse aorta, peripheral blood, bone marrow, mesenteric lymph node (MLN), and spleen, as previously described.5 Briefly, mice were anaesthetized with ketamine/xylazine cocktail through i.p. injection (ketamine, 60 mg/kg; xylazine, 8 mg/kg) and the death was confirmed by cervical dislocation. Whole mouse aortas were collected and digested with Liberase TM and collagenase B (Roche Applied Science, Indianapolis, IN; Cat#: 05401119001 and 11088807001) in RPMI at 37°C for 30 min and then passed through 70 µm cell strainer (BD Biosciences, San Jose, CA, USA). Anti-coagulated peripheral blood (collected in EDTA tubes by mandibular vein puncture) and MLNs were filtered through 70 µm cell strainer and were treated with lysing buffer (BD Biosciences, San Jose, CA, USA) to remove erythrocytes. Cells were first stained with a Live/Dead marker (Invitrogen, Carlsbad, CA, USA) for 20 min and blocked with rat anti-mouse CD16/32 for 15 min. Then cells were stained 30 min at 4°C with the following mouse-specific antibodies according to the manufacturer's instructions: CD45-PE (eBioscience, Cat#: 12-0451-83), CD45-Pacific Blue (BioLegend, Cat#: 103126), CD3-FITC (BD Biosciences, Cat#: 555274), CD4-APC-Cy7 (eBioscience, Cat#: 47-0042-82), CD8-APC (eBioscience, Cat#: 17-0081-83), CD8-PE-Cy7 (eBioscience, Cat#: 25-0081-82), CD11c-APC (eBioscience, Cat#: 17-0114-82), CD19-PerCP-Cy5.5 (BioLegend, Cat#: 115534), CD25-PerCP-Cy5.5 (eBioscience, Cat#: 45-0251-82), MHCII-Pacific Blue (BioLegend, Cat#: 116422). For intracellular staining, cells were stimulated with Leukocyte Activation Cocktail (BD Biosciences, Cat#: 550583) and were fixed/permeabilized with BD cytofix/cytoperm solution (BD Biosciences, Cat#: 554722) before specific cytokine antibodies were added: IFN-γ-PE-Cy7 (eBioscience, Cat#: 25-7311-82), IL4-APC (eBioscience, Cat#: 17-7041-82), IL10-PerCP-Cy5.5 (eBioscience, Cat#: 45-7101-82), IL17A-PE (BD Biosciences, Cat#: 559502), and Foxp3-PE-Cy7 (eBioscience, Cat#: 25-5773-82). Also, corresponding isotype controls were used in both the surface staining and intracellular staining. Cells were analysed on a LSRII flow cytometer (BD Biosciences, San Jose, CA, USA) and the data were analysed with the FlowJo software (version 9.4.2; Treestar, Ashland, OR, USA).
2.7. In vivo T cell adoptive transfer
T cells were first purified from spleens of Ccr7−/−ApoE−/− mice and Ccr7+/+ApoE−/− mice by negative selection (Pan T Cell Isolation Kit, Cat#: 130-095-130; Miltenyi Biotec). Then they were labelled by CMFDA Cell Tracker Orange or CMTMR Cell Tracker Green (Invitrogen, Carlsbad, CA, USA) in a water bath for 10 min at 37°C (final concentration, 5 μM). Cells were washed two times with cold RPMI + 10% FBS + penicillin/streptomycin and resuspended in sterile PBS at a final concentration of 50 × 106 cells/mL, and mixed at a 1:1 ratio. 5 × 106 labelled cells of each population were injected intravenously into Ccr7+/+ApoE−/− mice. After 24 h, the whole aorta, blood, bone marrow, MLNs, and spleen were collected for flow cytometry analysis. In vivo T cell migration was calculated based on the ratio detected in different organs compared with the initial input ratio of labelled cells at the time of adoptive transfer after gating on CD45+ cells.
2.8. mRNA expression analysis
Mouse aorta RNA was isolated using the Trizol (Invitrogen, Carlsbad, CA, USA) and RNeasy kit (Qiagen, Valencia, CA, USA). The levels of mRNA were analysed by Real-time PCR (ABI Prism 7900HT, Applied Biosystems), using either SYBR Green or Taqman primers (Applied Biosystems, Carlsbad, CA, USA), as previously described.5 Relative gene expression changes were calculated by the ΔΔCT method.
2.9. Cytokine assays
Mouse serum was frozen at −20°C and thawed prior to measurement. The protein level of IL-4 was determined using the murine Quantikine ELISA kit (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's instructions. Other cytokines were analysed by the Luminex assay, using the Bio-Plex200 (Luminex) Instrument (Bio-Rad Laboratories, Hercules, CA, USA) and the following kits: Bio-Plex Mouse Cytokine Group I 10-plex Assay (Cat#: L60-0000CWQ) for detecting IL-1α, IL-1β, IL-5, IL-6, IL-10, IL-12 (p40), IL-12 (p70), IL-17A, IFN-γ, TNF-α; Bio-Plex Pro™ TGF-β 3-plex Assay (Cat#: 171-W4001M) for the detection of TGF-β1, TGF-β2, TGF-β3; Bio-Plex Mouse Cytokine Th17 Group III 3-plex Assay (Cat#: YJ0000000M) for detecting IL-17F, IL-21, IL-23p19 (Bio-Rad Laboratories, Hercules, CA). The luminex assay was carried out by the Protein Expression Laboratory (Rachel Bagni, SAIC-Frederick, Inc., National Cancer Institute, Frederick, MD, USA).
2.10. Statistical analysis
Results were analysed using unpaired parametric t-tests (two-tailed) with Prism 6 (GraphPad Software) and are presented as the mean ± SEM of summary data (the data were approximately normally distributed). Non-parametric Mann–Whitney tests (not shown) were also performed, and confirmed the results of parametric t-tests in all cases but two. In those two cases (Figures 1B and 3A for CD8+ cells), the results of both tests were very close to 0.05, as indicated in the figure legends. Only the results of parametric t-tests are presented, except in those two figures. The cut-off for statistical significance was defined as P < 0.05 (****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05; ns, P ≥ 0.05).
Figure 1.

Ccr7 deficiency on bone marrow cells exacerbates atherogenesis in ApoE−/− mice. (A) Representative photographs of Sudan IV-stained mouse aortic arch of Ccr7+/+ApoE−/− mice and Ccr7−/−ApoE−/− mice on a Western diet for 8 weeks (upper part), and quantification of the atherosclerotic lesions, shown as percentage of the whole aorta (lower part) (female, n = 17 mice per group, **P = 0.001). (B) Representative frozen aortic root sections stained with Oil Red O (upper part, bar = 500 μm), and quantification of aortic root lesion size (mean area) by the IVision software (female, 8 weeks on Western diet, n = 8 mice per group; *P = 0.023 by the parametric t-test and P = 0.083 by the Mann–Whitney test). (C) Quantification of atherosclerotic lesion size in the whole aorta of mice on Western diet for 8 weeks (male, n = 12 mice per group,*P = 0.0165). (D) Quantification of the atherosclerotic lesions in ApoE−/− recipient mice after either ApoE−/− or Ccr7−/−ApoE−/− bone marrow transplantation (female, n = 10 mice in each group, *P = 0.012).
Figure 3.

Ccr7 deficiency results in increased T cells in the aorta, blood, bone marrow, and spleen of ApoE−/− mice. Percentages of CD4+ T cells, CD8+ T cells, CD11c+ DCs and CD19+ B cells in the whole aorta (A), circulating blood (B), bone marrow (C), spleen (D), and mesenteric lymph nodes (E) of Ccr7+/+ApoE−/− mice and Ccr7−/−ApoE−/− mice (female, 8 weeks on Western diet) were determined by FACS analysis (each experiment was repeated two to three times; n = 3–6 mice/group in each experiment) [For the aorta CD4+ **P = 0.002 and for CD8+ *P = 0.042 (P = 0.056 by the Mann–Whitney test); for the blood CD4+ ***P = 0.0007; for the bone marrow CD4+ ****P < 0.0001, CD8+ ****P < 0.0001, and CD19+ ***P = 0.0009; for the spleen CD4+ ****P < 0.0001, CD8+ **P = 0.0025, and CD19+ **P = 0.0049; for mesenteric lymph nodes CD4+ ****P < 0.0001, CD8+ ***P = 0.0008, and CD19+ ****P < 0.0001].
3. Results
3.1. Ccr7 deficiency increases atherosclerosis in ApoE−/− mice
To clarify the role of Ccr7 in atherogenesis, we generated Ccr7−/−ApoE−/− and Ccr7+/+ApoE−/− littermates by crossing ApoE−/− mice with Ccr7−/− mice. There was no significant difference in body weight or serum levels of total cholesterol, HDL, LDL/VLDL, and triglycerides between the two groups of age- and diet- matched mice (Supplementary material online, Table S1). Both Ccr7−/−ApoE−/− mice and Ccr7+/+ApoE−/− mice had the most atherosclerotic lesions at the lesser curvature of the aortic arch and their lesions distributed similarly along the whole aorta. However, female Ccr7−/−ApoE−/− mice developed 80% more lesions than Ccr7+/+ApoE−/− mice in the whole aorta after being fed a Western diet for 8 weeks (Figure 1A), and the atherosclerotic lesion size in the aortic root of Ccr7−/−ApoE−/− mice was increased by about two times compared with Ccr7+/+ApoE−/− mice (Figure 1B). The atherosclerotic lesion size in the whole aorta of male Ccr7−/−ApoE−/− mice had also increased ∼50% in comparison with Ccr7+/+ApoE−/− mice (8 weeks on Western diet; Figure 1C), indicating that the effect of Ccr7 deficiency on atherogenesis is not sex-specific. A similar difference was also obtained using female mice stratified by the same genotypes and fed a Chow diet for 14 weeks (Supplementary material online, Figure S1). To define the underlying mechanism of how Ccr7 deficiency accelerated the progression of atherosclerosis, bone marrow cells from Ccr7+/+ApoE−/− mice and Ccr7−/−ApoE−/− mice were transplanted into irradiated age- and sex- matched Ccr7+/+ApoE−/− mice. Transplantation of Ccr7−/−ApoE−/− bone marrow resulted in a 60% increase of the atherosclerotic lesion size in the whole aorta of Ccr7+/+ApoE−/− mice compared with Ccr7+/+ApoE−/− bone marrow (Figure 1D), suggesting that the phenotype was caused by bone marrow-derived Ccr7+ cell(s).
3.2. Disruption of Ccr7 increases T cell accumulation in atherosclerotic plaques
Atherosclerotic plaque is composed of collagen, lipids, platelets, fibroblasts, smooth muscle cells, and different subsets of leucocytes, such as monocytes, DCs, and T cells.12 Masson's Trichrome staining was used to assess the collagen content in the aortic root of Ccr7−/−ApoE−/− mice and Ccr7+/+ApoE−/− mice. No difference was found between the two groups (Figure 2A), suggesting that Ccr7 deletion did not affect the stability of atherosclerotic plaques in ApoE−/− mice. Previous studies have shown that Ccr7 may affect the egress of macrophages during atherosclerosis regression9,13 and Ccr7 deficiency may increase the infiltration of DCs and T cells into atherosclerotic plaques,10 so immunofluorescence staining was performed to examine the content of macrophages, DCs, and T cells in the plaques. No difference was found for the macrophage content (Figure 2B and E) and the CD11c+ DCs content (Figure 2C) in the plaques of Ccr7−/−ApoE−/− mice and Ccr7+/+ApoE−/− mice. However, a small but significant increase in CD3+ T cells was observed in the aortic roots of Ccr7−/−ApoE−/− mice; in particular, the number of CD3+ T cells in Ccr7−/−ApoE−/− mice was increased by 30% (Figure 2D and E).
Figure 2.

Ccr7 deficiency increases the T cell content in the aortic root of ApoE−/− mice. Frozen sections from the aortic root of Ccr7+/+ApoE−/− mice and Ccr7−/−ApoE−/− mice (female, 8 weeks on Western diet) were stained with Masson's Trichrome for collagen (A) MOMA-2 for macrophages (B) CD11c for DCs (C) and CD3 for T cells (D) and the staining was quantified by digital morphometry (n = 6–9 mice per group, *P = 0.011, ns P > 0.05). (E) Representative immunofluorescence photomicrographs of the aortic root sections stained with MOMA-2 (macrophages), CD3 (T cell), DNA (DAPI), and corresponding secondary antibodies (upper, bar = 200 μm; lower, bar = 25 μm).
3.3. Ccr7 deficiency increases T cell content in the aortas of ApoE−/− mice
It has been reported that Ccr7 deficiency may affect the distribution of T cells in different organs,14 so the aortas, blood, bone marrow, spleen, and MLNs from Ccr7+/+ApoE−/− mice and Ccr7−/−ApoE−/− mice were collected and analysed for their T cell content. The percentage and absolute number of CD4+ T cells in the whole aorta, blood, bone marrow, and spleen of Ccr7−/−ApoE−/− mice was significantly increased compared with Ccr7+/+ApoE−/− mice, ranging from 50 to 400% (Figure 3A–D and data not shown), but at the same time the content of CD4+ T cells in the lymph nodes of these mice was reduced by ∼40% (Figure 3E). The distribution of CD8+ T cells showed a similar pattern as CD4+ T cells, and there was no significant alteration of CD11c+ DCs distribution in these organs (Figure 3A–E). These results are consistent with the role of Ccr7 in organ-specific distribution of T cells6,14 and indicate that the increased T cell content in atherosclerotic aortas may be caused by their increase in the blood and bone marrow of Ccr7−/−ApoE−/− mice. We also checked the content of monocytes and neutrophils in different organs from Ccr7+/+ApoE−/− mice and Ccr7−/−ApoE−/− mice, but observed no significant differences for the distribution of either CD11b+Ly6Chi monocytes, CD11b+Ly6Clow monocytes or CD11b+Ly6G+ neutrophils between the two groups of mice (Supplementary material online, Figure S2 and data not shown).
3.4. Deletion of Ccr7 increases T cell migration into the atherosclerotic vessel wall
Previous studies have shown that Ccr7 is involved in both the entry of T cells into inflammatory sites and their egress into draining lymph nodes.15–17 Here, purified splenic CD3+ T cells from female Ccr7+/+ApoE−/− mice and Ccr7−/−ApoE−/− mice were labelled with cell tracker green and cell tracker orange, and then adoptively transferred into age- and sex- matched Ccr7+/+ApoE−/− mice (Figure 4A). After 24 h, the aortas, blood, bone marrow, spleen, and MLNs were collected for flow cytometry analysis. Compared with T cells from Ccr7+/+ApoE−/− mice, T cells from Ccr7−/−ApoE−/− mice had a two- to eight-fold increased migration into the whole aorta, bone marrow, and spleen (Figure 4A and B). More Ccr7−/−ApoE−/− T cells were found to be left in the blood, and their egress to the lymph nodes was reduced by 90% compared with Ccr7+/+ApoE−/− T cells (Figure 4B), which is consistent with the role of Ccr7 in T cell homing to the lymph nodes.6,15
Figure 4.
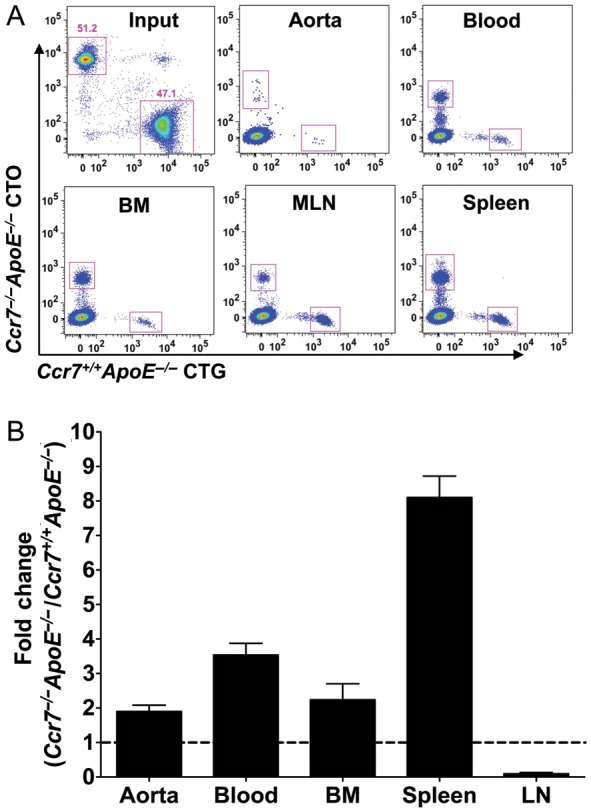
Trafficking of Ccr7−/−ApoE−/− T cells from the blood is abrogated to the lymph node but not to the aorta, spleen, or bone marrow. (A) Representative flow cytometry staining of Ccr7−/−ApoE−/− and Ccr7+/+ApoE−/− T cells. Purified spleen CD3+ T cells from female Ccr7+/+ApoE−/− mice and Ccr7−/−ApoE−/− mice were labelled with cell tracker green and cell tracker orange (input) and then were adoptively transferred into Ccr7+/+ApoE−/− mice. After 24 h, the aorta, blood, bone marrow, mesenteric lymph nodes, and spleen were collected and analysed by flow cytometry (n = 5 mice, the experiment was repeated two times). (B) Comparison of Ccr7−/−ApoE−/− and Ccr7+/+ApoE−/− T cells migrated to the aorta, blood, bone marrow, lymph node, and spleen of Ccr7+/+ApoE−/− mice after adoptive transfer, shown as fold change (Ccr7−/−ApoE−/− vs. Ccr7+/+ApoE−/−).
3.5. Ccr7 deficiency alters immunoregulatory cytokines in the blood of ApoE−/− mice
It has been reported that Ccr7 deficiency altered T cell responses in Ldlr−/− mice10 and that the lack of Ccr7 signalling favoured the development of Th2 cells in wild-type mice.18 To see how Ccr7 deficiency affects the activation of T cells in ApoE−/− mice, we measured the relevant serum cytokine levels for Th1 (IFN-γ, TNF-α, IL-12p40, IL-12p70), Th2 (IL-4, IL-5), Th17 (IL-17A, IL-17F, IL-21, IL-1α, IL-1β, IL-6), and Treg (TGFβ1-3, IL-10) cells. The levels of IL-12p40 and IL-17F were significantly increased in Ccr7−/−ApoE−/− mice, whereas the levels of IL-4 and TGF-β3 were markedly reduced (Figure 5A–D), suggesting an increased response of Th1 and Th17 cells and a diminished response of Th2 and Treg cells in those mice. However, the serum levels of IFN-γ, TNF-α, TGF-β1, TGF-β2, IL-1α, IL-1β, IL-5, IL-6, IL-10, IL-12p70, and IL-17A were similar in the two groups of mice (Supplementary material online, Figure S3A–D and data not shown). The distribution of different T cell subsets (Th1, Th2, Th17 and Treg cells) in the blood and aorta of these mice was measured by intracellular cytokine staining. In the blood, the percentages of IFN-γ+, IL4+, IL10+, IL17+ and CD25+Foxp3+ cells within the CD4+ subset were similar between the two groups of mice but the percentage of aortic CD3+IFN-γ+ T cells was significantly increased in Ccr7−/−ApoE−/− mice (Figure 6A and B), suggesting an increased Th1 response localized within the vessel wall of those mice. The RNA expression of other chemokines and chemokine receptors (Ccl1–25, Ccr1–10, Cxcl1–16, Cxcr1–7, Cx3cl1, and Cx3cr1) in the whole mouse aorta was also examined in order to further characterize the differences between Ccr7+/+ApoE−/− mice and Ccr7−/−ApoE−/− mice, but no significant difference was observed for any of these genes (Supplementary material online, Figure S4 and data not shown).
Figure 5.

Ccr7 deficiency alters immunoregulatory cytokines in the blood of ApoE−/− mice. Serum IL-12p40 (A), IL-4 (B), TGF-β3 (C), and IL-17F (D) levels were measured in Ccr7+/+ApoE−/− and Ccr7−/−ApoE−/− mice (female, 8 weeks on Western diet, n = 8–10 mice per group) by either the luminex assay or ELISA (for IL-12p40, ****P < 0.0001; for IL-4, **P = 0.0017; for TGF-β3, *P = 0.021; for IL-17F, ****P < 0.0001).
Figure 6.

Ccr7 deficiency does not affect the distribution of blood T cell subsets but increases aortic CD3+IFN-γ+ T cells in ApoE−/− mice (female, 8 weeks on Western diet, n = 4–8 mice per group). (A) Percentages of IFN-γ+, IL4+, IL10+, IL17+, and CD25+Foxp3+ cells in the blood CD4+ T cell subset of Ccr7+/+ApoE−/− and Ccr7−/−ApoE−/− mice. (B) Percentages of IFN-γ+, IL4+, IL10+, IL17+ cells in the aorta CD3+ T cell subset, determined by FACS intracellular staining (for aorta IFN-γ+ cells, **P = 0.0075).
4. Discussion
Atherosclerosis is a chronic inflammatory disease in which both innate and adaptive immunity are involved.2 Ccr7 is a chemokine receptor known to be important for the maintenance of adaptive immune system homoeostasis, e.g. it regulates the homing of T cells into lymph nodes, where activated DCs interact with T cells and enable their efficient priming.6 Recent studies suggested that Ccr7 may be an essential factor in the development of atherosclerosis since both the receptor and its ligands Ccl19, Ccl21 were shown to be expressed in atherosclerotic plaques.8 In the current study, we found that Ccr7 deficiency in ApoE−/− mice significantly increased atherosclerosis development, which was associated with selectively increased T cell accumulation in atherosclerotic plaque and blood. The phenotype is caused by bone marrow-derived cell(s) and Ccr7−/−ApoE−/− mice had increased Th1 and Th17 responses but reduced Th2 and Treg responses.
The role of Ccr7 in atherogenesis is controversial and our results provide the first detailed examination of this question in the classic ApoE deficient mouse model. In humans, originally it was reported that the expression of CCR7, CCL19 and CCL21 was increased within human atherosclerotic carotid plaques,8 but in contrast a recent report showed that the expression of CCR7, CCL19 and CCL21 was significantly down-regulated in human atherosclerotic carotid plaques compared with non-atherosclerotic controls (81, 99, and 90% lower, respectively).19 In mice, one group suggested that Ccr7 is atheroprotective in a regression model,9 whereas another group found that Ccr7 is atherogenic in the Ldlr−/− model.10 These findings suggest a complex role for Ccr7 signalling in different experiment mouse models. However, the former study was only done in a transplantation model and was not confirmed in Ccr7 knockout mice; while the latter study only used a limited number of male mice at one time point, and it is not clear whether the effect is sex-specific or time-dependent. To further clarify the role of Ccr7 in atherogenesis and to overcome these limitations, Ccr7+/+ApoE−/− and Ccr7−/−ApoE−/− mice were generated in our study by directly deleting Ccr7 in the ApoE−/− mouse model; both male and female mice were used in the study, and a large number of animals were subjected to atherosclerosis lesion analysis (n = 17 for female mice, n = 12 for male mice). We found that Ccr7 deficiency in the ApoE−/− mouse model significantly increased the size of atherosclerotic plaques in the whole aorta of both male and female mice (50 and 80%, respectively), which is comparable with the effect of Cxcr4 blockade and Ccr1 deletion in the same mouse model (60 and 100% increase, respectively).20,21 Bone marrow transplantation of Ccr7−/−ApoE−/− cells into Ccr7+/+ApoE−/− mice increased the whole aorta atherosclerotic lesion size by 60%, which is also similar to the increase caused by Ccr1−/− bone marrow transplantation into Ldlr−/− mice (70%).22 However, the effect of Ccr7 in atherogenesis seems to be independent from other chemokines or chemokine receptors since Ccr7 deficiency did not affect the expression of any other chemokines or chemokine receptors in the whole aorta of ApoE−/− mice (Supplementary material online, Figure S4 and data not shown).
Ccr7 is critical for the appropriate migration and localization of naïve T cells within secondary lymphoid organs (e.g. lymph node) and Ccr7 deficiency was found to result in delayed induction of adaptive immune responses.6,14,23 It has been reported that the number of CD4+ T cells within the MLNs of Ccr7−/− mice was reduced by ∼50–70% while at the same time there was a two- to six-fold increase of CD4+ cells in the peripheral blood, the spleen, and the bone marrow of these mice.14,24 Here, in the ApoE−/− background, we found that the content of CD4+ T cells was increased from 50% to four-fold in the blood, BM, and spleen of Ccr7−/−ApoE−/− mice but was reduced by 40% in the MLNs compared with Ccr7+/+ApoE−/− mice (Figure 3B–E). Also, our adoptive transfer experiment showed that T cells from Ccr7−/−ApoE−/− mice have increased migration ability into the mouse aorta, BM, and spleen, although their homing to the MLNs is impaired compared with T cells from Ccr7+/+ApoE−/− mice (Figure 4). More importantly, the T cell content in the atherosclerotic aortic root and whole aorta of Ccr7−/−ApoE−/− mice was also significantly increased (Figures 2D and 3A), suggesting that Ccr7 deficiency resulted in increased accumulation of T cells in atherosclerotic plaques. It is still not clear how Ccr7 deletion increased the mobilization of T cells into atherosclerotic lesions, one possibility is that this is a net effect of increased T cells in the blood and reduced T cell exit from plaque, since Ccr7 is also critical for T cell exit from peripheral tissues.16,17
In addition to controlling homoeostasis of the adaptive immune system, recent evidence suggests that Ccr7 might also affect T cell activation and polarization during chronic inflammation.18,25 We found that Ccr7 deficiency in ApoE−/− mice significantly increased the serum levels of IL-12p40 and IL-17F, but notably was associated with reduced serum levels of IL-4 and TGF-β (Figure 5), which suggested that the Th1 and Th17 responses were enhanced, whereas the Th2 and Treg responses were impaired in those mice. The former was confirmed by FACS intracellular cytokine staining since more aortic CD3+IFN-γ+ T cells were found in Ccr7−/−ApoE−/− mice than in Ccr7+/+ApoE−/− mice (Figure 6). IL-12 is critical for the differentiation of Th1 cells and it has been shown that IL-12-treated ApoE−/− mice had increased atherosclerotic lesions, whereas genetic deletion of IL-12 in ApoE−/− mice resulted in reduced lesions,26,27 indicating that IL-12 is a pathogenic factor for atherosclerosis development. IL-4 and IL-17F are the cytokines secreted by Th2 and Th17/γδ T cells, respectively, but their roles in atherogenesis are still controversial since contradictory results have been reported depending on the stage/site of the lesion and the experimental mouse models.28,29 TGF-β inhibits the differentiation of T cells towards Th1 and Th2 cells, but facilitates Treg cell differentiation.30 Both systemic neutralization and genetic deletion of TGF-β resulted in increased atherosclerotic lesion development in the ApoE−/− mice,31–33 suggesting that TGF-β plays a protective role in atherogenesis. Thus, Ccr7 deficiency in ApoE−/− mice generated a pro-atherogenic inflammatory environment (more IL-12/Th1, less TGF-β/Treg), which may together exacerbate the development of atherosclerosis.
In conclusion, genetic deletion of Ccr7 in ApoE−/− mice significantly increased atherogenesis, increased T cell levels in the circulating blood, and enhanced T cell accumulation in atherosclerotic plaques. Also, Ccr7 deficiency in ApoE−/− mice generated a pro-atherogenic inflammatory cytokine environment. However, additional work will be needed to define the mechanism of T cell priming/recruitment in the absence of Ccr7. Also, considering that Ccr7 may be expressed on DCs, B cells, and neutrophils, cell type-specific Ccr7 knockout mice are needed to test which cell type is driving the phenotype. Compared with other chemokine receptors known to regulate atherogenesis in the ApoE knockout model, Ccr7 is unusual in being protective in the model. Overall, the data suggest that activating Ccr7 pharmacologically may be beneficial in the context of cardiovascular disease.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Supplementary Material
Acknowledgments
We thank Pamela Shaw, Ph.D. for statistical advice.
Conflict of interest: none declared.
References
- 1.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. doi:10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 2.Packard RR, Lichtman AH, Libby P. Innate and adaptive immunity in atherosclerosis. Semin Immunopathol. 2009;31:5–22. doi: 10.1007/s00281-009-0153-8. doi:10.1007/s00281-009-0153-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–1422. doi: 10.1038/nm.2538. doi:10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- 4.Koenen RR, Weber C. Chemokines: established and novel targets in atherosclerosis. EMBO Mol Med. 2011;3:713–725. doi: 10.1002/emmm.201100183. doi:10.1002/emmm.201100183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wan W, Lim JK, Lionakis MS, Rivollier A, McDermott DH, Kelsall BL, et al. Genetic deletion of chemokine receptor Ccr6 decreases atherogenesis in ApoE-deficient mice. Circ Res. 2011;109:374–381. doi: 10.1161/CIRCRESAHA.111.242578. doi:10.1161/CIRCRESAHA.111.242578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Förster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol. 2008;8:362–371. doi: 10.1038/nri2297. doi:10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- 7.Beauvillain C, Cunin P, Doni A, Scotet M, Jaillon S, Loiry ML, et al. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood. 2011;117:1196–1204. doi: 10.1182/blood-2009-11-254490. doi:10.1182/blood-2009-11-254490. [DOI] [PubMed] [Google Scholar]
- 8.Damås JK, Smith C, Øie E, Fevang B, Halvorsen B, Waehre T, et al. Enhanced expression of the homeostatic chemokines CCL19 and CCL21 in clinical and experimental atherosclerosis: possible pathogenic role in plaque destabilization. Arterioscler Thromb Vasc Biol. 2007;27:614–620. doi: 10.1161/01.ATV.0000255581.38523.7c. doi:10.1161/01.ATV.0000255581.38523.7c. [DOI] [PubMed] [Google Scholar]
- 9.Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, et al. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci U S A. 2006;103:3781–3786. doi: 10.1073/pnas.0511043103. doi:10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luchtefeld M, Grothusen C, Gagalick A, Jagavelu K, Schuett H, Tietge UJ, et al. Chemokine receptor 7 knockout attenuates atherosclerotic plaque development. Circulation. 2010;122:1621–1628. doi: 10.1161/CIRCULATIONAHA.110.956730. doi:10.1161/CIRCULATIONAHA.110.956730. [DOI] [PubMed] [Google Scholar]
- 11.Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe-/- mice during disease regression. J Clin Invest. 2011;121:2025–2036. doi: 10.1172/JCI43802. doi:10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. doi:10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feig JE, Shang Y, Rotllan N, Vengrenyuk Y, Wu C, Shamir R, et al. Statins promote the regression of atherosclerosis via activation of the CCR7-dependent emigration pathway in macrophages. PLoS One. 2011;6:e28534. doi: 10.1371/journal.pone.0028534. doi:10.1371/journal.pone.0028534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. doi:10.1016/S0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 15.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. doi:10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 16.Debes GF, Arnold CN, Young AJ, Krautwald S, Lipp M, Hay JB, et al. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat Immunol. 2005;6:889–894. doi: 10.1038/ni1238. doi:10.1038/ni1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bromley SK, Thomas SY, Luster AD. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat Immunol. 2005;6:895–901. doi: 10.1038/ni1240. doi:10.1038/ni1240. [DOI] [PubMed] [Google Scholar]
- 18.Moschovakis GL, Bubke A, Dittrich-Breiholz O, Braun A, Prinz I, Kremmer E, et al. Deficient CCR7 signaling promotes TH2 polarization and B-cell activation in vivo. Eur J Immunol. 2012;42:48–57. doi: 10.1002/eji.201141753. doi:10.1002/eji.201141753. [DOI] [PubMed] [Google Scholar]
- 19.Nickel T, Pfeiler S, Summo C, Kopp R, Meimarakis G, Sicic Z, et al. oxLDL downregulates the dendritic cell homing factors CCR7 and CCL21. Mediators Inflamm. 2012;2012:320953. doi: 10.1155/2012/320953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braunersreuther V, Zernecke A, Steffens S, Shagdarsuren E, Bidzhekov K, Burger F, et al. Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2007;27:373–379. doi: 10.1161/01.ATV.0000253886.44609.ae. doi:10.1161/01.ATV.0000253886.44609.ae. [DOI] [PubMed] [Google Scholar]
- 21.Zernecke A, Bot I, Djalali-Talab Y, Shagdarsuren E, Bidzhekov K, Meiler S, et al. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res. 2008;102:209–217. doi: 10.1161/CIRCRESAHA.107.160697. doi:10.1161/CIRCRESAHA.107.160697. [DOI] [PubMed] [Google Scholar]
- 22.Potteaux S, Combadiere C, Esposito B, Casanova S, Merval R, Ardouin P, et al. Chemokine receptor CCR1 disruption in bone marrow cells enhances atherosclerotic lesion development and inflammation in mice. Mol Med. 2005;11:16–20. doi: 10.2119/2005-00028.Potteaux. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wendland M, Willenzon S, Kocks J, Davalos-Misslitz AC, Hammerschmidt SI, Schumann K, et al. Lymph node T cell homeostasis relies on steady state homing of dendritic cells. Immunity. 2011;35:945–957. doi: 10.1016/j.immuni.2011.10.017. doi:10.1016/j.immuni.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 24.Schneider MA, Meingassner JG, Lipp M, Moore HD, Rot A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J Exp Med. 2007;204:735–745. doi: 10.1084/jem.20061405. doi:10.1084/jem.20061405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moschovakis GL, Förster R. Multifaceted activities of CCR7 regulate T-cell homeostasis in health and disease. Eur J Immunol. 2012;42:1949–1955. doi: 10.1002/eji.201242614. doi:10.1002/eji.201242614. [DOI] [PubMed] [Google Scholar]
- 26.Lee TS, Yen HC, Pan CC, Chau LY. The role of interleukin 12 in the development of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:734–742. doi: 10.1161/01.atv.19.3.734. doi:10.1161/01.ATV.19.3.734. [DOI] [PubMed] [Google Scholar]
- 27.Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163:1117–1125. doi: 10.1016/S0002-9440(10)63471-2. doi:10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:969–979. doi: 10.1161/ATVBAHA.110.207415. doi:10.1161/ATVBAHA.110.207415. [DOI] [PubMed] [Google Scholar]
- 29.Butcher M, Galkina E. Current views on the functions of interleukin-17A-producing cells in atherosclerosis. Thromb Haemost. 2011;106:787–795. doi: 10.1160/TH11-05-0342. doi:10.1160/TH11-05-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall BM, Verma ND, Tran GT, Hodgkinson SJ. Distinct regulatory CD4+T cell subsets: differences between naïve and antigen specific T regulatory cells. Curr Opin Immunol. 2011;23:641–647. doi: 10.1016/j.coi.2011.07.012. doi:10.1016/j.coi.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 31.Mallat Z, Gojova A, Marchiol-Fournigault C, Esposito B, Kamaté C, Merval R, et al. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ Res. 2001;89:930–934. doi: 10.1161/hh2201.099415. doi:10.1161/hh2201.099415. [DOI] [PubMed] [Google Scholar]
- 32.Gojova A, Brun V, Esposito B, Cottrez F, Gourdy P, Ardouin P, et al. Specific abrogation of transforming growth factor-β signaling in T cells alters atherosclerotic lesion size and composition in mice. Blood. 2003;102:4052–4058. doi: 10.1182/blood-2003-05-1729. doi:10.1182/blood-2003-05-1729. [DOI] [PubMed] [Google Scholar]
- 33.Robertson AK, Rudling M, Zhou X, Gorelik L, Flavell RA, Hansson GK. Disruption of TGF-β signaling in T cells accelerates atherosclerosis. J Clin Invest. 2003;112:1342–1350. doi: 10.1172/JCI18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.